
Forward Precision Medicine: Micelles for Active Targeting Driven by Peptides


Abstract
:1. Introduction
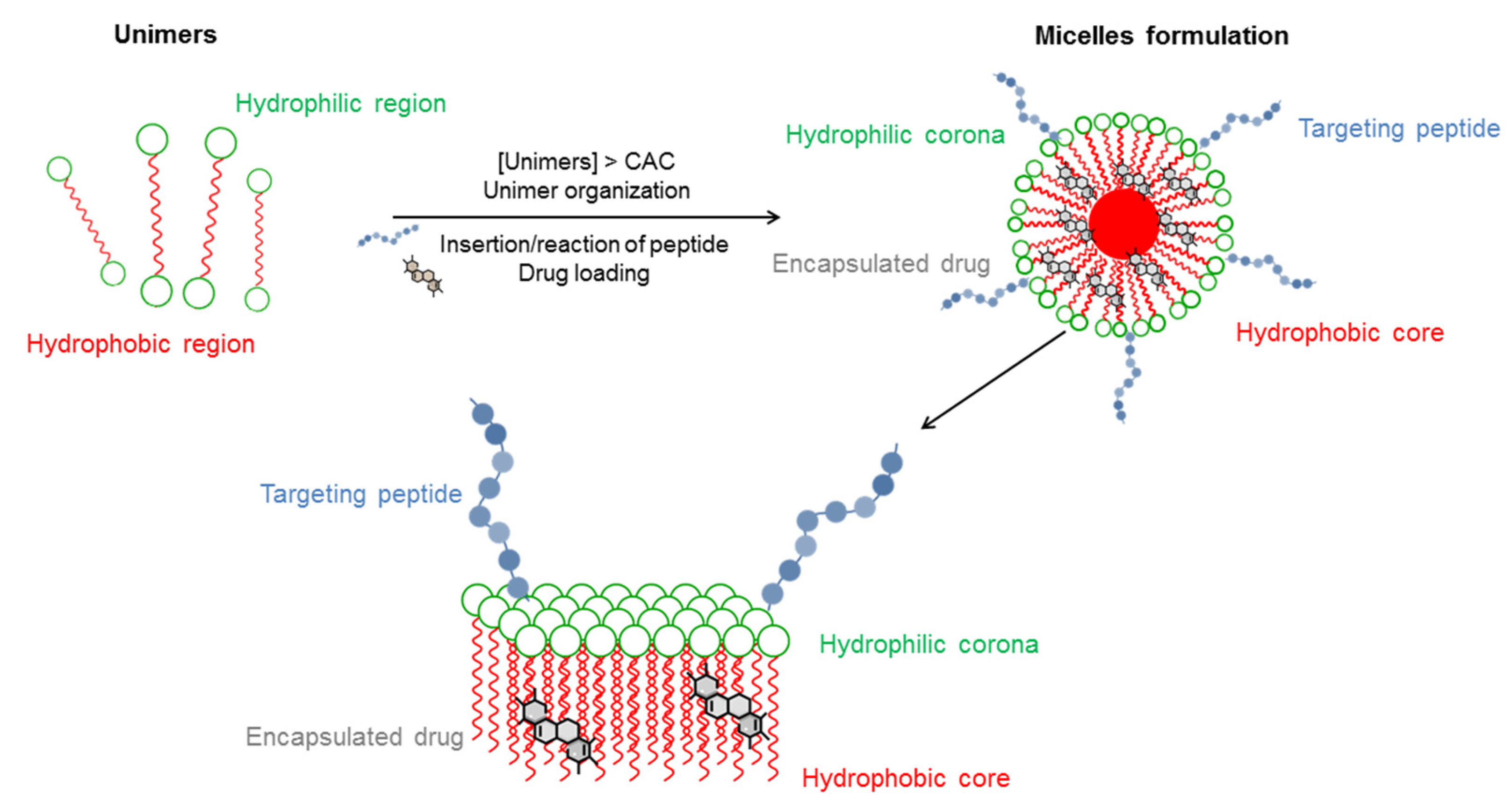
1.1. Micelle-Like Carriers
1.2. Micelle Composition
2. Passive and Active Targeting
2.1. Cell-Penetrating Peptides (CPPs)
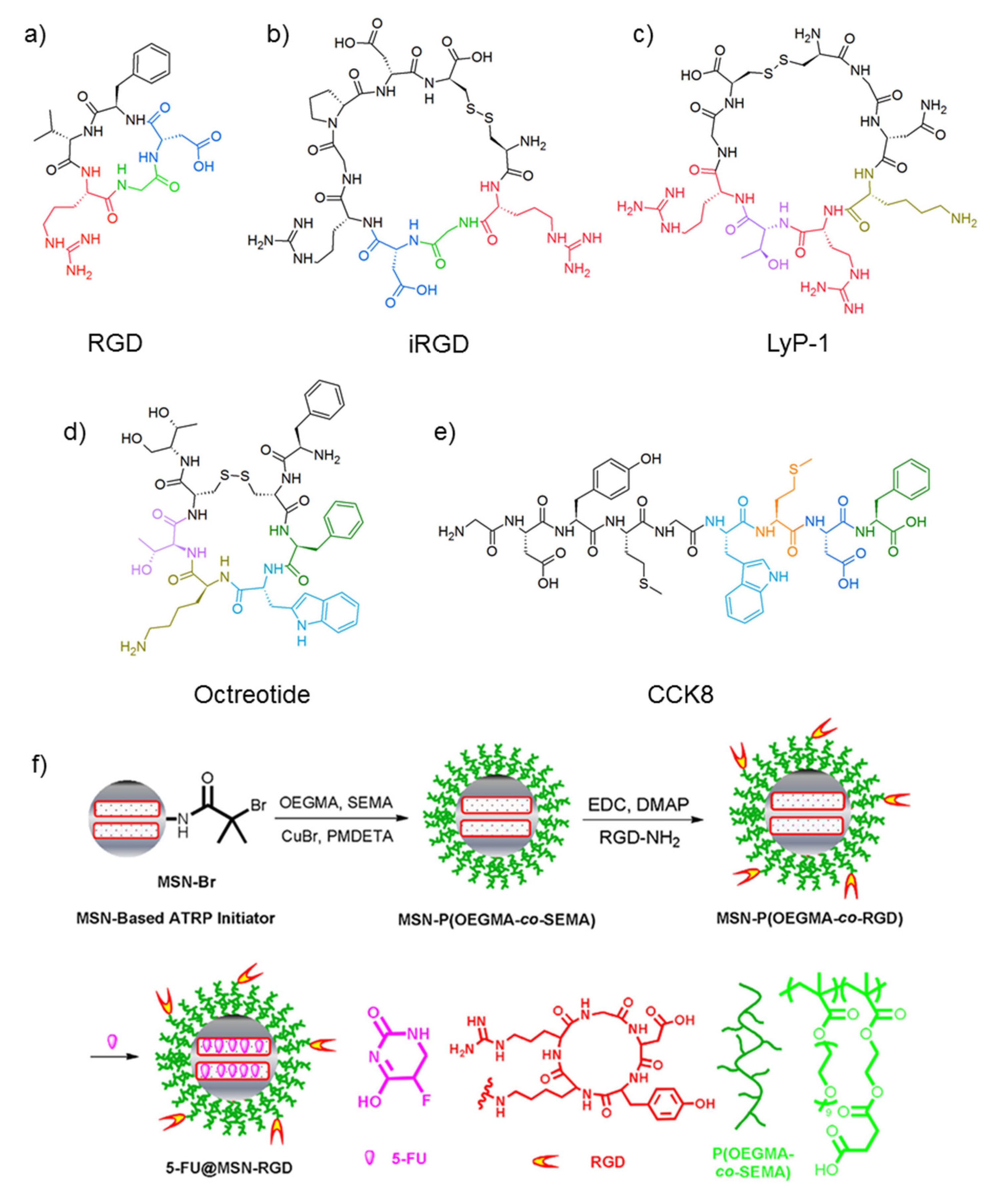
2.2. Receptor-Recognizing Peptides
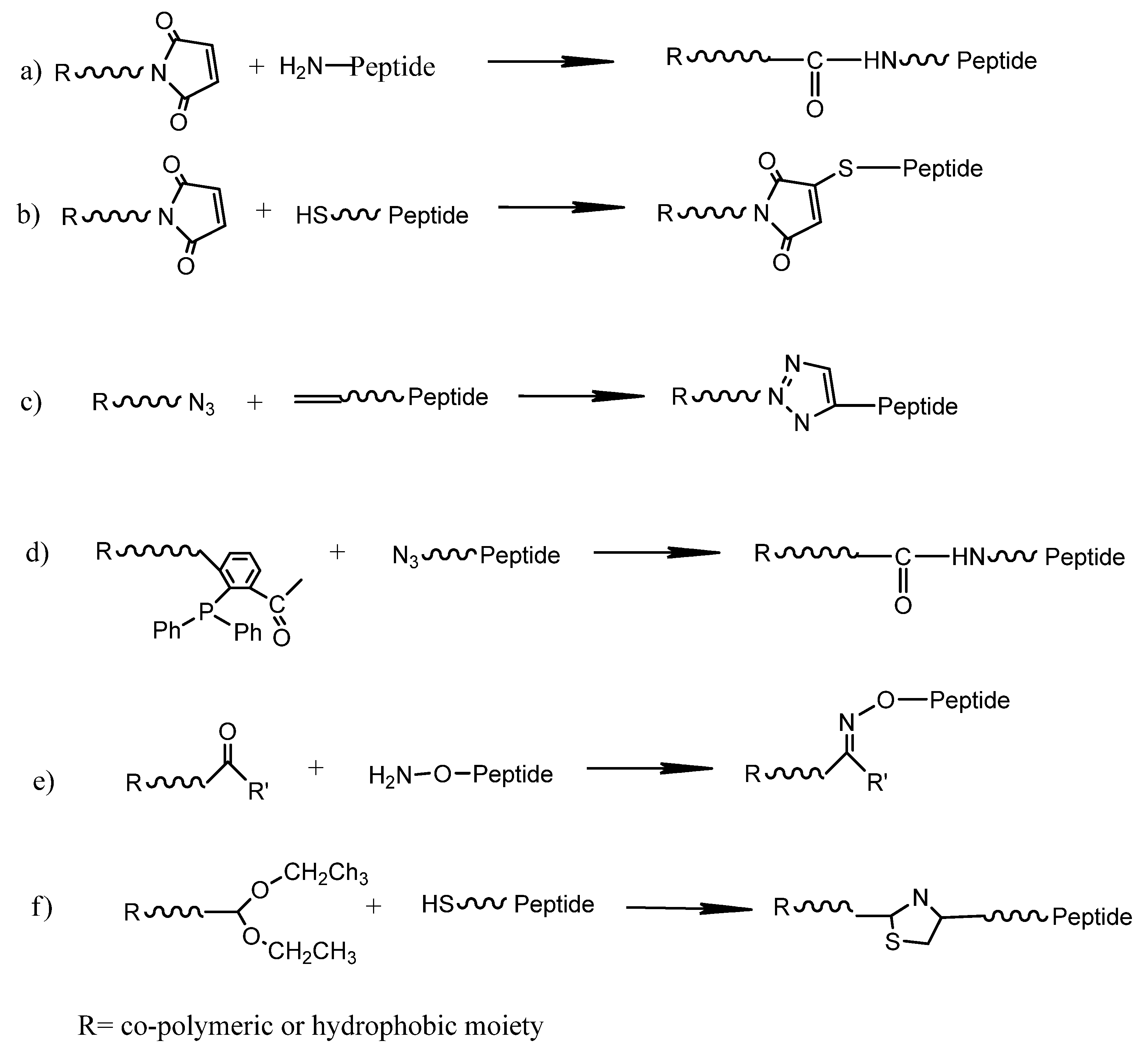
3. Peptide Amphiphilic Micelles (PAs)
4. Polymeric Micelles
4.1. PEG-PLA Micelles
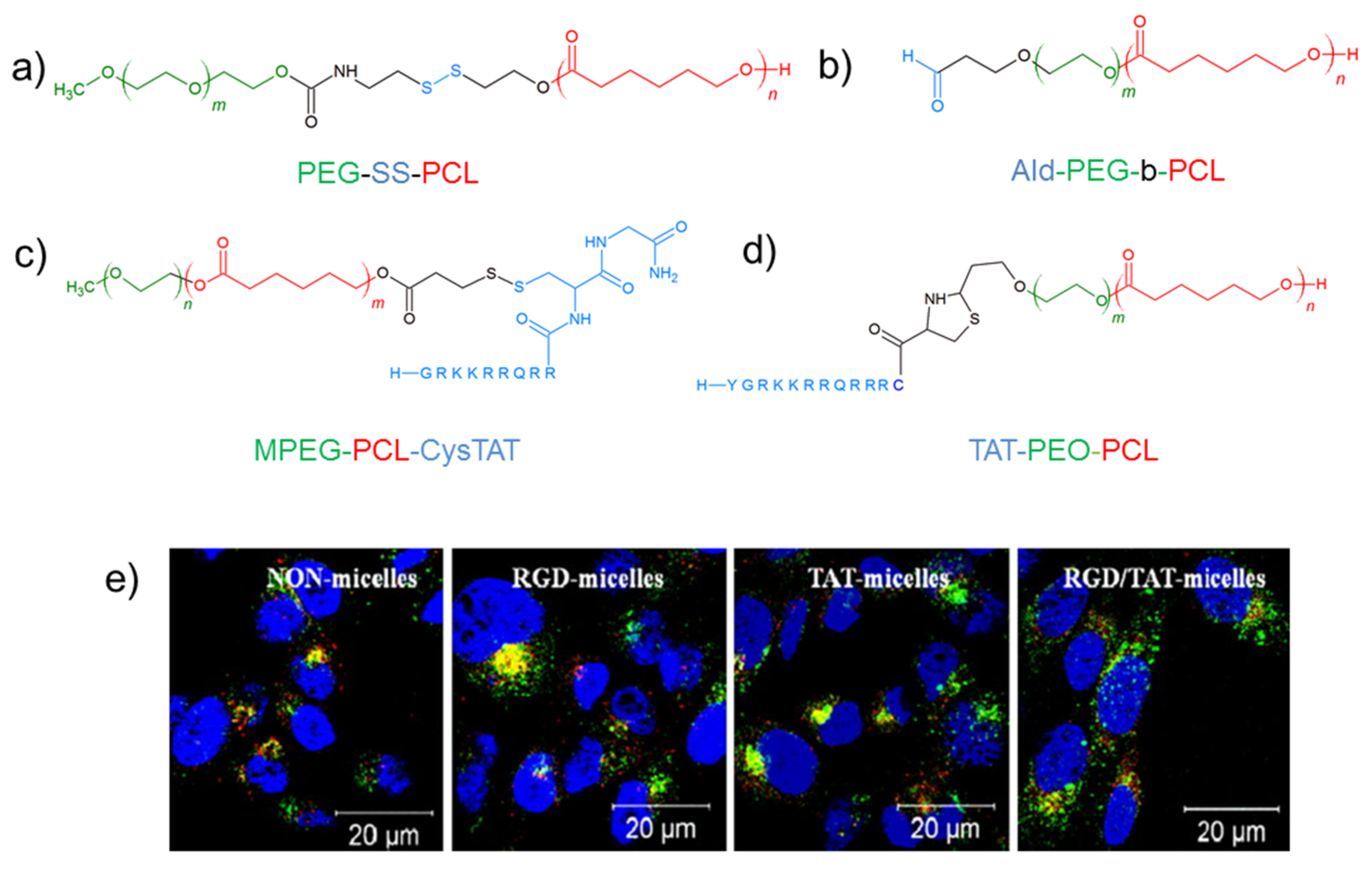
4.2. PEG-PCL
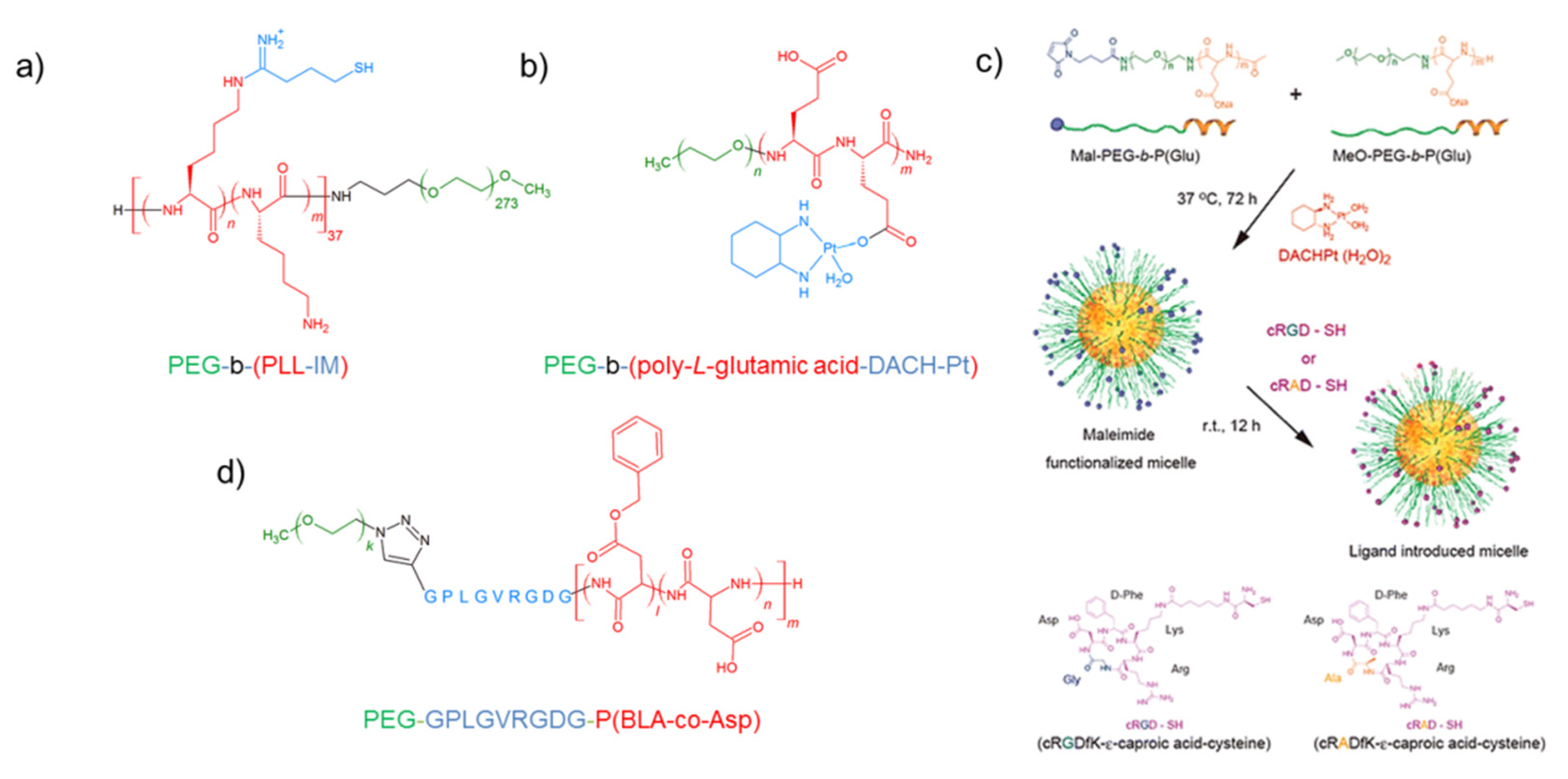
4.3. PEG-Poly(l-amino Acid)
4.4. Pluronics
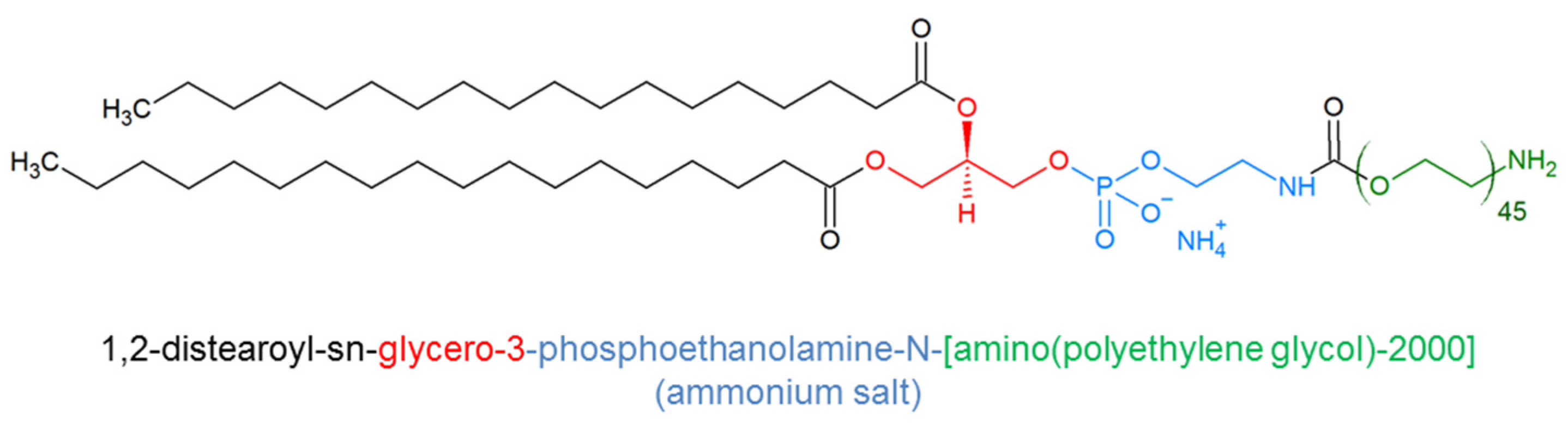
4.5. DSPE-PEG Micelles
4.6. Others Micelles
5. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Singh, M.N.; Hemant, K.S.Y.; Ram, M.; Shivakumar, H.G. Microencapsulation: A promising technique for controlled drug delivery. Res. Pharm Sci. 2010, 5, 65–77. [Google Scholar]
- Chaudhary, J.; Bower, J.; Corbin, I.R. Lipoprotein drug delivery vehicles for cancer: Rationale and reason. Int. J. Mol. Sci. 2019, 20, 6327. [Google Scholar] [CrossRef] [Green Version]
- Kazi, K.M.; Mandal, A.S.; Biswas, N.; Guha, A.; Chatterjee, S.; Behera, M.; Kuotsu, K. Niosome: A future of targeted drug delivery systems. J. Adv. Pharm. Technol. Res. 2010, 1, 374–380. [Google Scholar] [CrossRef]
- Duan, Y.; Dhar, A.; Patel, C.; Khimani, M.; Neogi, S.; Sharma, P.; Siva Kumar, N.; Vekariya, R.L. A brief review on solid lipid nanoparticles: Part and parcel of contemporary drug delivery systems. RSC Adv. 2020, 10, 26777–26791. [Google Scholar] [CrossRef]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, A.M.; Torchilin, V.P. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front. Pharmacol. 2014, 5, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haimhoffer, Á.; Rusznyák, Á.; Réti-Nagy, K.; Vasvári, G.; Váradi, J.; Vecsernyés, M.; Bácskay, I.; Fehér, P.; Ujhelyi, Z.; Fenyvesi, F. Cyclodextrins in drug delivery systems and their effects on biological barriers. Sci. Pharm. 2019, 87, 33. [Google Scholar] [CrossRef] [Green Version]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, T.; Li, Y.; Huang, G.; White, M.A.; Gao, J. Investigation of endosome and lysosome biology by ultra ph-sensitive nanoprobes. Adv. Drug. Deliv. Rev. 2017, 113, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Atanase, L.I. Micellar Drug Delivery Systems Based on Natural Biopolymers. Polymers 2021, 13, 477. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.B.; Huang, Y.; Lu, W.L.; Zhang, X.; Zhang, H.; Nagai, T.; Zhang, Q. Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J. Control. Release 2005, 107, 262–275. [Google Scholar] [CrossRef]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyer, J.; Neundorf, I. Peptide vectors for the nonviral delivery of nucleic acids. Acc. Chem. Res. 2012, 45, 1048–1056. [Google Scholar] [CrossRef]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, U. Cellpenetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Gupta, B.; Levchenko, T.S.; Torchilin, V.P. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliv. Rev. 2005, 57, 637–651. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The 3rd helix of the antennapedia homeodomain translocates through biological-membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Traub, L.M. Tickets to ride: Selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell. Biol. 2009, 10, 583–596. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Steinman, L.; Kim, D.T.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationichomopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; Mcmaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A New Potent Secondary Amphipathic Cell–penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding cell penetration of cyclic peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, A.; Tornesello, A.; Tornesello, M.; Buonaguro, F. Cell penetrating peptides as molecular carriers for anti-cancer agents. Molecules 2018, 23, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, D.; Xu, W.; Pan, R.; Ding, Y.; Sui, W.; Chen, P. Rational modification of oligoarginine for highly efficient siRNA delivery: Structure–activity relationship and mechanism of intracellular trafficking of siRNA. Nanotechnol. Biol. Med. 2015, 11, 435–446. [Google Scholar] [CrossRef]
- Verdurmen, W.P.; Brock, R. Biological responses towards cationic peptides and drug carriers. Trends Pharmacol. Sci. 2011, 32, 116–124. [Google Scholar] [CrossRef]
- Sani, S.; Messe, M.; Fuchs, Q.; Pierrevelcin, M.; Laquerriere, P.; Entz-Werle, N.; Reita, D.; Etienne-Selloum, N.; Bruban, V.; Choulier, L.; et al. Biological relevance of RGD-integrin subtype-specific ligands in cancer. ChemBioChem 2020, 22, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef]
- Schiffelers, R.M.; Koning, G.A.; ten Hagen, T.L.M.; Fens, M.H.A.M.; Schraa, A.J.; Janssen, A.P.C.A.; Kok, R.J.; Molema, G.; Storm, G. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J. Control. Release 2003, 91, 115–122. [Google Scholar] [CrossRef]
