
Insertion of Phosphenium Ions into a Bicyclo[1.1.0]Tetraphosphabutane Iron Complex


Abstract
:1. Introduction
2. Results
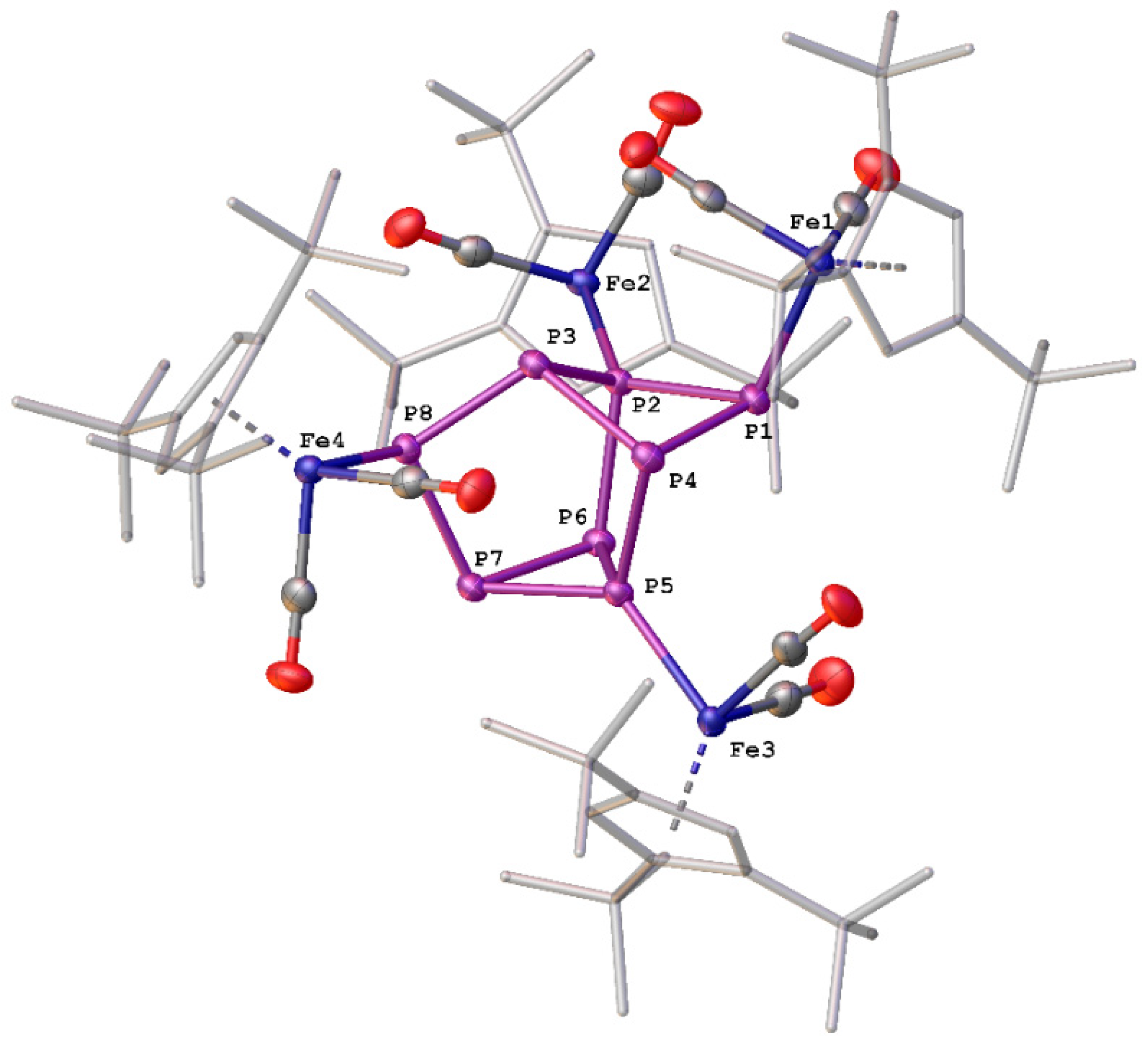
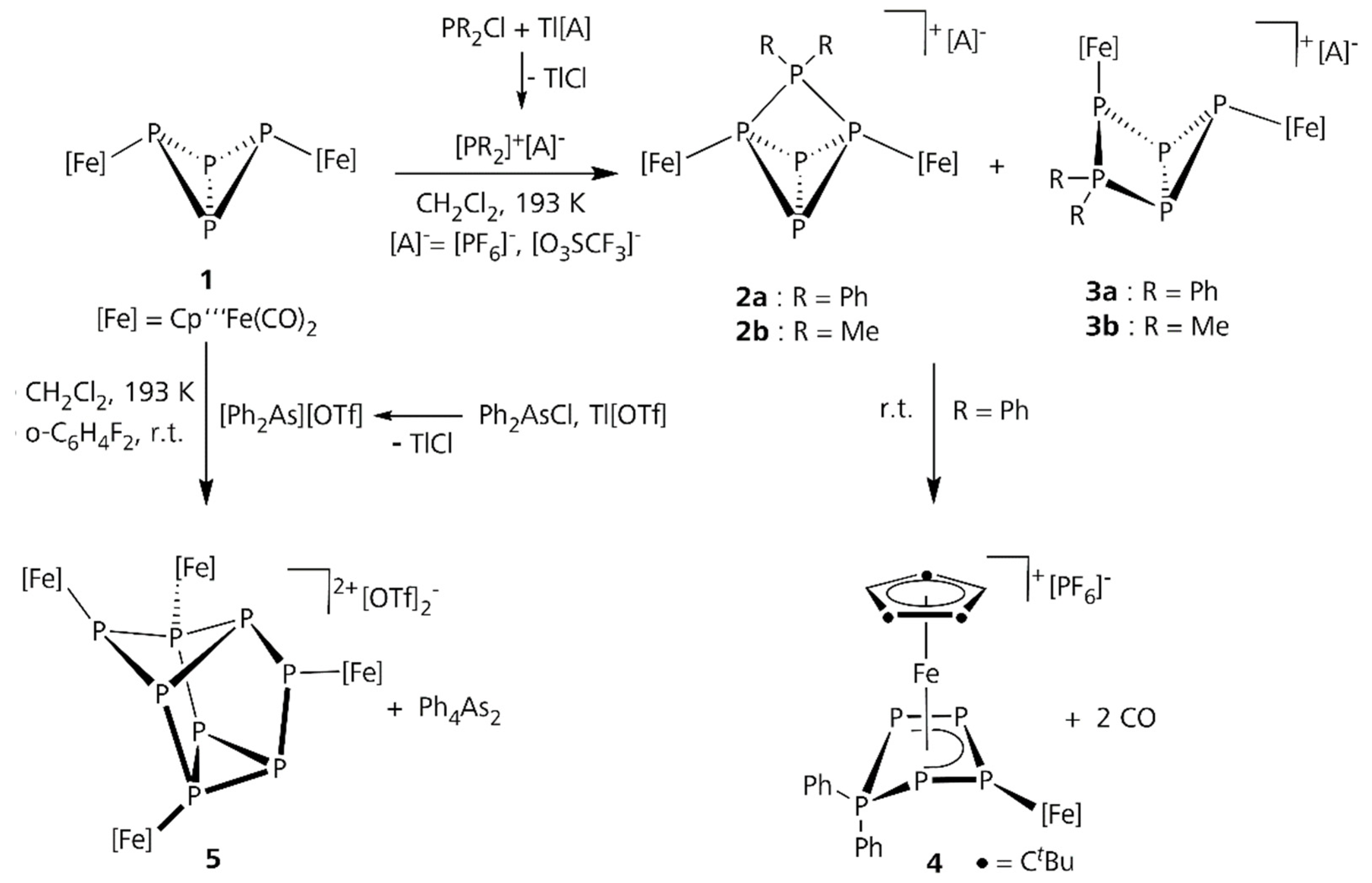
2.1. Reactions of 1 with [Ph2P]+[A]−
2.2. Reactions of 1 with [Ph2As]+[OTf]−
3. Discussion
4. Materials and Methods
4.1. General Techniques and Materials
4.2. Reaction of 1 with in Situ Generated [Ph2P][PF6] at Room Temperature
4.3. Low Temperature NMR Experiments
4.4. Reaction of 1 with in Situ Generated [Ph2As][OTf] at 193 K
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cossairt, B.M.; Piro, N.A.; Cummins, C.C. Early-transition-metal-mediated activation and transformation of white phosphorus. Chem. Rev. 2010, 110, 4164–4177. [Google Scholar] [CrossRef] [Green Version]
- Scheer, M.; Balázs, G.; Seitz, A. P4 activation by main group elements and compounds. Chem. Rev. 2010, 110, 4236–4256. [Google Scholar] [CrossRef]
- Caporali, M.; Gonsalvi, L.; Rossin, A.; Peruzzini, M. P4 activation by late-transition metal complexes. Chem. Rev. 2010, 110, 4178–4235. [Google Scholar] [CrossRef]
- Khan, S.; Sen, S.S.; Roesky, H.W. Activation of phosphorus by group 14 elements in low oxidation states. Chem. Commun. 2012, 48, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Borger, J.E.; Jongkind, M.K.; Ehlers, A.W.; Lutz, M.; Slootweg, J.C.; Lammertsma, K. Metalate-Mediated Functionalization of P4 by Trapping Anionic Cp*Fe(CO)2(η1-P4)− with Lewis Acids. ChemistryOpen 2017, 6, 350–353. [Google Scholar] [CrossRef]
- Ginsberg, A.P.; Lindsell, W.E. Rhodium complexes with the molecular unit P4 as a ligand. J. Am. Chem. Soc. 1971, 93, 2082–2084. [Google Scholar] [CrossRef]
