
Assessment of Antidiabetic Activity of the Shikonin by Allosteric Inhibition of Protein-Tyrosine Phosphatase 1B (PTP1B) Using State of Art: An In Silico and In Vitro Tactics

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents, and Cell Culture
2.2. In Silico Docking Studies of Shikonin
2.3. Identification of Compounds Similar to Shikonin
2.3.1. Pharmacophore Modeling and Virtual Screening
2.3.2. Drug Likeness and In-Silico ADMET Prediction
2.3.3. Molecular-Dynamics Simulation Studies
2.4. In Vitro Anti-Diabetic Activity
2.4.1. Protein Tyrosine Phosphate 1B (PTP1B) Inhibitory Assay
2.4.2. Kinetic Study Against PTP1B
2.5. Statistical Analysis
3. Results and Discussion
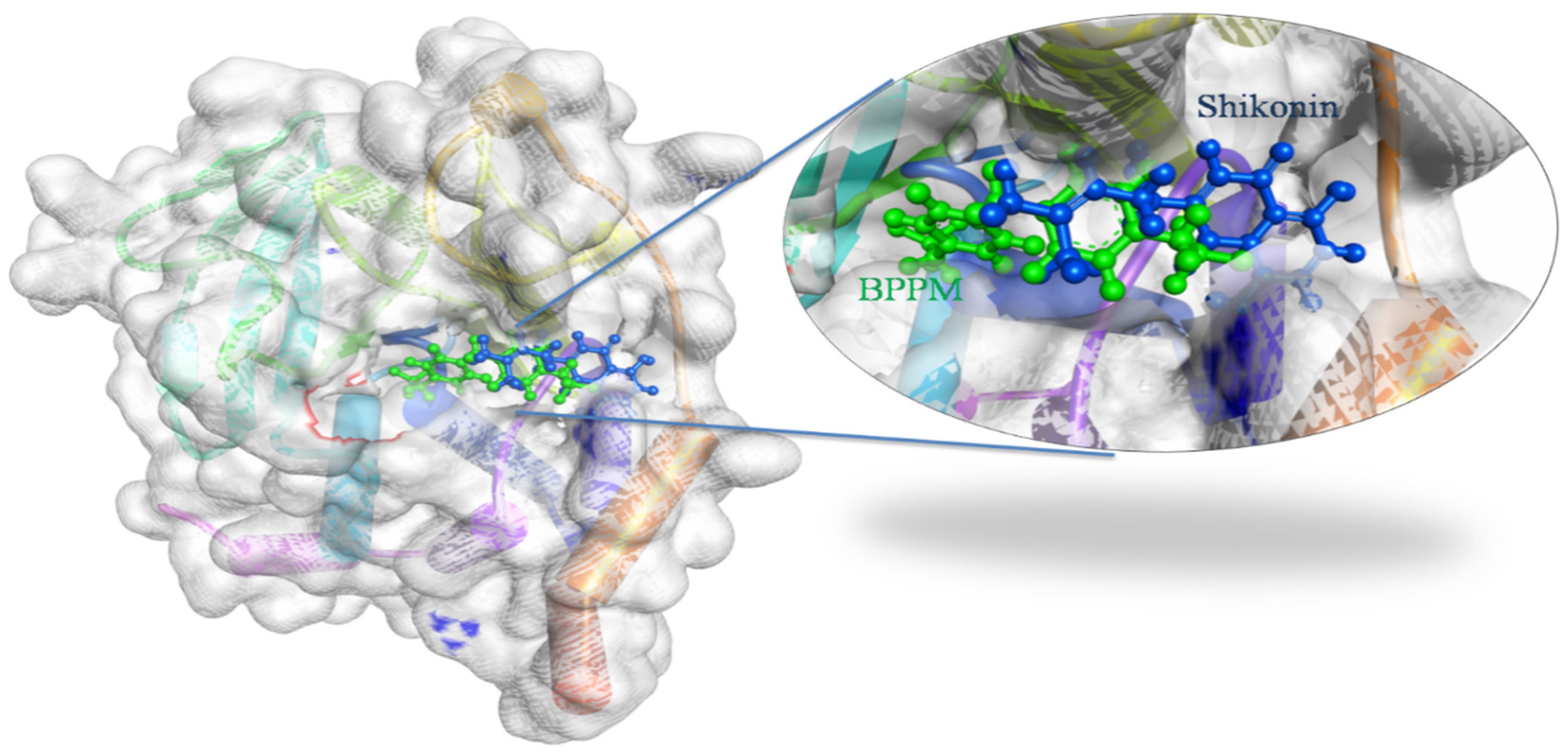
3.1. In Silico Docking Studies of Shikonin
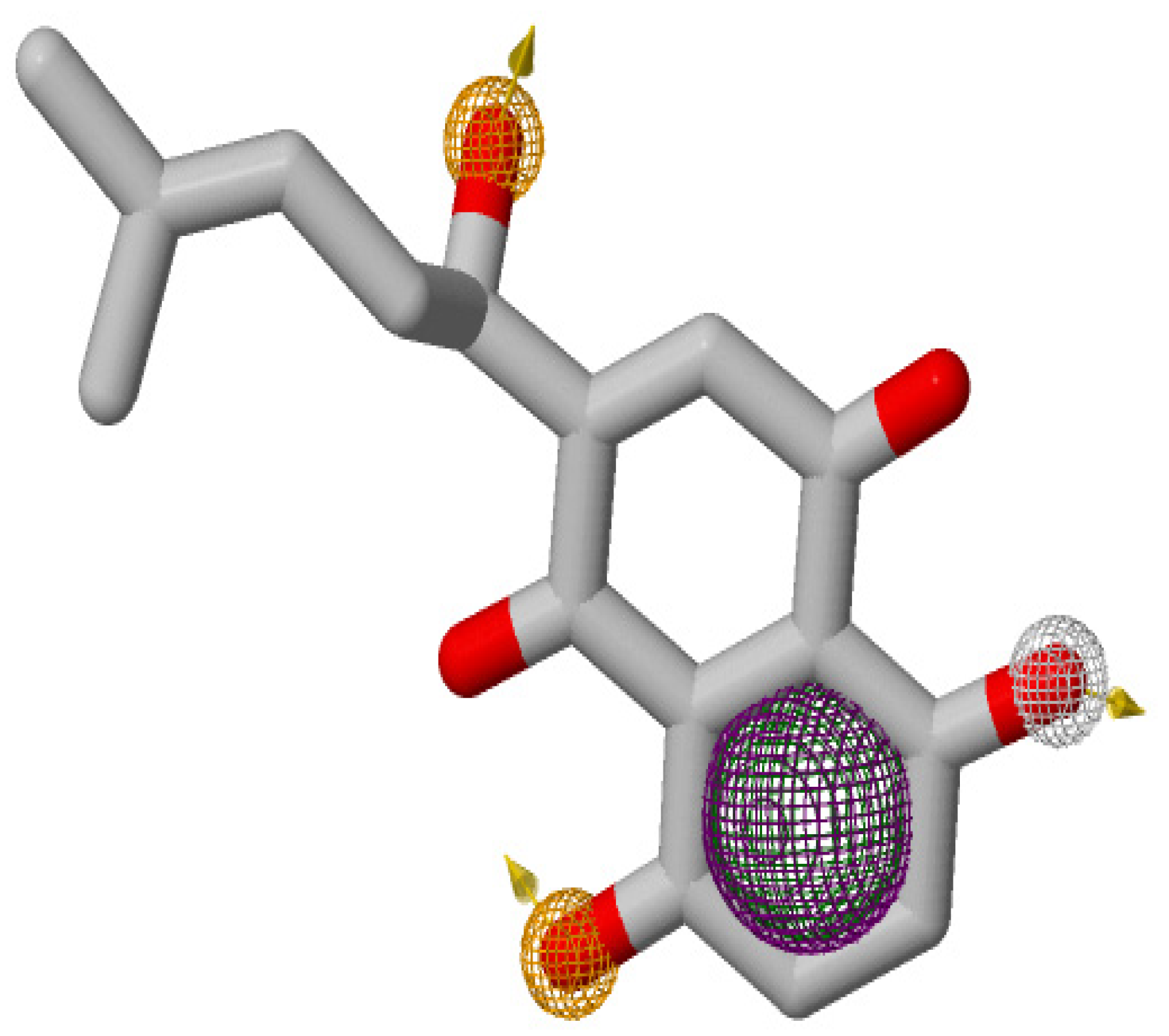
3.2. Pharmacophore Modeling and Virtual Screening
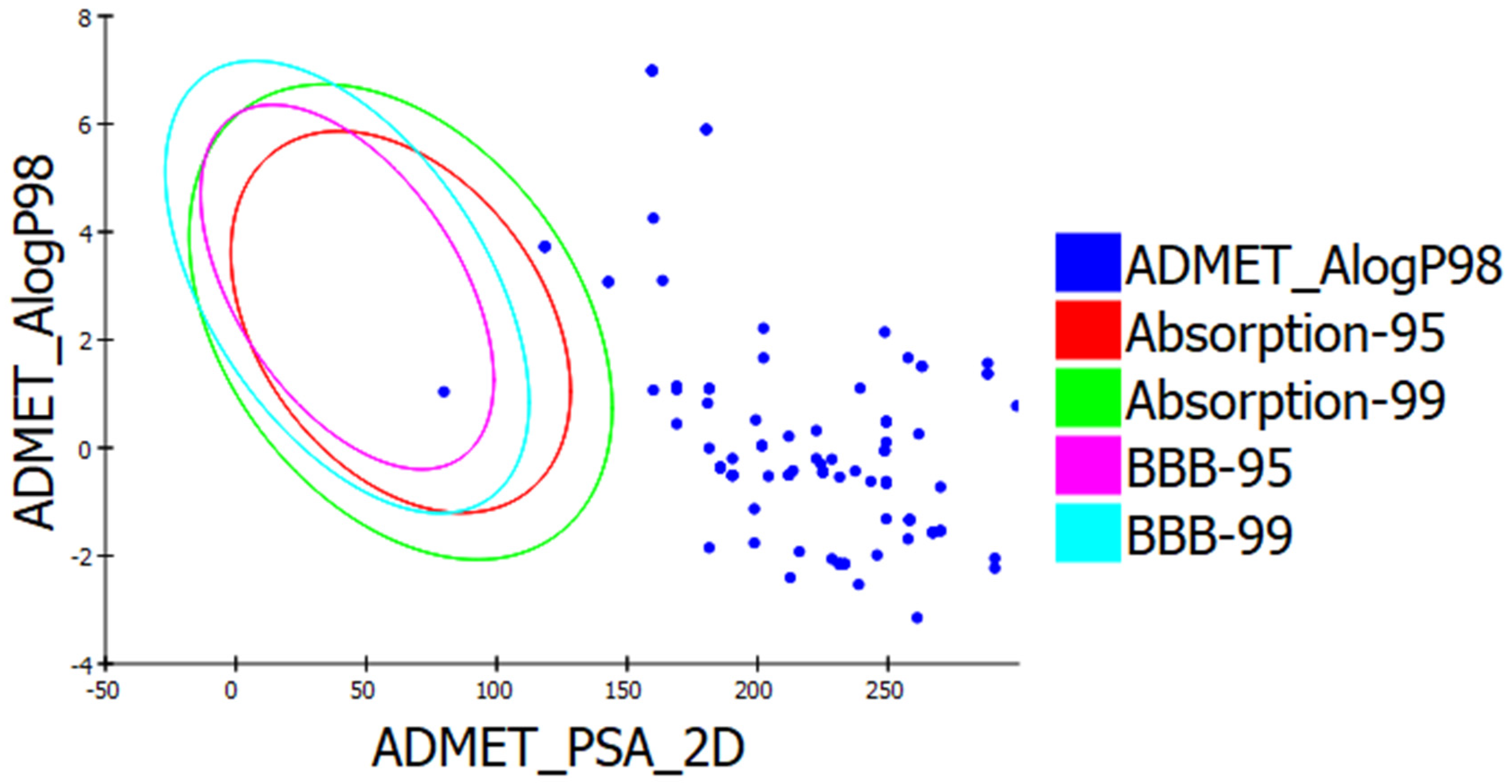
3.3. Drug Likeness and In Silico ADMET Prediction
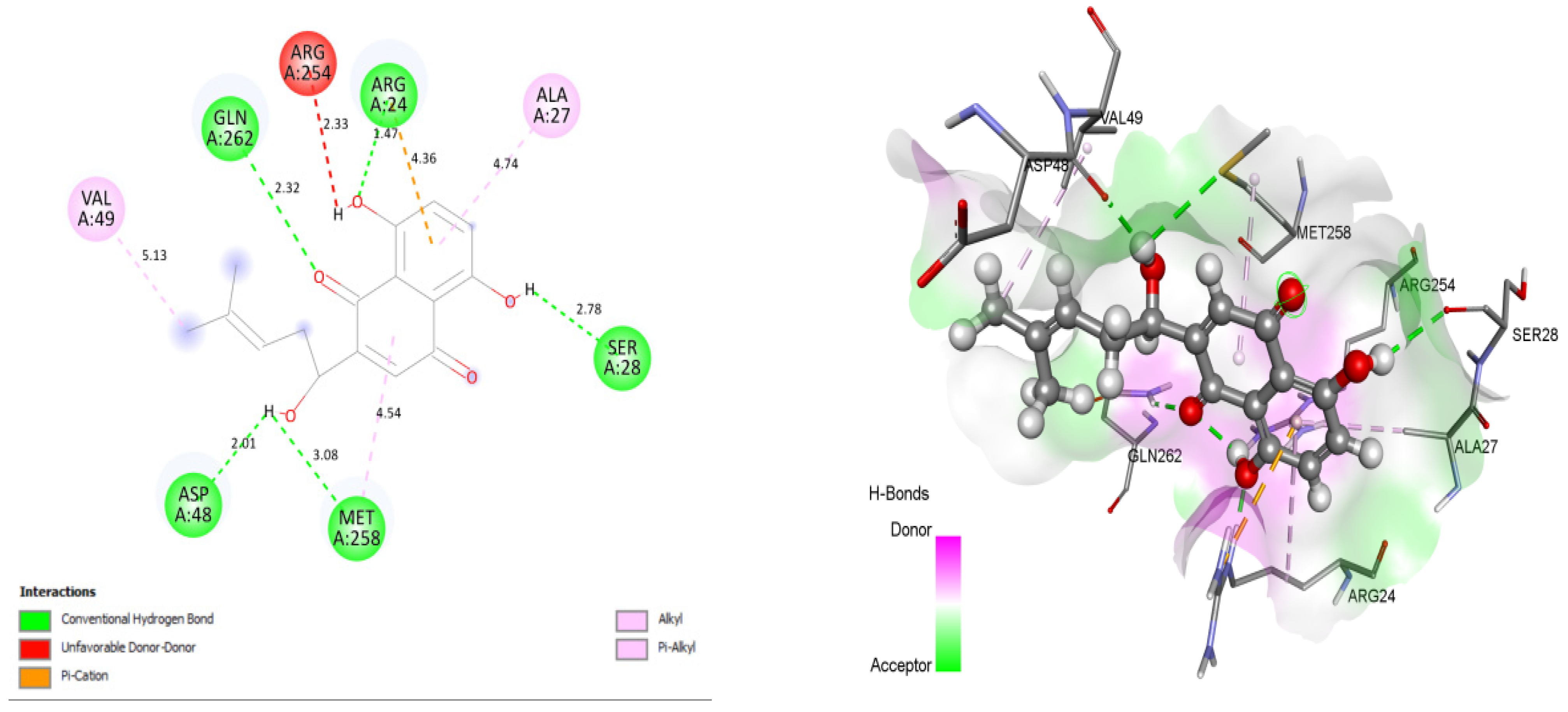
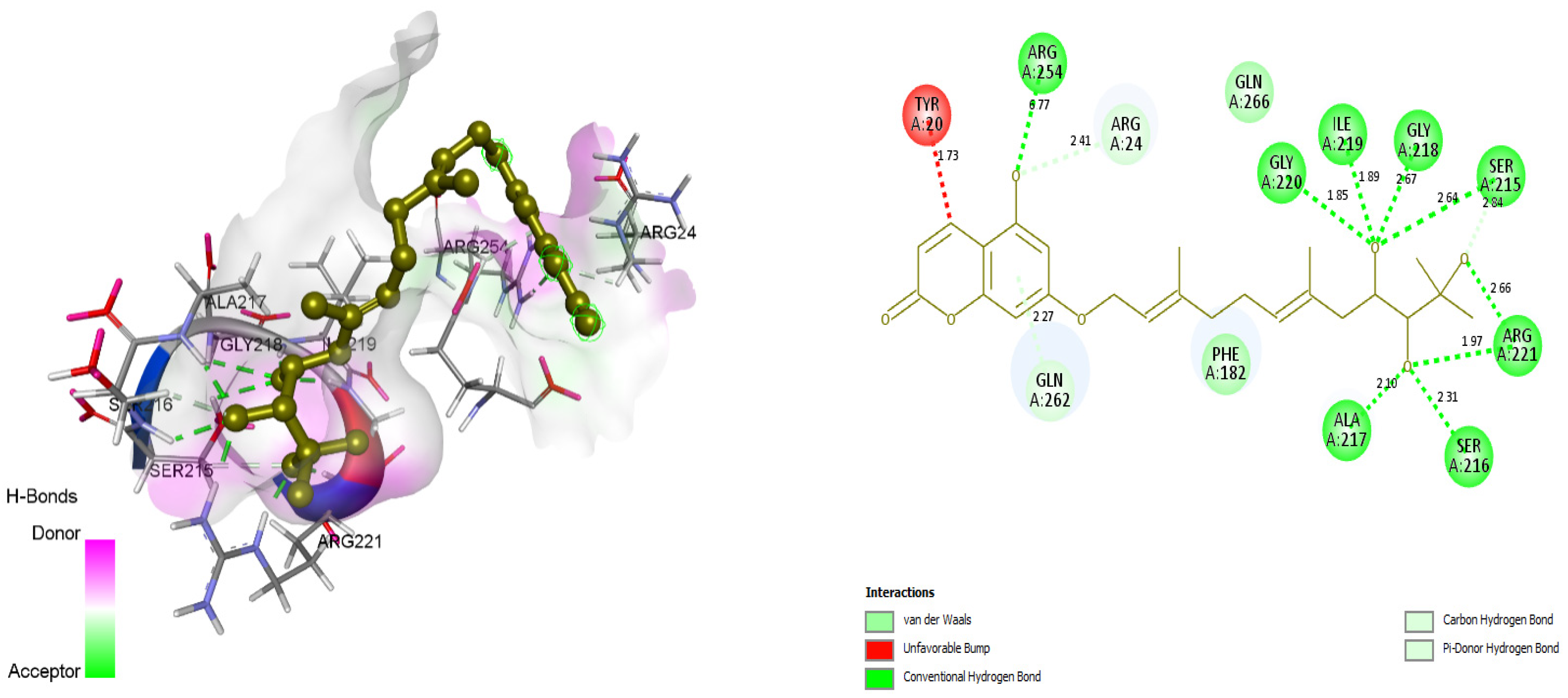
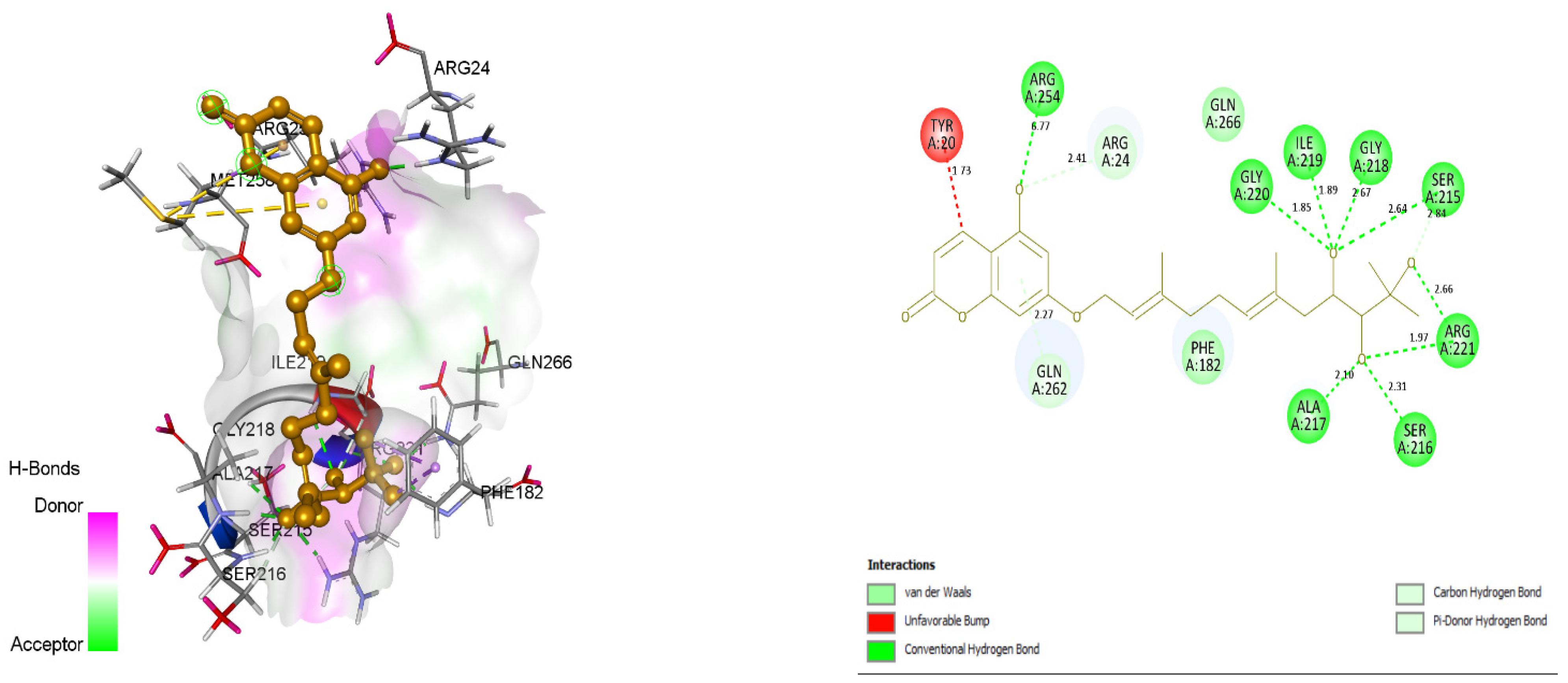
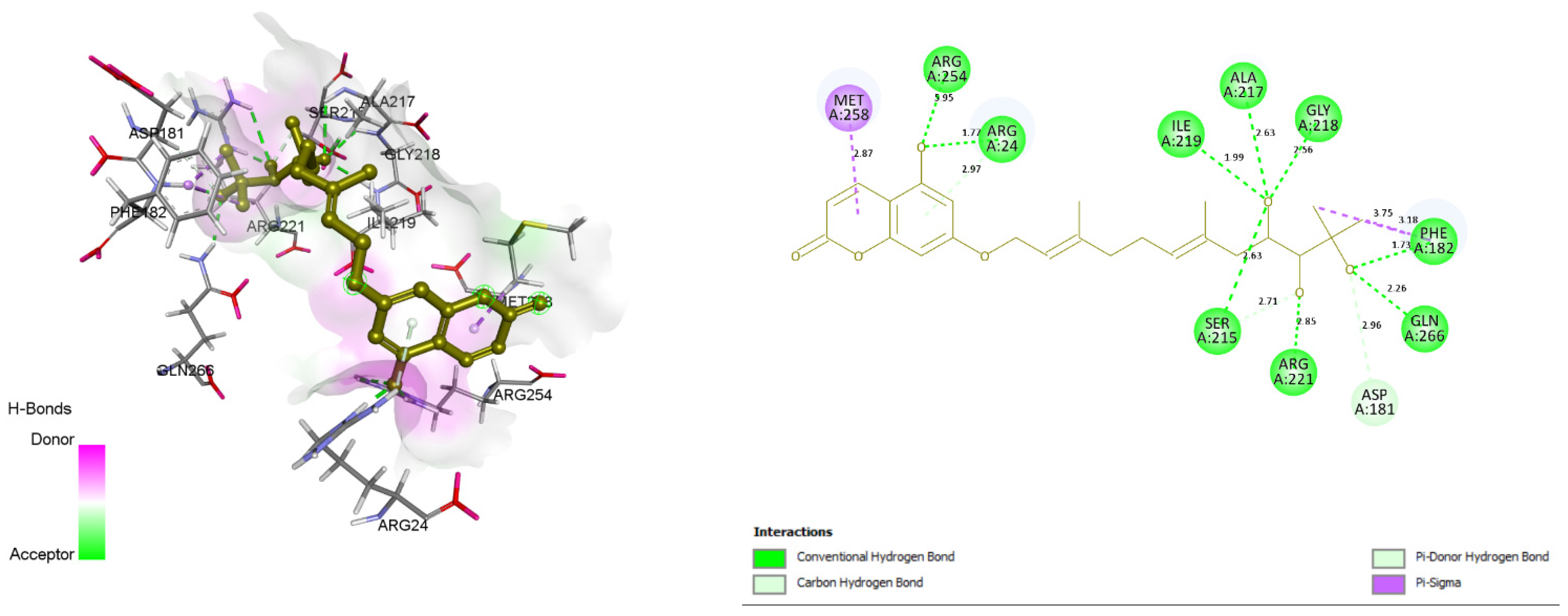
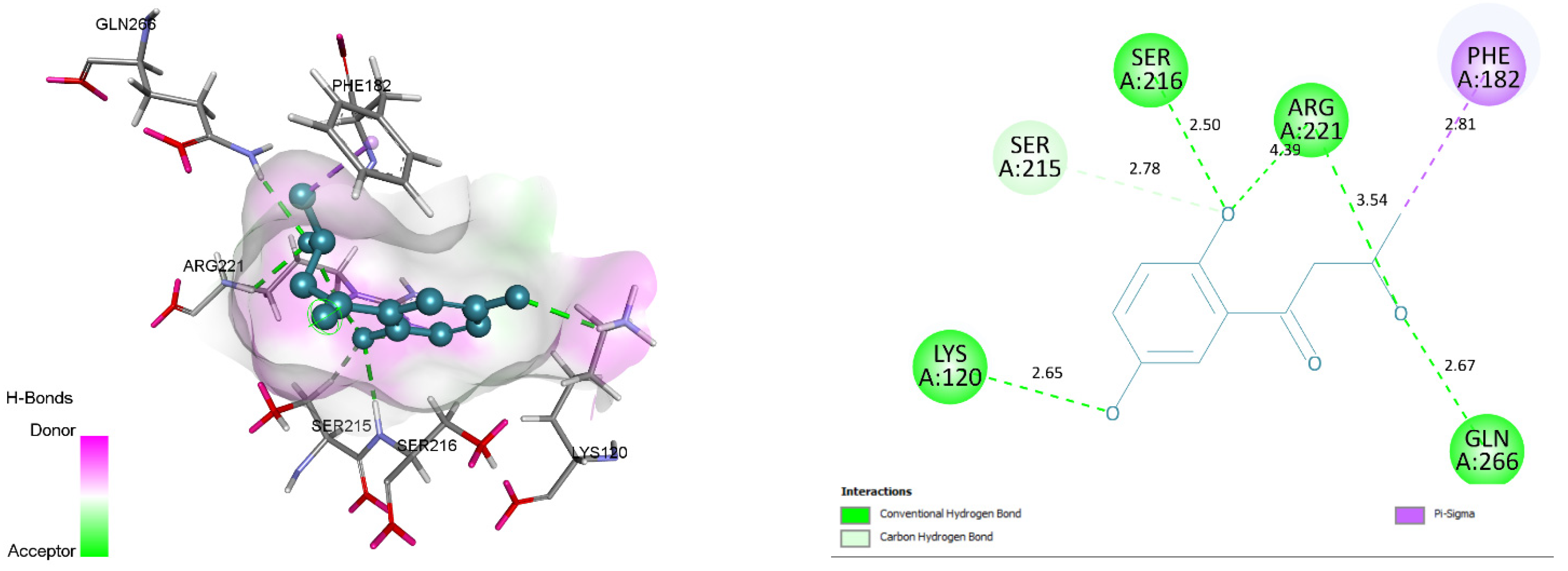
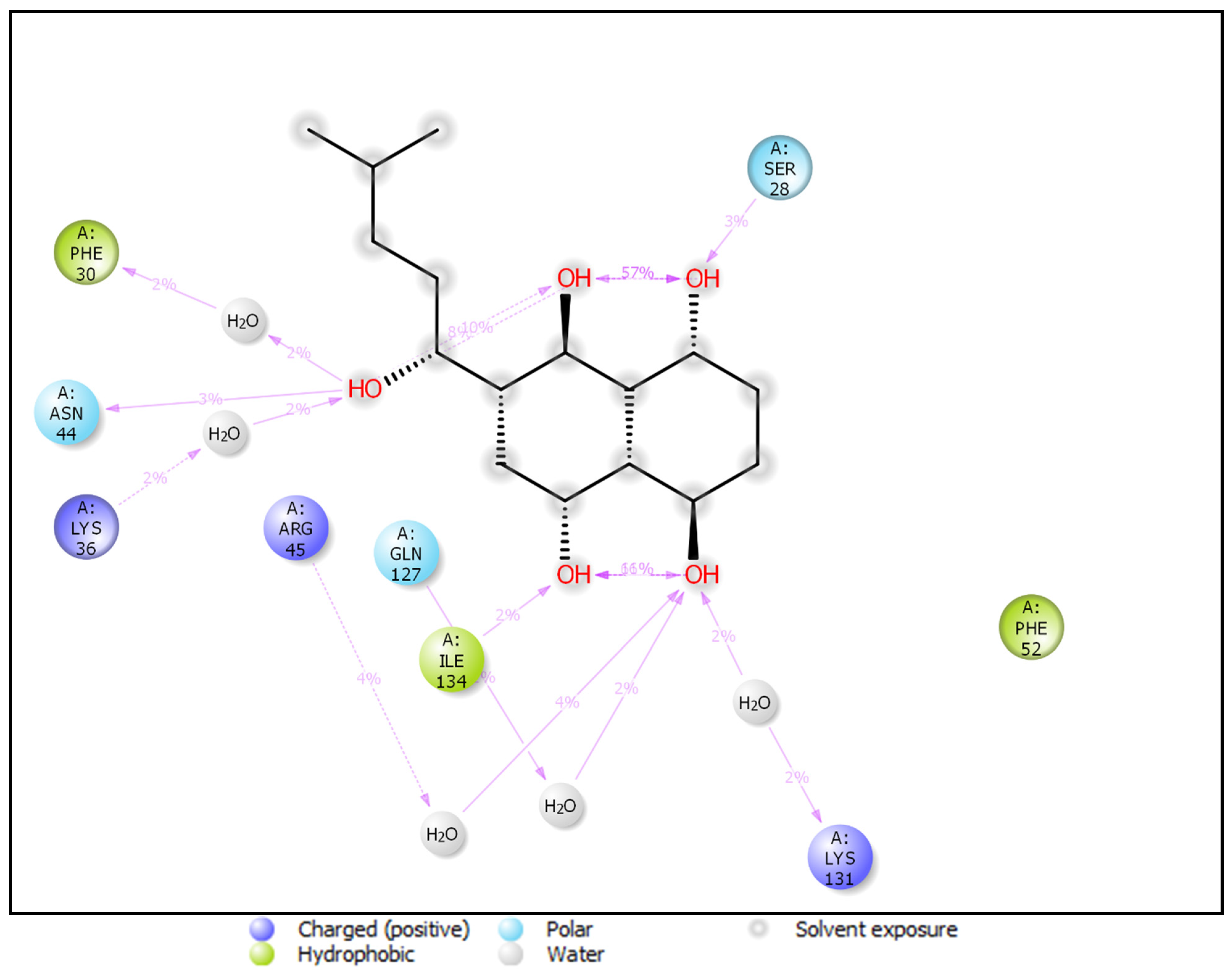
3.4. Docking and Intra-Molecular Interaction Analysis of Top 4 Compounds
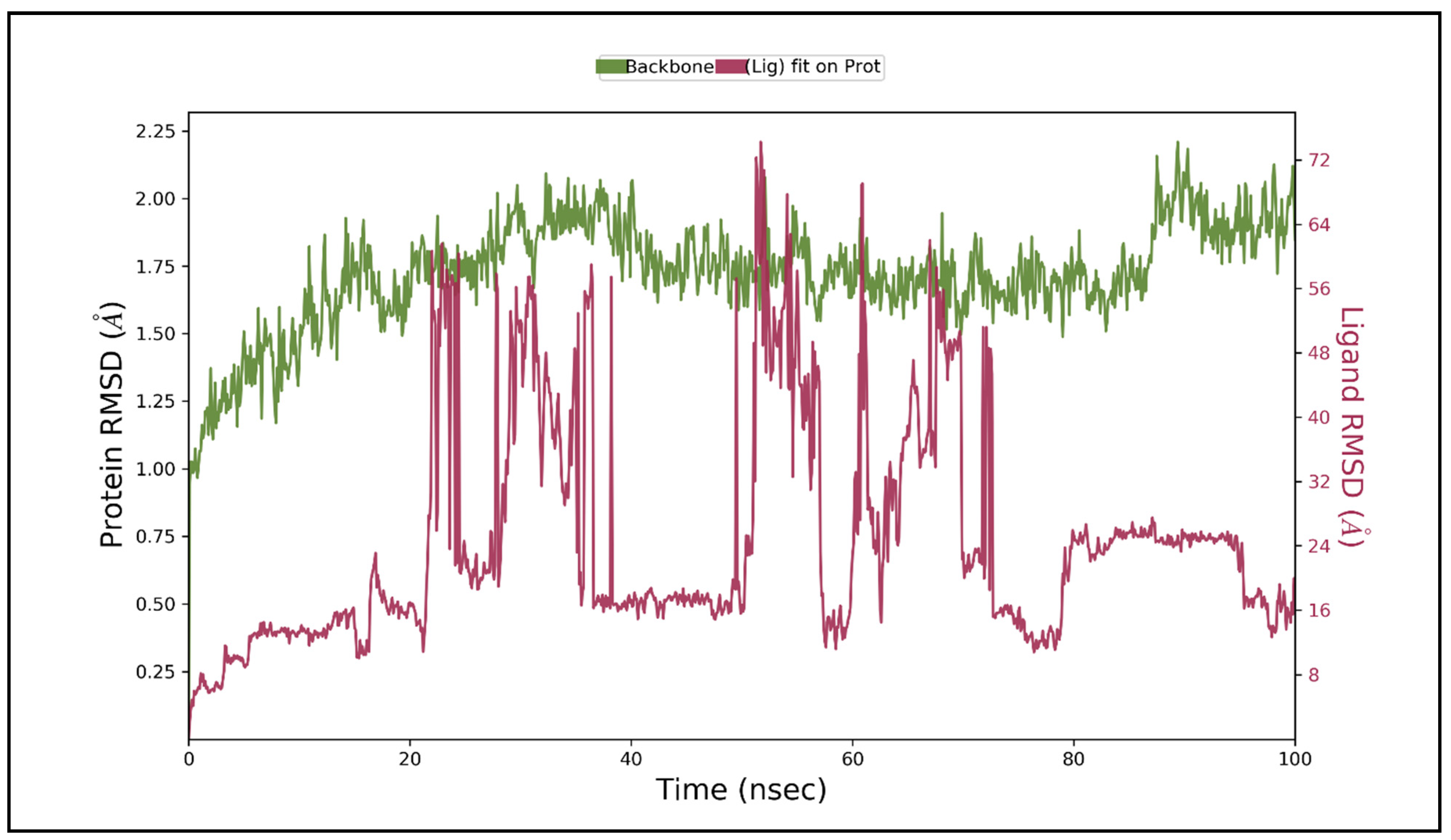