- Bibby, D.C.; Talmadge, J.E.; Dalal, M.K.; Kurz, S.G.; Chytil, K.M.; Barry, S.E.; Shand, D.G.; Steiert, M. Pharmacokinetics and biodistribution of RGD-targeted doxorubicin-loaded nanoparticles in tumor-bearing mice. Int. J. Pharm. 2005, 293, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kang, C.; Liu, F.; Zhou, Y.; Luo, L.; Qiao, H. RGD peptide-based target drug delivery of doxorubicin nanomedicine. Drug Dev. Res. 2017, 78, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hu, X.; Liu, S.; Sun, X.; Gai, X. Cyclic RGD targeting cisplatin micelles for near-infrared imaging-guided chemotherapy. RSC Adv. 2016, 6, 1151–1157. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.-C.; Sun, Q.-S.; Luo, C.-L.; Zhang, Q. RGD-based strategies for improving antitumor activity of paclitaxel-loaded liposomes in nude mice xenografted with human ovarian cancer. J. Drug Target. 2009, 17, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Su, B.; Li, W.; Ding, Y.; Tang, L.; Zhou, W.; Song, Y.; Li, H.; Zhou, C. Enhanced antitumor effect of novel dual-targeted paclitaxel liposomes. Nanotechnology 2010, 21. [Google Scholar] [CrossRef]
- Zhang, X.; He, Z.; Xiang, L.; Li, L.; Zhang, H.; Lin, F.; Cao, H. Codelivery of GRP78 siRNA and docetaxel via RGD-PEG-DSPE/DOPA/CaP nanoparticles for the treatment of castration-resistant prostate cancer. Drug Des. Dev. Ther. 2019, 13, 1357–1372. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, J.; Bian, D.; Zhang, X.; Zhang, Q. Targeted delivery of RGD-modified liposomes encapsulating both combretastatin A-4 and doxorubicin for tumor therapy: In vitro and in vivo studies. Eur. J. Pharm. Biopharm. 2010, 74, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Jia, T.T.; Huang, Q.X.; Qiu, Y.Y.; Xu, J.; Yin, P.H.; Liu, T. Mesoporous silica nanoparticles (MSNs)-based organic/inorganic hybrid nanocarriers loading 5-Fluorouracil for the treatment of colon cancer with improved anticancer efficacy. Colloids Surf. B 2017, 159, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; You, Y.; Li, X.; Liu, L.; Guo, F.; Zhang, Q.; Liu, D.; Wu, S.; Zhang, W.; He, Y.; et al. Preparation of RGD peptide/folate acid double-targeted mesoporous silica nanoparticles and its application in human breast cancer MCF-7 cells. Front. Pharmcol. 2020, 11, 00898. [Google Scholar] [CrossRef]
- Garanger, E.; Boturyn, D.; Dumy, P. Tumor targeting with RGD peptide ligands-design of new molecular conjugates for imaging and therapy of cancers. Anti-Cancer Agents Med. Chem. 2007, 7, 552–558. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [Green Version]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef]
- Roth, L.; Agemy, L.; Kotamraju, V.; Braun, G.; Teesalu, T.; Sugahara, K.N.; Hamzah, J.; Ruoslahti, E. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene 2012, 31, 3754–3763. [Google Scholar] [CrossRef] [Green Version]
- Guiellemin, R. Peptides in the brain: The new endocrinology of the neurons. Science 1978, 202, 390–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberts, S.W.J. Octreotide: The Next Decade; BioScientifica: Bristol, UK, 1999. [Google Scholar]
- Lamberts, S.W.J.; Hofland, L.J. Octreotide, 40 years later. Eur. J. End. 2019, 181, R173–R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, J.O.; Pozderac, R.V.; Hinkle, G.; Hill, T.; O’Dorisio, T.M.; Schirmer, W.J.; Ellison, E.C.; O’Dorisio, M.S. Somatostatin receptor imaging of neuroendocrine tumors with indium-111 pentetreotide (Octreoscan). Semin. Nucl. Med. 1995, 25, 251–261. [Google Scholar] [CrossRef]
- Duijzentkunst, D.A.S.; Kwekkeboom, D.J.; Bodei, L. Somatostatin receptor 2-targeting compounds. J. Nucl. Med. 2017, 58, 54S–60S. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jin, W.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. A novel octreotide modified lipid vesicle improved the anticancer efficacy of doxorubicin in somatostatin receptor 2 positive tumor models. Mol. Pharm. 2010, 7, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhou, Z.; Li, L.; Yang, Q.; Yang, Y.; Guan, S.; Zhang, J.; Zhu, X.; Jin, Y.; Huang, Y. Comparison of active and passive targeting of doxorubicin for somatostatin receptor 2 positive tumor models by octreotide-modified HPMA copolymer-doxorubicin conjugates. Drug Deliv. 2016, 23, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Aloj, L.; Aurilio, M.; Rinaldi, V.; D’Ambrosio, L.; Tesauro, D.; Peitl, P.K.; Maina, T.; Mansi, R.; von Guggenberg, E.; Joosten, L.; et al. Comparison of the binding and internalization properties of 12 DOTA-coupled and ¹¹¹In-labelled CCK2/gastrin receptor binding peptides: A collaborative project under COST ActionBM0607. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, M.; Mierke, D.F. Molecular complex of cholecystokinin-8 and N-terminus of the cholecystokinin A receptor by NMR spectroscopy. Biochemistry 1999, 38, 14775–14783. [Google Scholar] [CrossRef] [PubMed]