- Scherer, O.J.; Hilt, T.; Wolmershäuser, G. P4 Activation with [{Cp‴(OC)2Fe}2](Cp‴ = C5H2tBu3-1,2,4): Exclusive Formation of the Exo/ Exo-Butterfly Complex [{Cp‴(OC)2Fe}2(μ-η1:η1-P4)]. Organometallics 1998, 17, 4110–4112. [Google Scholar] [CrossRef]
- Schwarzmaier, C.; Timoshkin, A.Y.; Balázs, G.; Scheer, M. Selective Formation and Unusual Reactivity of Tetraarsabicyclo[1.1.0]butane Complexes. Angew. Chem. Int. Ed. 2014, 53, 9077–9081. [Google Scholar] [CrossRef]
- Müller, J.; Balázs, G.; Scheer, M. From a P4 butterfly scaffold to cyclo- and catena-P4 units. Chem. Commun. 2021, 57, 2257–2260. [Google Scholar] [CrossRef]
- Müller, J.; Heinl, S.; Schwarzmaier, C.; Balázs, G.; Keilwerth, M.; Meyer, K.; Scheer, M. Rearrangement of a P4 Butterfly Complex-The Formation of a Homoleptic Phosphorus-Iron Sandwich Complex. Angew. Chem. Int. Ed. Engl. 2017, 56, 7312–7317. [Google Scholar] [CrossRef] [PubMed]
- Grünbauer, R.; Schwarzmaier, C.; Eberl, M.; Balázs, G.; Scheer, M. The reactivity of the P4-butterfly ligand [{Cp‴Fe(CO)2}2(µ,η1:1-P4)] towards transition metal complexes: Coordination versus rearrangement. Inorg. Chim. Acta 2021, 518, 120234. [Google Scholar] [CrossRef]
- Schwarzmaier, C.; Heinl, S.; Balázs, G.; Scheer, M. E4 Butterfly Complexes (E = P, As) as Chelating Ligands. Angew. Chem. Int. Ed. Engl. 2015, 54, 13116–13121. [Google Scholar] [CrossRef]
- Reichl, S.; Grünbauer, R.; Balázs, G.; Scheer, M. Reactivity of P4 butterfly complexes towards NHCs - generation of a metal-bridged P2 dumbbell complex. Chem. Commun. 2021, 57, 3383–3386. [Google Scholar] [CrossRef] [PubMed]