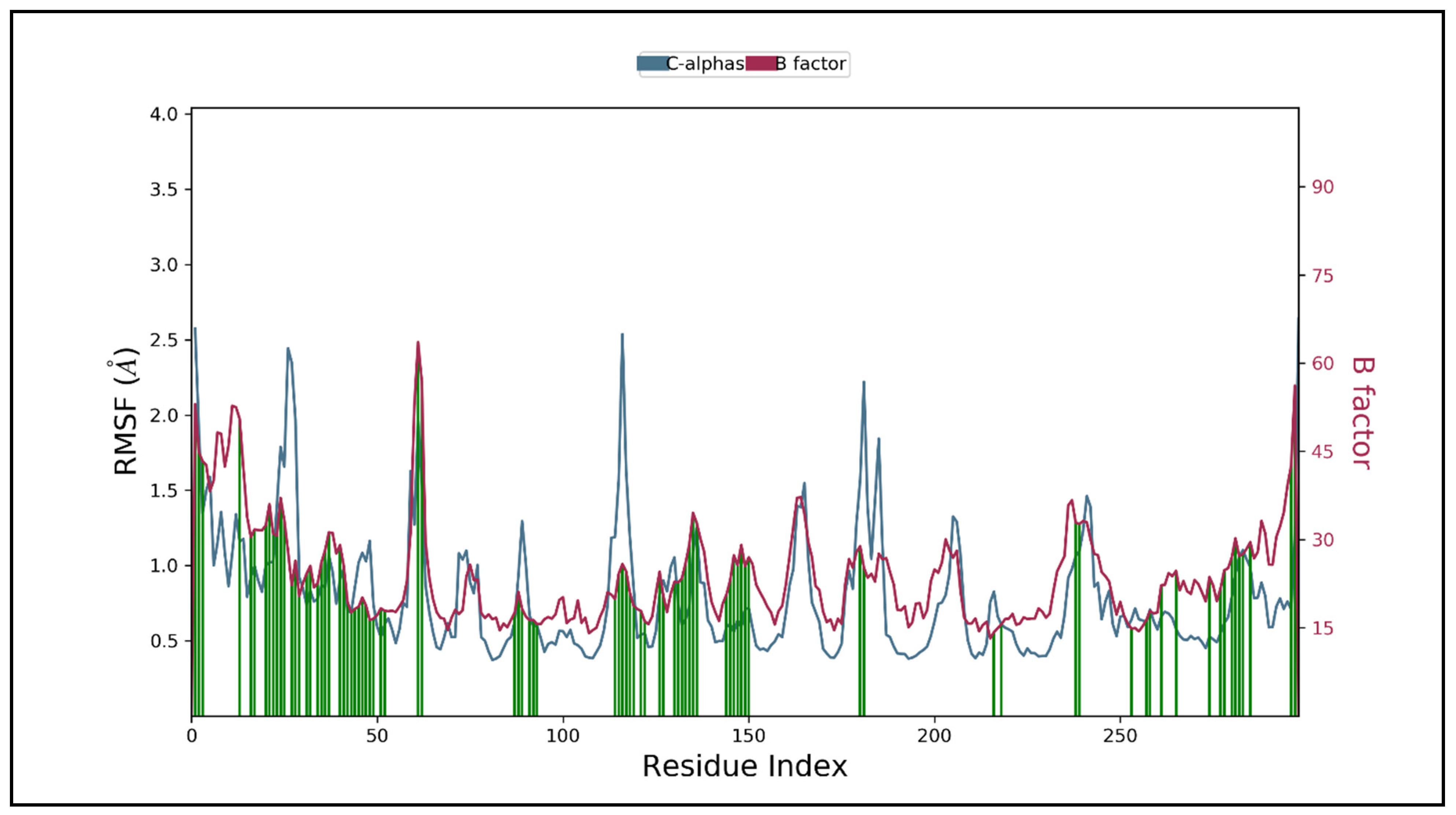
3.5. Molecular Dynamic Simulation Studies
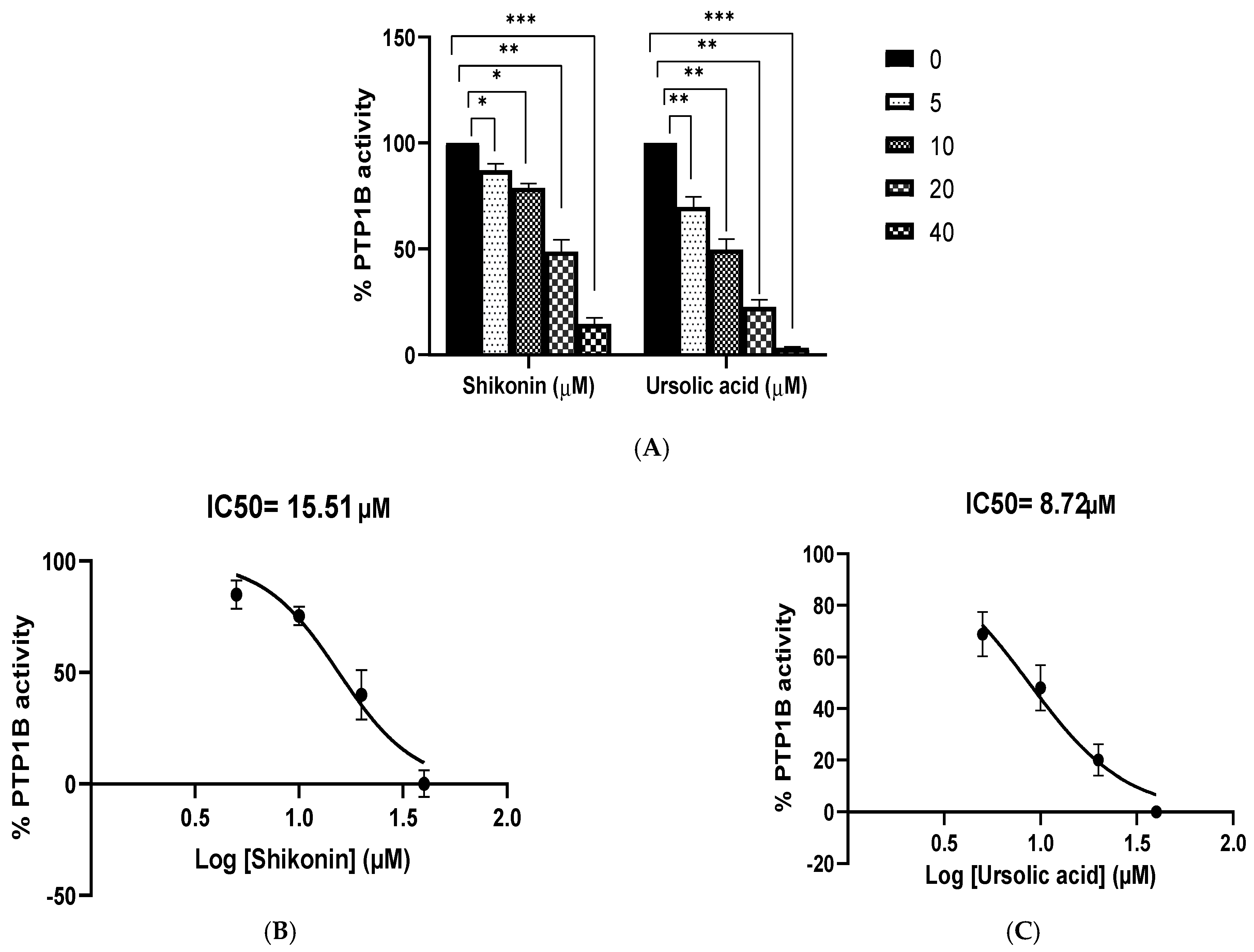
3.6. Inhibitory Activity of Shikonin against PTP1B
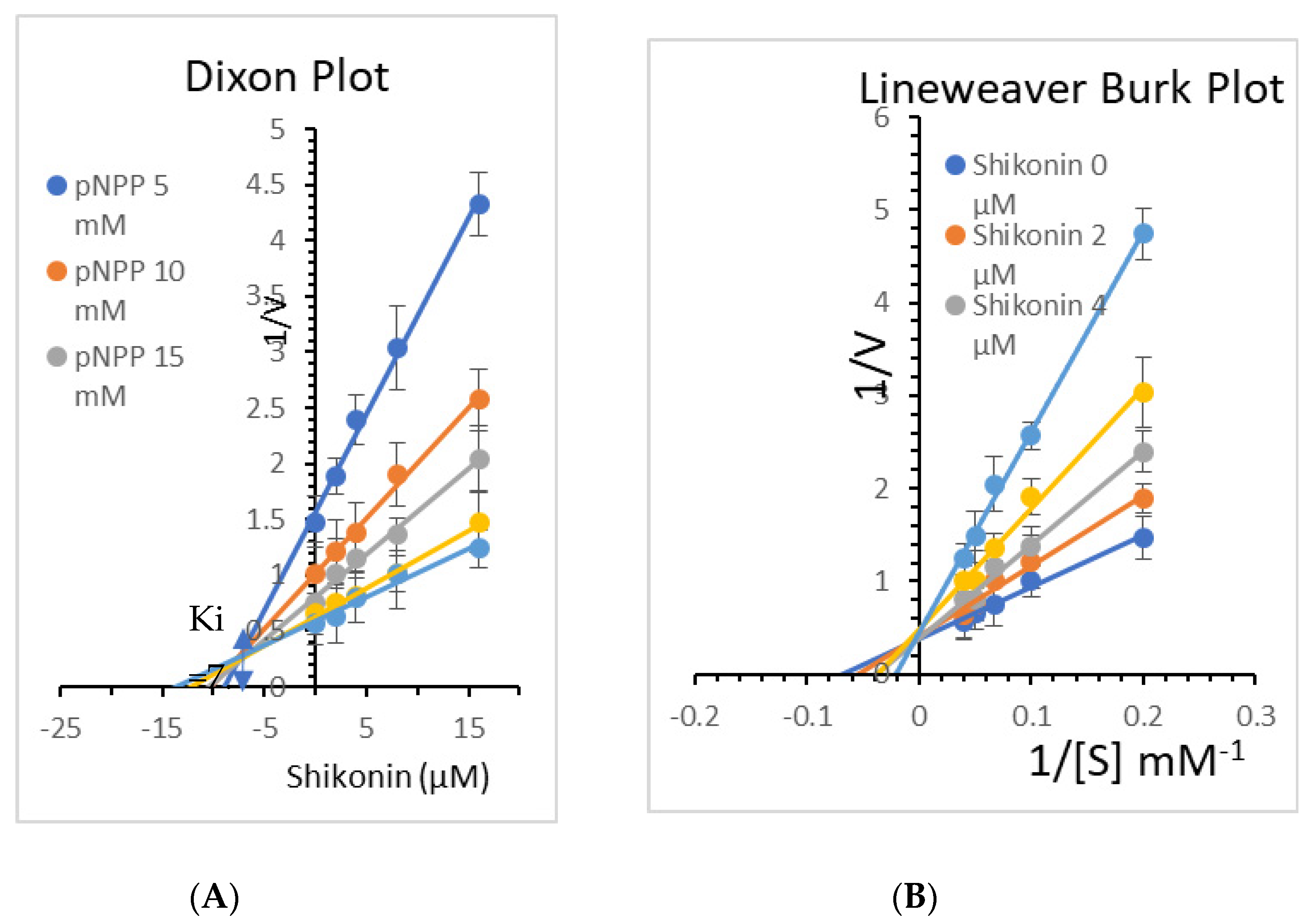
3.7. Enzyme Kinetics of PTP1B Inhibition by Shikonin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Shoaib, A.; Salem-Bekhit, M.M.; Siddiqui, H.H.; Dixit, R.K.; Bayomi, M.; Khalid, M.; Badruddeen; Shakeel, F. Antidiabetic activity of standardized dried tubers extract of Aconitum napellus in streptozotocin-induced diabetic rats. 3 Biotech. 2020, 10, 56. [Google Scholar] [CrossRef]
- Rowley, W.R.; Bezold, C. Creating public awareness: State 2025 diabetes forecasts. Popul. Health Manag. 2012, 15, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Al Dawish, M.A.; Robert, A.A.; Braham, R.; Al Hayek, A.A.; Al Saeed, A.; Ahmed, R.A.; Al Sabaan, F.S. Diabetes Mellitus in Saudi Arabia: A Review of the Recent Literature. Curr. Diabetes Rev. 2016, 12, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.A.; Al-Dawish, A.; Mujammami, M.; Dawish, M.A.A. Type 1 Diabetes Mellitus in Saudi Arabia: A Soaring Epidemic. Int. J. Pediatr. 2018, 2018, 9408370. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, P.; Lal Khokra, S.; Rana, A.C.; Kaushik, D. Pharmacophore Modeling and Molecular Docking Studies on Pinus roxburghii as a Target for Diabetes Mellitus. Adv. Bioinform. 2014, 2014, 903246. [Google Scholar] [CrossRef] [Green Version]