- Morelli, G.; De Luca, S.; Tesauro, D.; Saviano, M.; Pedone, C.; Dolmella, A.; Visentin, R.; Mazzi, U. CCK8 peptide derivatized with diphenylphosphine for rhenium labelling: Synthesis and molecular mechanics calculations. J. Pept. Sci. 2002, 8, 373–381. [Google Scholar] [CrossRef]
- Feger, G.; Angelov, B.; Angelova, A. Prediction of amphiphilic cell-penetrating peptide building blocks from protein-derived amino acid sequences for engineering of drug delivery nanoassemblies. J. Phys. Chem. B 2020, 124, 4069–4078. [Google Scholar] [CrossRef]
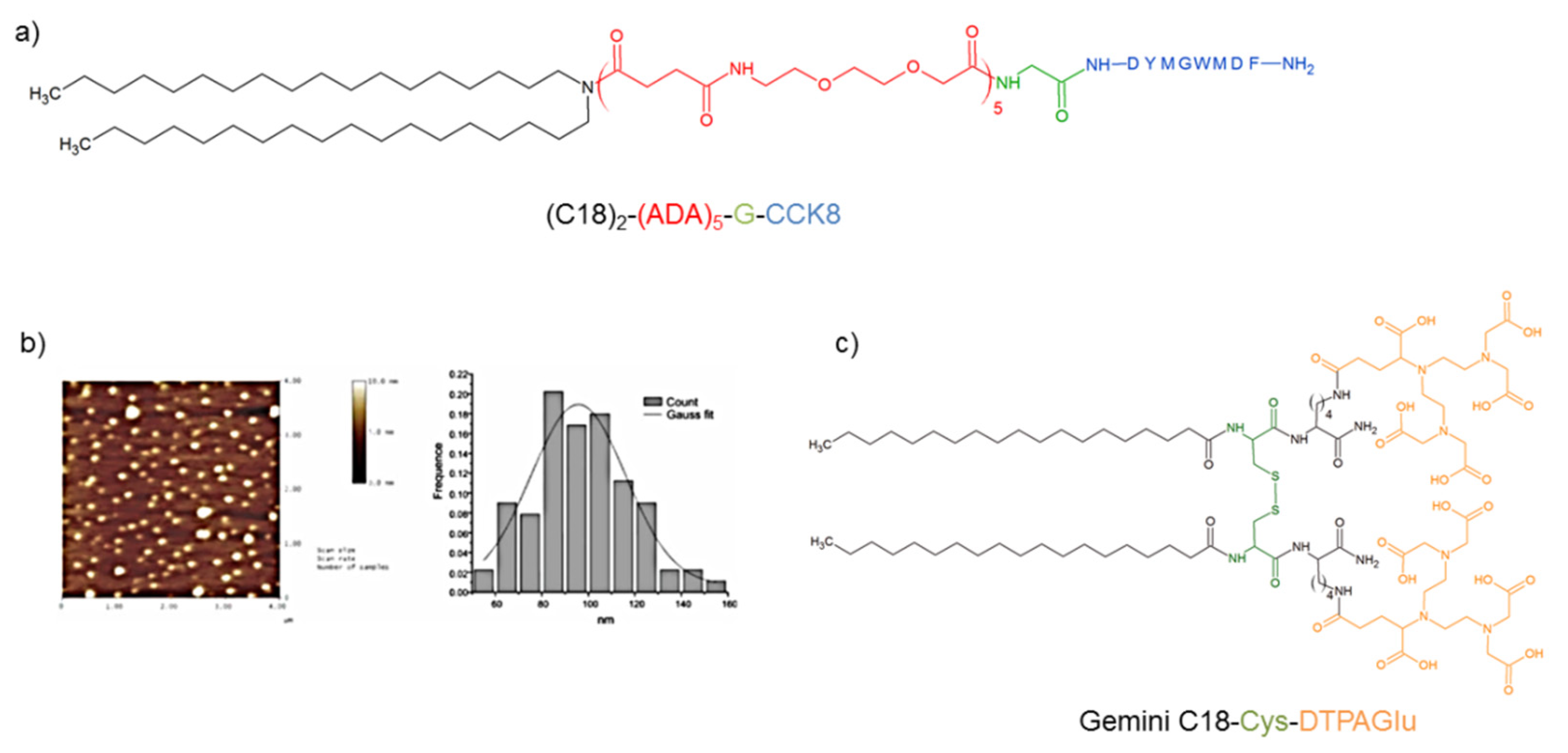
- Accardo, A.; Tesauro, D.; Mangiapia, G.; Pedone, C.; Morelli, G. Nanostructures by self-assembling peptide amphiphile as potential selective drug carriers. Pept. Sci. 2007, 88, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Tesauro, D.; Morelli, G.; Gianolio, E.; Aime, S.; Vaccaro, M.; Mangiapia, G.; Paduano, L.; Schillen, K. High-relaxivity supramolecular aggregates containing peptide and Gd complexes agents in MRI. J. Biol. Inorg. Chem. 2007, 12, 267–276. [Google Scholar] [CrossRef]
- Accardo, A.; Morisco, A.; Palladino, P.; Palumbo, R.; Tesauro, D.; Morelli, G. Amphiphilic CCK peptides assembled in supramolecular aggregates: Structural investigations and in vitro studies. Mol. Biosyst. 2011, 7, 862–870. [Google Scholar] [CrossRef]
- Accardo, A.; Tesauro, D.; Morisco, A.; Mangiapia, G.; Vaccaro, M.; Gianolio, E.; Heenan, R.K.; Paduano, L.; Morelli, G. Micelles Obtained by Aggregation of Gemini Surfactants Containing the CCK8 Peptide and a Gadolinium Complex. J. Biol. Inorg. Chem. 2009, 14, 577–589. [Google Scholar] [CrossRef]
- Morisco, A.; Accardo, A.; Gianolio, E.; Tesauro, D.; Benedetti, E.; Morelli, G. Micelles derivatized with octreotide as potential target-selective contrast agents in MRI. J. Pept. Sci. 2009, 15, 242–250. [Google Scholar] [CrossRef]
- Accardo, A.; Morisco, A.; Gianolio, E.; Tesauro, D.; Mangiapia, G.; Radulescu, A.; Brandte, A.; Morelli, G. Nanoparticles containing octreotide peptides and gadolinium complexes for MRI applications. J. Pept Sci. 2011, 16, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Javali, N.M.; Raj, J.; Saraf, P.; Li, X.; Jasti, B. Fatty Acid–RGD peptide amphiphile micelles as potential paclitaxel delivery carriers to avß3integrin overexpressing tumors. Pharm. Res. 2012, 29, 3347–3361. [Google Scholar] [CrossRef]
- Saraf, P.; Li, X.; Wrischnik, L.; Jasti, B. In vitro and in vivo efficacy of self-assembling RGD peptide amphiphiles for targeted delivery of paclitaxel. Pharm. Res. 2015, 32, 3087–3101. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wu, W.; Lai, D.; Li, J.; Fang, C. Enhanced solubility and targeted delivery of curcumin by lipopeptide micelles. J. Biomater. Sci. Polym. Ed. 2015, 26, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Xuan, M.; Liang, J.; Li, J.; Wu, W. Multi-functional lipopeptide micelles as a vehicle for curcumin delivery. Colloids Surf. A Phys. Eng. Asp. 2021, 616, 126208. [Google Scholar] [CrossRef]
- Yang, X.; Li, Z.; Wang, N.; Li, L.; Song, L.; He, T.; Sun, L.; Wang, Z.; Wu, Q.; Luo, N.; et al. Curcumin-encapsulated polymeric micelles suppress the development of colon cancer in vitro and in vivo. Sci. Rep. 2015, 5, 10322. [Google Scholar] [CrossRef] [PubMed]
- Rakotoariso, M.; Angelov, B.; Garamus, V.M.; Angelova, A. Curcumin- and fish oil-loaded spongosome and cubosome nanoparticles with neuroprotective potential against H2O2-induced oxidative stress in differentiated human SH-SY5Y cells. ACS Omega 2019, 4, 3061–3073. [Google Scholar] [CrossRef] [Green Version]
- Vaccaro, M.; Mangiapia, G.; Accardo, A.; Tesauro, D.; Gianolio, E.; Frielinghaus, H.; Morelli, G.; Paduano, L. Polymerized mixed aggregates containing gadolinium complex and CCK8 peptide. Colloid Polym. Sci. 2008, 286, 1643–1652. [Google Scholar] [CrossRef]
- Yao, D.; Li, S.; Zhu, X.; Wu, J.; Tian, H. Tumor-cell targeting polydiacetylene micelles encapsulated with an antitumor drug for the treatment of ovarian cancer. Chem. Commun. 2017, 53, 1233–1236. [Google Scholar] [CrossRef]
- Angelova, A.; Drechsler, M.; Garamus, V.M.; Angelov, B. Pep-lipid cubosomes and vesicles compartmentalized by micelles from self-assembly of multiple neuroprotective building blocks including a large peptide hormone PACAP-DHA. ChemNanoMat. 2019, 5, 1381–1389. [Google Scholar] [CrossRef]
- Riess, G. Micellization of block copolymers. Prog. Polym. Sci. 2003, 28, 1107–1170. [Google Scholar] [CrossRef] [Green Version]
- Nasongkla, N.; Bey, E.; Ren, J.; Ai, H.; Khemtong, C.; Guthi, G.S.; Chin, S.F.; Sherry, A.D.; Boothman, D.A.; Gao, J. Multifunctional polymeric micelles as cancer-targeted, MRI-ultrasensitive drug delivery systems. Nano Lett. 2006, 6, 2427–2430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, T.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. Targeted polymeric micelle system for delivery of combretastatin A4 to tumor vasculature in vitro. Pharm. Res. 2010, 27, 1861–1868. [Google Scholar] [CrossRef]