- Grünbauer, R.; Balázs, G.; Scheer, M. The Butterfly Complex {Cp*Cr(CO)3}2(μ,η1:1-P4) as a Versatile Ligand and Its Unexpected P1/P3 Fragmentation. Chemistry 2020, 26, 11722–11726. [Google Scholar] [CrossRef] [PubMed]
- Giffin, N.A.; Masuda, J.D. Reactivity of white phosphorus with compounds of the p-block. Coord. Chem. Rev. 2011, 255, 1342–1359. [Google Scholar] [CrossRef]
- Weigand, J.J.; Holthausen, M.; Fröhlich, R. Formation of [Ph2P5]+, [Ph4P6]2+, and [Ph6P7]3+ Cationic Clusters by Consecutive Insertions of [Ph2P]+ into P-P Bonds of the P4 Tetrahedron. Angew. Chem. Int. Ed. Engl. 2009, 48, 295–298. [Google Scholar] [CrossRef]
- Burford, N.; Cameron, T.S.; Ragogna, P.J.; Ocando-Mavarez, E.; Gee, M.; McDonald, R.; Wasylishen, R.E. Phosphine ligand exchange at a phosphine lewis acceptor: The first structural characterization of homoleptic phosphinophosphonium salts. J. Am. Chem. Soc. 2001, 123, 7947–7948. [Google Scholar] [CrossRef]
- Burford, N.; Cameron, T.S.; LeBlanc, D.J.; Losier, P.; Sereda, S.; Wu, G. Structural Alternatives in R2(Cl)P: GaCl3 Systems (R = Alkyl, Phenyl), Including Examples of Intermolecular P → P Coordination. Organometallics 1997, 16, 4712–4717. [Google Scholar] [CrossRef]
- Dyker, C.A.; Burford, N. Catena-phosphorus cations. Chem. Asian J. 2008, 3, 28–36. [Google Scholar] [CrossRef]
- Weigand, J.J.; Burford, N.; Lumsden, M.D.; Decken, A. A Melt Approach to the Synthesis ofcatena-Phosphorus Dications To Access Derivatives of [P6Ph4R4]2+. Angew. Chem. 2006, 118, 6885–6889. [Google Scholar] [CrossRef]
- Chitnis, S.S.; MacDonald, E.; Burford, N.; Werner-Zwanziger, U.; McDonald, R. P-P Menschutkin preparation of prototypical phosphinophosphonium salts. Chem. Commun. 2012, 48, 7359–7361. [Google Scholar] [CrossRef] [PubMed]