- Rocha, S.; Lucas, M.; Silva, V.L.M.; Gomes, P.M.O.; Silva, A.M.S.; Araujo, A.N.; Aniceto, N.; Guedes, R.C.; Corvo, M.L.; Fernandes, E.; et al. Pyrazoles as novel protein tyrosine phosphatase 1B (PTP1B) inhibitors: An in vitro and in silico study. Int. J. Biol. Macromol. 2021, 181, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, K.; Hegde, R.S. The Eyes Absent Proteins: Unusual HAD Family Tyrosine Phosphatases. Int. J. Mol. Sci. 2021, 22, 3925. [Google Scholar] [CrossRef]
- Belete, T.M. A recent achievement in the discovery and development of novel targets for the treatment of type-2 diabetes mellitus. J. Exp. Pharmacol. 2020, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kerru, N.; Singh-Pillay, A.; Awolade, P.; Singh, P. Current anti-diabetic agents and their molecular targets: A review. Eur. J. Med. Chem. 2018, 152, 436–488. [Google Scholar] [CrossRef]
- Asante-Appiah, E.; Kennedy, B.P. Protein tyrosine phosphatases: The quest for negative regulators of insulin action. Am. J. Physiol.-Endocrinol. Metab. 2003, 284, E663–E670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012, 40, D302–D305. [Google Scholar] [CrossRef] [PubMed]