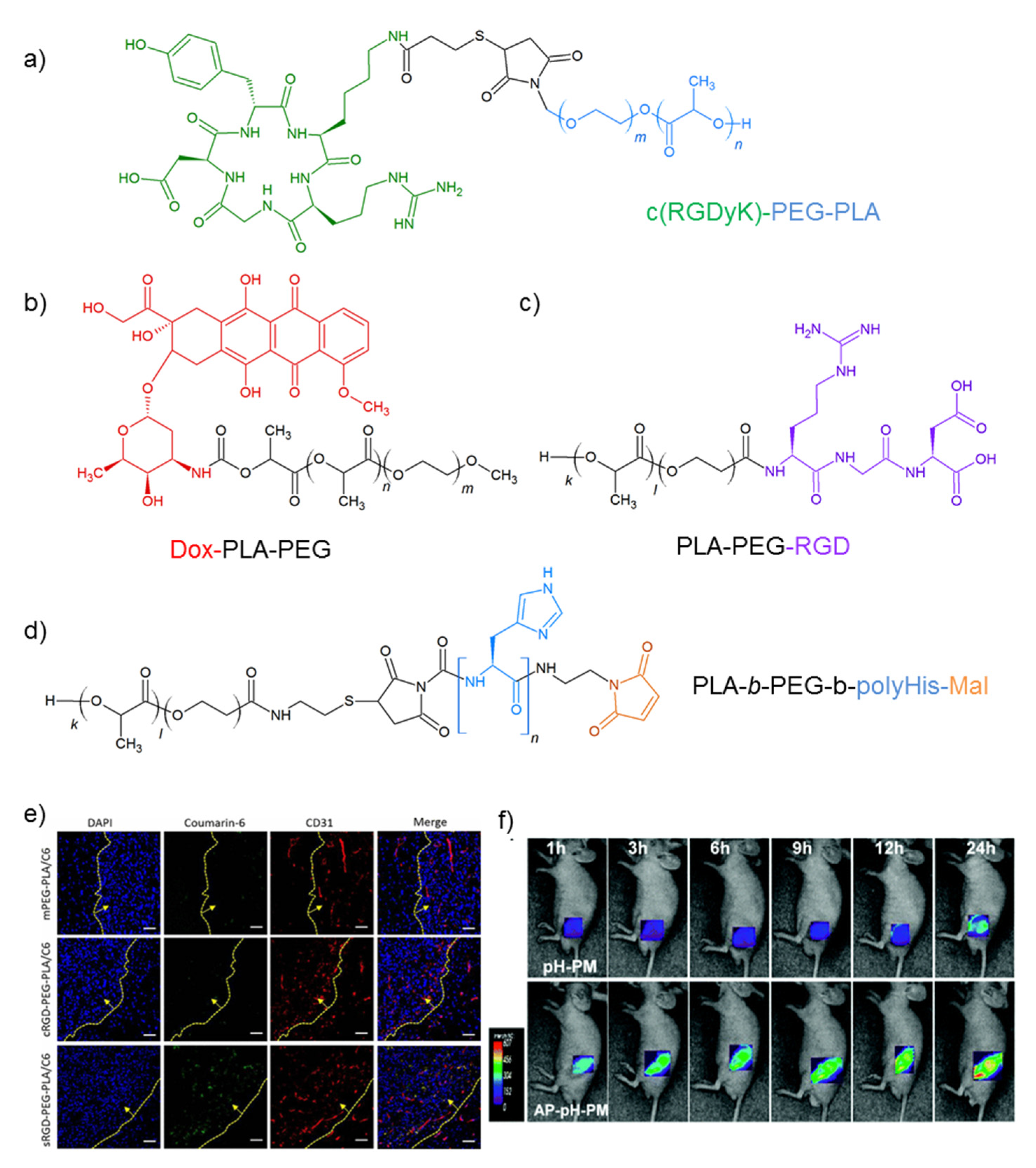
- Guan, X.; Guan, X.; Tong, H.; Ma, J.; Sun, X. Target delivery of daunorubicin to glioblastoma by cyclic RGD-linked PEG-PLA micelles. J. Macromol. Sci. Part A Pure Appl. Chem. 2015, 52, 401–406. [Google Scholar] [CrossRef]
- Zhan, C.; Gu, B.; Xie, C.; Li, J.; Liu, Y.; Lu, W. Cyclic RGD conjugated poly(ethylene glycol)-co-poly(lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J. Control. Release 2010, 143, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Chen, X.; Xie, C.; Li, B.; Ying, M.; Liu, Y.; Zhang, M.; Zhang, X.; Zhan, C.; Lu, W.; et al. Stapled RGD peptide enables glioma-targeted drug delivery by overcoming multiple barriers. ACS Appl. Mater. Interfaces 2017, 9, 17745–17756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, T.; Wang, X.; Dai, W.; Wang, J.; Zhang, X.; Li, Z.; Zhang, Q. Materializing sequential killing of tumor vasculature and tumor cells via targeted polymeric micelle system. J. Control. Release 2011, 149, 299–306. [Google Scholar] [CrossRef]
- Hu, Q.; Gao, X.; Gu, G.; Kang, T.; Tu, Y.; Liu, Z.; Song, Q.; Yao, L.; Pang, Z.; Jiang, X. Glioma therapy using tumor homing and penetrating peptide-functionalized PEG-PLA nanoparticles loaded with paclitaxel. Biomaterials 2013, 34, 5640–5650. [Google Scholar] [CrossRef]
- Wu, X.L.; Kim, J.H.; Koo, H.; Bae, S.M.; Shin, H.; Kim, M.S.; Lee, B.-H.; Park, R.-W.; Kim, I.-S.; Choi, K. Tumor-targeting peptide conjugated pH-responsive micelles as a potential drug carrier for cancer therapy. Bioconjug. Chem. 2010, 21, 208–213. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. Octreotide-modified polymeric micelles as potential carriers for targeted docetaxel delivery to somatostatin receptor overexpressing tumor cells. Pharm. Res. 2011, 28, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Chen, X.; Wang, J.; Zhang, X.; Zhang, Q. NGR-modified micelles enhance their interaction with CD13-overexpressing tumor and endothelial cells. J. Control. Release 2009, 139, 56–62. [Google Scholar] [CrossRef]
- Lee, E.S.; Gao, Z.; Kim, D.; Park, K.; Kwon, I.C.; Bae, Y.H. Super pH-sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J. Control. Release 2008, 129, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Gao, Z.; Liu, S.; Fan, B.; Kang, L.; Huang, W.; Jin, M. Hybrid polymeric micelles based on bioactive polypeptides as pH-responsive delivery systems against melanoma. Biomaterials 2014, 35, 7008–7021. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, J.; Meng, F.; Deng, C.; Cheng, R.; Feijen, J.; Zhong, Z. cRGD-functionalized reduction-sensitive shell-sheddable biodegradable micelles mediate enhanced doxorubicin delivery to human glioma xenografts in vivo. J. Control. Release 2016, 233, 29–38. [Google Scholar] [CrossRef]
- Fang, Y.; Jiang, Y.; Zou, Y.; Meng, F.; Zhang, J.; Deng, C.; Sun, H.; Zhong, Z. Targeted glioma chemotherapy by cyclic RGD peptide-functionalized reversibly core-crosslinked multifunctional poly(ethylene glycol)-b-poly(ε-caprolactone) micelles. ActaBiomater 2017, 50, 396–406. [Google Scholar] [CrossRef]
- Shahin, M.; Ahmed, S.; Kaur, K.; Lavasanifar, A. Decoration of polymeric micelles with cancer-specific peptide ligands for active targeting of paclitaxel. Biomaterials 2011, 32, 5123–5133. [Google Scholar] [CrossRef] [PubMed]
- Mathews, A.S.; Ahmed, S.; Shahin, M.; Lavasanifar, A.; Kaur, K. Peptide modified polymeric micelles specific for breast cancer cells. Bioconjug. Chem. 2013, 24, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Mathews, A.S.; Byeon, N.; Lavasanifar, A.; Kaur, K. Peptide arrays for screening cancer specific peptides. Anal. Chem. 2010, 82, 7533–7541. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.B.; Lavasanifar, A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano 2011, 5, 5202–5213. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. The eradication of breast cancer and cancer stem cells using octreotide modified paclitaxel active targeting micelles and salinomycin passive targeting micelles. Biomaterials 2012, 33, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Dai, W.; Du, W.; Zhang, H.; Lei, L.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Gao, J.; et al. A novel lanreotide-encoded micelle system targets paclitaxel to the tumors with overexpression of somatostatin receptors. Mol. Pharm. 2012, 9, 1175–1188. [Google Scholar] [CrossRef]
- Gu, G.; Xia, H.; Hu, Q.; Liu, Z.; Jiang, M.; Kang, T.; Miao, D.; Tu, Y.; Pang, Z.; Song, Q.; et al. PEG-co-PCL nanoparticles modified with MMP-2/9 activatable low molecular weight protamine for enhanced targeted glioblastoma therapy. Biomaterials 2013, 34, 196–208. [Google Scholar] [CrossRef]
- Tanaka, K.; Kanazawa, T.; Shibata, Y.; Suda, Y.; Fukuda, T.; Takashima, Y.; Okada, H. Development of cell-penetrating peptide modified MPEG-PCL diblock copolymeric nanoparticles for systemic gene delivery. Int. J. Pharm. 2010, 396, 229–238. [Google Scholar] [CrossRef]
- Kanazawa, T.; Sugawara, K.; Tanaka, K.; Horiuchi, S.; Takashima, Y.; Okada, H. Suppression of tumor growth by systemic delivery of anti-VEGF siRNA with cell-penetrating peptide-modified MPEG-PCL nanomicelles. Eur. J. Pharm. Biopharm. 2012, 81, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kanazawa, T.; Horiuchi, S.; Ando, T.; Sugawara, K.; Takashima, Y.; Seta, Y.; Okada, H. Cytoplasm-responsive nanocarriers conjugated with a functional cell-penetrating peptide for systemic siRNA delivery. Int. J. Pharm. 2013, 455, 40–47. [Google Scholar] [CrossRef]
- Ibaraki, H.; Kanazawa, T.; Owada, M.; Iwaya, K.; Takashima, Y.; Seta, Y. Anti-metastatic effects on melanoma via intravenous administration of anti-NF-κB siRNA complexed with functional peptide-modified nano-micelles. Pharmaceutics 2020, 12, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazawa, T.; Taki, H.; Takashima, Y.; Okada, H. Cell-penetrating peptide modified block copolymer micelles promote direct brain delivery via intranasal administration. Pharm. Res. 2011, 28, 2130–2139. [Google Scholar] [CrossRef]
- Kanazawa, T.; Morisaki, K.; Suzuki, S.; Takashima, Y. Prolongation of Life in Rats with malignant glioma by intranasal siRNA/drug codelivery to the brain with cell-penetrating peptide-modified micelles. Mol. Pharm. 2014, 11, 1471–1478. [Google Scholar] [CrossRef]
- Kanazawa, T.; Kurano, T.; Ibaraki, H.; Takashima, Y.; Suzuki, T.; Seta, Y. Therapeutic effects in a transient middle cerebral artery occlusion rat model by nose-to-brain delivery of anti-TNF-alpha siRNA with cell-penetrating peptide-modified polymer micelles. Pharmaceutics 2019, 11, 478. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, T.; Taki, H.; Okada, H. Nose-to-brain drug delivery system with ligand/cell-penetrating peptide modified. Eur. J. Pharm. Biopharm. 2020, 152, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Salsano, G.; Morisco, A.; Aurilio, M.; Parisi, A.; Maione, F.; Cicala, C.; Tesauro, D.; Aloj, L.; De Rosa, G.; et al. Peptide-modified liposomes for selective targeting of bombesin receptors overexpressed by cancer cells: A potential theranostic agent. Int. J. Nanomed. 2012, 7, 2007–2017. [Google Scholar] [CrossRef] [Green Version]
- Accardo, A.; Mansi, R.; Salzano, G.; Morisco, A.; Aurilio, M.; Parisi, A.; Maione, F.; Cicala, C.; Ziaco, B.; Tesauro, D.; et al. Bombesin peptide antagonist for target-selective delivery of liposomal doxorubicin on cancer cells. J. Drug Target. 2013, 21, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.B.; Uludag, H.; Lavasanifar, A. Biodegradable amphiphilic poly(ethylene oxide)-block-polyesters with grafted polyamines as supramolecular nanocarriers for efficient siRNA Delivery. Biomaterials 2009, 30, 242–253. [Google Scholar] [CrossRef]
- Xiong, X.B.; Uludag, H.; Lavasanifar, A. Virus-mimetic polymeric micelles for targeted siRNA delivery. Biomaterials 2010, 31, 5886–5893. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, J.; Meng, F.; Deng, C.; Cheng, R.; Feijen, J.; Zhong, Z. RGD/TAT dual-ligand reversibly cross-linked micelles loaded with docetaxel penetrate deeply into tumor tissue and show high antitumor efficacy in vivo. ACS Appl. Mater. Interfaces 2017, 9, 35651–35663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Meng, L.; Huang, Q.; Zhu, Y.; Cui, W.; Cheng, Y.; Liu, R. Targeted micelles with chemotherapeutics and gene drugs to inhibit the G1/S and G2/M mitotic cycle of prostate cancer. J. Nanobiotechnol. 2021, 19, 17. [Google Scholar] [CrossRef]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Kumar, A.R.; Murphy, D.G.; et al. [(177)Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Matsumoto, S.; Christie, R.J.; Nishiyama, N.; Miyata, K.; Ishii, A.; Oba, M.; Koyama, H.; Yamasaki, Y.; Kataoka, K. Environment-responsive block copolymer micelles with a disulfide cross-linked core for enhanced siRNA delivery. Biomacromolecules 2009, 10, 119–127. [Google Scholar] [CrossRef]
- Christie, R.J.; Matsumoto, Y.; Miyata, K.; Nomoto, T.; Fukushima, S.; Osada, K.; Halnaut, J.; Pittella, F.; Kim, H.J.; Nishiyama, N.; et al. Targeted polymeric micelles for sirna treatment of experimental cancer by intravenous injection. ACS Nano 2012, 6, 5174–5189. [Google Scholar] [CrossRef]
- Oe, Y.; Christie, R.J.; Naito, M.; Low, S.A.; Fukushima, S.; Toh, K.; Miura, Y.; Matsumoto, Y.; Nishiyama, N.; Miyata, K.; et al. Actively-targeted polyion complex micelles stabilized by cholesterol and disulfide cross-linking for systemic delivery of siRNA to solid tumors. Biomaterials 2014, 35, 7887–7895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Zhou, X.; Xu, M.; Tian, H.; Chen, X.; Chen, M. Dimeric camptothecin-loaded RGD-modified targeted cationic polypeptide-based micelles with high drug loading capacity and redox-responsive drug release capability. Biomater. Sci. 2017, 5, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Li, Z.Y.; Zhu, J.Y.; Han, K.; Zeng, Z.Y.; Hong, W.; Li, W.X.; Jia, H.Z.; Liu, Y.; Zhuo, R.X.; et al. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small 2015, 11, 2543–2554. [Google Scholar] [CrossRef]