- de Castro, E.; Sigrist, C.J.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Larvol, B.L.; Wilkerson, L.J. In silico drug discovery: Tools for bridging the NCE gap. Nat. Biotechnol. 1998, 16, 33–34. [Google Scholar] [CrossRef]
- Samad, A.; Ahammad, F.; Nain, Z.; Alam, R.; Imon, R.R.; Hasan, M.; Rahman, M.S. Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef]
- Balaramnavar, V.M.; Ahmad, K.; Saeed, M.; Ahmad, I.; Kamal, M.; Jawed, T. Pharmacophore-based approaches in the rational repurposing technique for FDA approved drugs targeting SARS-CoV-2 Mpro. RSC Adv. 2020, 10, 40264–40275. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–414. [Google Scholar] [CrossRef] [PubMed]
- Kumalo, H.M.; Soliman, M.E. Per-Residue Energy Footprints-Based Pharmacophore Modeling as an Enhanced in Silico Approach in Drug Discovery: A Case Study on the Identification of Novel b-Secretase1 (BACE1) Inhibitors as AntiAlzheimer Agents. Cell. Mol. Bioeng. 2016, 9, 175–189. [Google Scholar] [CrossRef]
- Agrawal, R.; Jain, P.; Dikshit, S.N.; Bahare, R.S.; Ganguly, S. Ligand-based pharmacophore detection, screening of potential pharmacophore and docking studies, to get effective glycogen synthase kinase inhibitors. Med. Chem. Res. 2013, 22, 5504–5535. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Al-Mohammadi, M.M.; Alamry, K.A.; Khan, K.A. Synthesis and biological evaluation of fluoropyrazolesulfonylurea and thiourea derivatives as possible antidiabetic agents. J. Enzym. Inhib. Med. Chem. 2016, 31, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Paudel, P.; Yu, T.; Seong, S.H.; Kuk, E.B.; Jung, H.A.; Choi, J.S. Protein Tyrosine Phosphatase 1B Inhibition and Glucose Uptake Potentials of Mulberrofuran G, Albanol B, and Kuwanon G from Root Bark of Morus alba L. in Insulin-Resistant HepG2 Cells: An In Vitro and In Silico Study. Int. J. Mol. Sci. 2018, 19, 1542. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Hirayama, T.; Ikeda, N. Design, synthesis and bioactivities of novel diarylthiophenes: Inhibitors of tumor necrosis factor-alpha (TNF-alpha) production. Bioorganic Med. Chem. 2002, 10, 3113–3122. [Google Scholar] [CrossRef]
- Barford, D.; Das, A.K.; Egloff, M.P. The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 133–164. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore | AlogP | PSA_2D | Plasma Protein Binding | Hepatotoxicity | CYP2D6 Binding | Aqueous Solubility | BBB Penetration | Intestinal Absorption |
|---|---|---|---|---|---|---|---|---|
| Shikonin | 2.444 | 97.048 | True (highly bound) | True (toxic) | False (non-inhibitor) | 3 (good) | 3 (low) | 0 (good) |
| ZINC31168041 | 3.725 | 79.747 | False (poorly bounded) | False (non-toxic) | False (non-inhibitor) | 3 (good) | 4 (undefined) | 1 (moderate) |
| ZINC31168045 | 3.725 | 118.422 | False (poorly bounded) | False (non-toxic) | False (non-inhibitor) | 3 (good) | 4 (undefined) | 1 (moderate) |
| ZINC31168048 | 3.725 | 118.422 | False (poorly bounded) | False (non-toxic) | False (non-inhibitor) | 3 (good) | 4 (undefined) | 1 (moderate) |
| ZINC31168554 | 1.039 | 118.422 | False (poorly bounded) | True (toxic) | False (non-inhibitor) | 4 (optimal) | 3 (low) | 0 (good) |
| Compounds | Jurs TPSA (<140 Å2) | MW (<500) | ALog P (≤5) | H-Bond Donor (≤5) | H-Bond Acceptor (≤10) | Rule of 5 Violations |
|---|---|---|---|---|---|---|
| Shikonin | 164.344 | 288.295 | 2.444 | 3 | 5 | 1 |
| ZINC31168041 | 192.687 | 432.507 | 3.725 | 4 | 7 | 1 |
| ZINC31168045 | 218.542 | 432.507 | 3.725 | 4 | 7 | 1 |
| ZINC31168048 | 186.02 | 432.507 | 3.725 | 4 | 7 | 1 |
| ZINC31168554 | 162.648 | 196.2 | 1.039 | 3 | 4 | 1 |
| Parameters | Shikonin | ZINC31168041 | ZINC31168045 | ZINC31168048 | ZINC31168554 |
|---|---|---|---|---|---|
| Rate of Oral LD50 (g/kg body weight) | 1.33207 | 4.97354 | 4.97354 | 4.97354 | 1.84165 |
| Rat inhalation LC50 (mg/m3/h) | 2359.66 | 1288.65 | 1288.65 | 1288.65 | 7376.76 |
| Daphnia EC50 (mg/mL) | 25.4641 | 1.89946 | 1.89946 | 1.89946 | 506.316 |