- Cabral, H.; Nishiyama, N.; Okazaki, S.; Kato, Y.; Kataoka, K. Preparation and Biological Properties of Dichloro(1,2-diaminocyclohexane)platinum (II) (DACHPt)-loaded polymeric micelles. J. Control. Release 2005, 101, 223–232. [Google Scholar] [CrossRef]
- Miura, Y.; Takenaka, T.; Toh, K.; Wu, S.; Nishihara, H.; Kano, M.R.; Ino, Y.; Nomoto, T.; Matsumoto, Y.; Koyama, H.; et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano 2013, 7, 8583–8592. [Google Scholar] [CrossRef]
- Makino, J.; Cabral, H.; Miura, Y.; Matsumoto, Y.; Wang, M.; Kinoh, H.; Mochida, Y.; Nishiyama, N.; Kataoka, K. RGD-installed polymeric micelles loading platinum anticancer drugs enable cooperative treatment against lymph node metastasis. J. Control. Release 2015, 220, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Miyano, K.; Cabral, H.; Miura, Y.; Matsumoto, Y.; Mochida, Y.; Kinoh, H.; Iwata, C.; Nagano, O.; Saya, H.; Nishiyama, N.; et al. RGD peptide installation on cisplatin-loaded nanomedicines enhances efficacy against locally advanced head and neck squamous cell carcinoma bearing cancer stem-like cells. J. Control. Release 2017, 261, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Tang, Z.; Zhang, D.; Zhang, Y.; Yu, H.; Li, M.; Lv, S.; Sun, H.; Deng, M.; Chen, X. Anti-tumor efficacy of c(RGDfK)-decorated polypeptide-based micelles co-loaded with docetaxel and cisplatin. Biomaterials 2014, 35, 3005–3014. [Google Scholar] [CrossRef]
- Guan, X.; Hu, X.; Li, Z.; Zhang, H.; Xie, Z. RGD targeted and charge conversion-controlled release micelles for doxorubicin delivery. RSC Adv. 2015, 5, 22957–22964. [Google Scholar] [CrossRef]
- Liu, X.; Cui, W.; Li, B.; Hong, Z. Targeted therapy for glioma using cyclic RGD-entrapped polyionic complex nanomicelles. Int. J. Nanomed. 2012, 7, 2853–2862. [Google Scholar] [CrossRef] [Green Version]
- Quader, S.; Liu, X.; Chen, Y.; Mi, P.; Chida, T.; Ishii, T.; Miura, Y.; Nishiyama, N.; Cabral, H.; Kataoka, K. RGD peptide-installed epirubicin-loaded polymeric micelles for effective targeted therapy against brain tumors. J. Control. Release 2017, 258, 56–66. [Google Scholar] [CrossRef]
- Ke, W.; Li, J.; Zhao, K.; Zha, Z.; Han, Y.; Wang, Y.; Yin, W.; Zhang, P.; Ge, Z. Modular design and facile synthesis of enzyme-responsive peptide linked block copolymers for efficient delivery of doxorubicin. Biomacromolecules 2016, 17, 3268–3276. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Ouyang, J.; Sun, H.; Meng, F.; Cheng, R.; Zhang, J.; Cheng, L.; Lan, Q.; Deng, C.; Zhong, Z. Biodegradable micelles based on poly(ethylene glycol)-b-polylipopeptide copolymer: A robust and versatile nanoplatform for anticancer drug delivery. ACS Appl. Mater. Interfaces 2017, 9, 27587–27595. [Google Scholar] [CrossRef]
- Qiu, M.; Wang, X.; Sun, H.; Zhang, J.; Deng, C.; Zhong, Z. Cyclic RGD-peptide-functionalized polylipopeptide micelles for enhanced loading and targeted delivery of monomethyl auristatin E. Mol. Pharm. 2018, 15, 4854–4861. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, M.Y.; Melik-Nubarov, N.S.; Batrakova, E.V.; Kabanov, A.V. Relationship between pluronic block copolymer structure, critical micellization concentration and partitioning coefficients of low molecular mass solutes. Macromolecules 2000, 33, 3305–3313. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, W.; Huang, Y.; Gao, F.; Fang, X. Dual-functional c(RGDyK)-decorated Pluronic micelles designed for antiangiogenesis and the treatment of drug-resistant tumor. Int. J. Nanomed. 2015, 10, 4863–4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.K.; Liu, W.; Gao, F.; Fang, X.; Chen, Y. c(RGDyK)-decorated Pluronic micelles for enhanced doxorubicin and paclitaxel delivery to brain glioma. Int. J. Nanomed. 2016, 11, 1629–1641. [Google Scholar] [CrossRef] [Green Version]
- Tesauro, D.; Mastro, R.; Cusimano, A.; Emma, M.R.; Cervello, M. Synthetic peptide-labelled micelles for active targeting of cells overexpressing EGF receptors. Amino Acids 2019, 51, 1177–1185. [Google Scholar] [CrossRef]
- Arleth, L.; Ashok, B.; Onyuksel, H.; Thiyagarajan, P.; Jacob, J.; Hjelm, R.P. Detailed structure of hairy mixed micelles formed by phosphatidylcholine and PEGylated phospholipids in aqueous media. Langmuir 2005, 21, 3279–3290. [Google Scholar] [CrossRef]
- Xu, P.; Wang, H.; Hu, H.; Ye, Y.; Dong, Y.; Li, S.; Mei, D.; Guo, Z.; Wang, D.; Sun, Y.; et al. cRGDfK-grafted small-size quercetin micelles for enhancing therapy efficacy of active ingredient from the chinese medicinal herb. Int. J. Nanomed. 2019, 14, 9173–9184. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, H.; Feng, N.; Xin, X.; Huo, X.P.; Wang, X.; Zhang, N. Construction and antitumor effects of antitumor micelles with cyclic RGD-modified anlotinib. Nanomed. Nanotechnol. Biol. Med. 2020, 28, 102224. [Google Scholar] [CrossRef]