| Rat chronic LOEAL (g/kg body weight) | 0.0825521 | 0.0370396 | 0.0370396 | 0.0370396 | 0.220851 |
| Fathead minnow LC50 (g/L) | 0.0185828 | 0.000967424 | 0.000967424 | 0.000967424 | 0.802847 |
| Carcinogenic potency TD50 (mg/kg body weight/day) | |||||
| Mouse | 132.774 | 176.288 | 176.288 | 176.288 | 339.579 |
| Rat | 25.1746 | 9.39947 | 9.39947 | 9.39947 | 7.86891 |
| Rat maximum tolerated dose (g/kg body weight) | 0.697318 | 0.938401 | 0.938401 | 0.938401 | 1.03762 |
| Ames mutagenicity | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen |
| Developmental toxicity potential | Toxic | Toxic | Toxic | Toxic | Toxic |
| Aerobic biodegradability | Degradable | Degradable | Degradable | Degradable | Degradable |
| Ocular irritancy | None | None | None | None | Moderate |
| Skin irritancy | None | Mild | Mild | Mild | None |
| S. No. | Compound | Hydrogen Bonds | Gold Fitness Score |
|---|---|---|---|
| 1. | Shikonin | ARG24, GLN262, ASP48, MET258, AND SER28 | 46.3057 |
| 2. | ZINC31168041 | SER215, SER216, ALA217, GLY218, ILE219, GLY220, ARG221, ARG254, ARG24, SER215, SER216, GLN262. | 116.001 |
| 3. | ZINC31168045 | ARG24, PHE182, SER215, SER216, ALA217, GLY220, ARG221, ARG254, GLN266. | 118.114 |
| 4. | ZINC31168048 | ARG24, PHE182, SER215, ALA217, GLY218, ILE219, ARG221, ARG254, GLN266, ASP181. | 115.39 |