- Tesauro, D.; Accardo, A.; Gianolio, E.; Paduano, L.; Teixeira, J.; Schillén, K.; Aime, S.; Morelli, G. Peptide derivatized lamellar aggregates as target specific MRI contrast agents. ChemBioChem 2007, 8, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Dai, W.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Wang, K.; Li, J.; Zhang, Q. Lanreotide-conjugated PEG-DSPE micelles: An efficient nanocarrier targeting to somatostatin receptor positive tumors. J. Drug Targ. 2015, 23, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Gao, C.; Zhou, L.; Liu, M.; Xie, C.; Lu, W. EGFR-targeted poly(ethyleneglycol)distearoylphosphatidylethanolamine micelle loaded with paclitaxel for laryngeal cancer: Preparation, characterization and in vitro evaluation. Drug Deliv. 2015, 22, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Liang, X.; Yang, D.; Pan, X.; Li, Z.; Wang, H.; Shi, B. Epidermal growth factor receptor-targeted peptide conjugated phospholipid micelles for doxorubicin delivery. J. Drug Target. 2016, 24, 111–119. [Google Scholar] [CrossRef]
- Vene, E.; Jarnouen, K.; Huang, W.Z.; Wahib, B.; Montier, T.; Cammas-Marion, S.; Loyer, P. In vitro toxicity evaluation and in vivo biodistribution of polymeric micelles derived from Poly(ethylene glycol)-b-poly(benzyl malate) Copolymers. Pharm. Nanotechnol. 2016, 4, 24–36. [Google Scholar] [CrossRef]
- Vert, M.; Lenz, R.W. Preparation and properties of poly-beta-malic acid: A functional polyester of potential biomedical importance. Am. Chem. Soc. Div. Polym. Chem. Prepr. 1979, 20, 608–611. [Google Scholar]
- Jiang, X.; Sha, X.; Xin, H.; Chen, L.; Gao, X.; Wang, X.; Law, K.; Gu, J.; Chen, Y.; Jiang, Y.; et al. Self-aggregated pegylated poly (trimethylene carbonate) nanoparticles decorated with c(RGDyK) peptide for targeted paclitaxel delivery to integrin-rich tumors. Biomaterials 2011, 32, 9457–9469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kuijer, R.; Bulstra, S.K.; Grijpma, D.W.; Feijen, J. The in vivo and in vitro degradation behavior of poly(trimethylene carbonate). Biomaterials 2006, 27, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Shia, J.; Liub, S.; Yu, Y.; He, C.; Tana, L.; Shena, Y.M. RGD peptide-decorated micelles assembled from polymer–paclitaxel conjugates towards gastric cancer therapy. Colloids Surf. B 2019, 180, 58–67. [Google Scholar] [CrossRef]
- Lu, L.; Zhao, X.; Fu, T.; Li, K.; Ye, H.; Luo, Z.; Dai, L.; Zeng, R.; Cai, K. An iRGD-conjugated prodrug micelle with blood-brain-barrier penetrability for anti-glioma therapy. Biomaterials 2020, 230, 119666. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Wu, Y.; Hu, Y.; Dong, Y.; Hao, T.; Zhang, C. TPE-based peptide micelles for targeted tumor therapy and apoptosis monitoring. ACS Appl. Bio Mater. 2021, 4, 1038–1044. [Google Scholar] [CrossRef]
- Bai, J.; Tian, Y.; Liu, F.; Li, X.; Shao, Y.; Lu, X.; Wang, J.; Zhu, G.; Xue, B.; Liu, M.; et al. Octreotide-conjugated core-cross-linked micelles with pH/redox responsivity loaded with etoposide for neuroendocrine neoplasms therapy and bioimaging with photoquenching resistance. ACS Appl. Mater. Interfaces 2019, 11, 18111–18122. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Zhan, C.; Wang, C.; Shi, X.; Yang, J.; He, X.; Ji, E.; Yu, Z.; Yan, C.; Wu, H. MMP-2 sensitive poly(malic acid) micelles stabilized by π-π stacking enable high drug loading capacity. J. Mater. Chem. B 2020, 8, 8527–8535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, Y.; Xie, X.; Wang, L.; Su, Z.; Wang, Y.; Leong, K.W.; Chen, M. Cleavable multifunctional targeting mixed micelles with sequential pH-triggered Tat peptide activation for improved antihepatocellular carcinoma efficacy. Mol. Pharm. 2017, 14, 3644–3659. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Amphiphilic Mol. | Tumor Target | Ref. |
|---|---|---|---|
| CCK8: DYMGWMDF | (C18)2(ADA)5CCK8 | CCK2-R-overexpressing cells | [58] |
| CCK8: DYMGWMDF | (C18)2(ADA)5CCK8 | CCK2-R-overexpressing cells | [59] |
| (C18H37)2Lys(DTPAGlu) | |||
| CCK8: GDYMGWMDF | (C18H37)(ADA)5-G-CCK8 | CCK2-R-overexpressing cells | [60] |
| (C18H37)2Lys(DTPAGlu) | |||
| CCK8: DYMGWMDF | [C18CysL5CCK8]2 | CCK2-R-overexpressing cells | [61] |
| OCT: (fCFwLTCT-ol) | OCA-DTPAGlu | SSTR-R-overexpressing cells | [62] |
| OCA-DOTA | |||
| OCT: (fCFwLTCT-ol) | (C18)2(ADA)5OCT | SSTR-R-overexpressing cells | [63] |
| (C18)2DTPAGlu/DOTA | |||
| RGD | (C18)2(ADA)5RGD | A2058 melanoma cells | [64] |
| (C16)2(ADA)5RGD | |||
| RGD:cRGDfK | (C18)2(ADA)5cRGD | A2058 melanoma cells | [65] |
| (C18)2(ADA)5RGD | |||
| RGD:KKGRGDS | (C12)KKGRGDS | HepG2-HeLa | [66] |
| RGD:R7RGDS | C18H5R7RGDS | HepG2 | [67] |
| CCK8: DYMGWMDF | (PDA)(ADA)2CCK8 | CCK2-R-overexpressing cells | [70] |
| (PDA)DTPAGlu | |||
| RGD:KGGGKRGDK | (PDA)(Nph)KGGGKRGDK | SKOV-3 cells | [71] |