| 5. | ZINC31168554 | LYS120, SER216, ARG221, GLN266, SER215 | 59.7591 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, M.; Shoaib, A.; Tasleem, M.; Alabdallah, N.M.; Alam, M.J.; Asmar, Z.E.; Jamal, Q.M.S.; Bardakci, F.; Alqahtani, S.S.; Ansari, I.A.; et al. Assessment of Antidiabetic Activity of the Shikonin by Allosteric Inhibition of Protein-Tyrosine Phosphatase 1B (PTP1B) Using State of Art: An In Silico and In Vitro Tactics. Molecules 2021, 26, 3996. https://doi.org/10.3390/molecules26133996
Saeed M, Shoaib A, Tasleem M, Alabdallah NM, Alam MJ, Asmar ZE, Jamal QMS, Bardakci F, Alqahtani SS, Ansari IA, et al. Assessment of Antidiabetic Activity of the Shikonin by Allosteric Inhibition of Protein-Tyrosine Phosphatase 1B (PTP1B) Using State of Art: An In Silico and In Vitro Tactics. Molecules. 2021; 26(13):3996. https://doi.org/10.3390/molecules26133996
Chicago/Turabian StyleSaeed, Mohd, Ambreen Shoaib, Munazzah Tasleem, Nadiyah M. Alabdallah, Md Jahoor Alam, Zeina El Asmar, Qazi Mohammad Sajid Jamal, Fevzi Bardakci, Saad S. Alqahtani, Irfan Ahmad Ansari, and et al. 2021. "Assessment of Antidiabetic Activity of the Shikonin by Allosteric Inhibition of Protein-Tyrosine Phosphatase 1B (PTP1B) Using State of Art: An In Silico and In Vitro Tactics" Molecules 26, no. 13: 3996. https://doi.org/10.3390/molecules26133996