| (PDA)KGGGKRGDK |
| Polymer | Peptide | Tumor Target | Drug | Ref. |
|---|---|---|---|---|
| RGD-PEG-PLA | c(RGDyK) | SLK endothelial cells | SPIO-DOX | [74] |
| cRGD PEG-PLA | c(RGDyK) | HUVEC | CA4 | [75] |
| PEG-PLA-Dau–cRGD PEG-PLA | c(RGDyK) | C6 glioma cells | DAU | [76] |
| c(RGDyK)-PEG-PLA | c(RGDyK) | U86MG glioblastoma | PTX | [77] |
| s-RGD-PEG-PLA | s-RGD | U86MG glioblastoma | PTX | [78] |
| RGD-PEG-PLA | RGD | B16F10 | DOX CA4 | [79] |
| tLyp-1-PEG-PLA | CGNKRTRGC | C6 glioma cells-HUVEC | PTX | [80] |
| AP-PEG-PLA/MPEG-PAE | AP peptide (CRKRLDRN) | MDA-MB231 | DOX | [81] |
| OCT-PEG-PLA | fCFwLTCT-ol | NCI-H446 | Dil-DTX | [82] |
| NGR-PEG-PLA | NGR | HT1080 | DTX | [83] |
| Tat-PEG-PLA-pHIS/pHIS-PEG | GCGGGTGRKRRQRRR | A2780/AD-MCF7 | DOX | [84] |
| A549-KB | ||||
| pGLU-pPLA/DSPE-PEG-Tat | GCGGGTGRKRRQRRR | A375 | DOX | [85] |
| cRGD-PEG-SS-PCL/PEG-SS-PCL | c(RGDyK) | U86MG glioblastoma | DOX | [86] |
| MCF 7 | ||||
| cRGD PEG-DTC-PCL | c(RGDyK) | U86MG glioblastoma | DOX | [87] |
| cRGD-PEG-b-PBCL/p160 | cRGDfK/VPWXEPAYQRFL | MDA-MB-435 | PTX | [88] |
| cRGD-PEG-b-PCL/p160 | ||||
| 11-PEG-b-PBCL 11–PEG-b-PCL | RGDPAYQGRFL | MDA-MB-435MDA-MB-231, MCF-7 | PTX | [89] |
| 18-PEG-b-PBCL 18–PEG-b-PCL | WXEAAYQRFL | |||
| cRGD4C-PEG-b-PCL | KACDCRGDCFCG | MDA-MB-435/LCC6MDR1 | SiRNA-DOX | [91] |
| TAT-PEG-b-PCL | CGRKKRRQRRR | |||
| OCT-PEG-b-PCL | fCFwLTCT-ol | MCF-7 | PTX-SAL | [92] |
| LAN-PEG-b-PCL | (D2NaI)CYWKVCT | H446-MCF-7 | PTX | [93] |
| ALMWP-PEG-PCL | ALMWP | C6 glioma | PTX | [94] |
| MPEG-PCL-SS-Tat | GRKKRRQRRR | COS7 S-180 | pDNA | [95] |
| MPEG-PCL-SS-Tat | GRKKRRQRRRCG | S-180 | siVEGF | [96] |
| MPEG-PCL–CH2R4H2C | CH2R4H2C | S-180 | siVEGF | [97] |
| MPEG-PCL-CH2R4H2C | CH2R4H2C | B16F10 | siRelA | [98] |
| MPEG-PCL-Tat | GRKKRRQRRRG | C6 glioma | CUM | [99] |
| MPEG-PCL-Tat | GRKKRRQRRRG | C6 glioma | siRNA-CPT | [100] |
| MPEG-PCL-Tat | GRKKRRQRRRG | RN33B | siRNA | [101] |
| MPEG-PCL-Tat; Steroyl-Bom | GRKKRRQRRRPPQ | C6 glioma | CPT CUM | [102] |
| GQWAVGHLM | ||||
| RGD4C-PEG-b-P(CL-g-DP) | KACDCRGDCFCG | MDA435/LCC6 | DOX-SiRNA | [106] |
| TAT-PEG-bP(CL-g-DP) | CGRKKRRQRRR | |||
| cRGD-PEG6k-P(DTC-co-CL), TAT-PEG2k-P(DTC-co-CL) | C(RGDfK) | U87MG glioma cells | DTX | [107] |
| YGRKKRRQRRRC | ||||
| DCL-PEG-b-PCL | YGRKKRRQRRR | PCa cells | SiRNA-DTX | [108] |
| TAT-PEG-b-P(CL-g-SP) | ||||
| cRGD-PEG-b-(PLL-IM) | c(RGDyK) | HeLa-luc | SiRNA | [111] |
| cRGD-PEG-PLL(MPA) | c(RGDyK) | HeLa-Luc | SiRNA | [112] |
| RGD-PEG-p(Lys)-p(Leu) | RGD | MDA-MB-231-MCF7 | DCPT | [113] |
| Tat-p(Lys)-p(Leu) | YGRKKRRQRRR | HELA | DOX | [114] |
| c-RGD-PEG–P(Glu) | c(RGDyK)-RAD | U87MG glioblastoma | DACHPt | [116] |
| cRGD-PEG–P(Glu) | c(RGDyK) | LNM | DACHPt | [117] |
| cRGD-PEG–P(Glu) | c(RGDyK) | HNSCC-CSC | CDDP | [118] |
| PLG-g-Ve/PEG | cRGD | B16F10 | DTX-CDDP | [119] |
| cRGD-PEG-PLA PEG-PGA)-API | c(RGDyK) | B16F10 | DOX | [120] |
| cRGD-PEG-PDEARGDPEGPAsp | c(RGDfC) | C6 | Cd-Te (QD) | [121] |
| cRGD PEG-b-PBLA | c(RGDfC) | U87MG | Epi | [122] |
| PEG-GPLGVRGDG-P(BLA-co-Asp) | GPLGVRGDG | HT1080 | DOX | [123] |
| cRGD-PEG-b-PAPA | c(RGDyK) | B16F10 | DTX | [124] |
| cRGD-Lipep-Ms | c(RGDyK) | HCT-116 | MMAE | [126] |
| c(RGD)-PF127 | c(RGDyK) | MDR KBv | DOX-PTX | [127] |
| c(RGD)-PF127 | c(RGDyK) | U87MG | DOX-PTX | [128] |
| Pf127-D4-(AdA)2(C18)2 | LARLLT | HepG2 | - | [130] |
| cRGDfK | cRGDfK | B16 t | Quercitin | [131] |
| cRGDfK | cRGDfK | B16F10 | Anlotinib | [132] |
| CCK8-DSPE-PEG2000 | DYMGWMDF | - | Gd | [133] |
| (C18)2DTPAGlu-Gd | ||||
| LAN-DPSE-PEG2000 | (D2NaI)CYWKVCT | H446 | PTX | [134] |
| GE11-DSPE-PEG2000 | YHWYGYTPQNVI | Hep-2 | PTX | [135] |
| GE11-L-DSPE-PEG2000 | YHWYGYTPQNVI | SMMC-7721 c | DOX | [136] |
| RGDBiot-StreptBiot-PEG62-b-PMLABe73 | c(RGDyK) | HepaRG | DiD DiR | [138] |
| cRGD-PEG-PTMC | c(RGDyK) | U87MG | PTX | [140] |
| c-RGD-PEG-SS-PTX | c(RGDyK) | SGC7901 | PTX | [141] |
| iRGD-PEG-SS-CPT | CRGDRGPDC | U87MG | CPT-IR780 | [142] |
| RGD-PEG-H9-DEVD-TPE | RGD | 4T1 | DOX-TPE | [143] |
| OCT-PEG-b-PGu(DA-TPE) | fCFwLTCT-ol | LCC-18-NCH-446 c | ETO | [144] |
| Tat-PEG-PBM | YGRKKRRQRRR | HT1080, MCF-7 MDA-MB-231 | DOX | [145] |
| Tat-PEG1k-PHHD-GA-P5kCV | AYGRKKRRQRRR | SMCC 7721 | DOX | [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prencipe, F.; Diaferia, C.; Rossi, F.; Ronga, L.; Tesauro, D. Forward Precision Medicine: Micelles for Active Targeting Driven by Peptides. Molecules 2021, 26, 4049. https://doi.org/10.3390/molecules26134049
Prencipe F, Diaferia C, Rossi F, Ronga L, Tesauro D. Forward Precision Medicine: Micelles for Active Targeting Driven by Peptides. Molecules. 2021; 26(13):4049. https://doi.org/10.3390/molecules26134049
Chicago/Turabian StylePrencipe, Filippo, Carlo Diaferia, Filomena Rossi, Luisa Ronga, and Diego Tesauro. 2021. "Forward Precision Medicine: Micelles for Active Targeting Driven by Peptides" Molecules 26, no. 13: 4049. https://doi.org/10.3390/molecules26134049