- Holthausen, M.H.; Weigand, J.J. The chemistry of cationic polyphosphorus cage—Syntheses, structure and reactivity. Chem. Soc. Rev. 2014, 43, 6639–6657. [Google Scholar] [CrossRef] [Green Version]
- Burford, N.; Ragogna, P.J.; McDonald, R.; Ferguson, M.J. Phosphine coordination complexes of the diphenylphosphenium cation: A versatile synthetic methodology for P-P bond formation. J. Am. Chem. Soc. 2003, 125, 14404–14410. [Google Scholar] [CrossRef]
- Weigand, J.J.; Burford, N.; Davidson, R.J.; Cameron, T.S.; Seelheim, P. New synthetic procedures to catena-phosphorus cations: Preparation and dissociation of the first cyclo-phosphino-halophosphonium salts. J. Am. Chem. Soc. 2009, 131, 17943–17953. [Google Scholar] [CrossRef]
- Krossing, I.; Raabe, I. P5X2+ (X=Br, I), a Phosphorus-Rich Binary P-X Cation with a C(2v)-Symmetric P5 Cage. Angew. Chem. Int. Ed. Engl. 2001, 40, 4406–4409. [Google Scholar] [CrossRef]
- Riesinger, C.; Dütsch, L.; Balázs, G.; Bodensteiner, M.; Scheer, M. Cationic Functionalisation by Phosphenium Ion Insertion. Chem. Eur. J. 2020, 26, 17165–17170. [Google Scholar] [CrossRef]
- Riesinger, C.; Balázs, G.; Bodensteiner, M.; Scheer, M. Stabilization of Pentaphospholes as η5-Coordinating Ligands. Angew. Chem. Int. Ed. Engl. 2020, 59, 23879–23884. [Google Scholar] [CrossRef]
- Kilah, N.L.; Petrie, S.; Stranger, R.; Wielandt, J.W.; Willis, A.C.; Wild, S.B. Triphenylphosphine-Stabilized Diphenyl-Arsenium, -Stibenium, and -Bismuthenium Salts. Organometallics 2007, 26, 6106–6113. [Google Scholar] [CrossRef]
- Braddock, J.M.F.; Coates, G.E. 628. Arsinophosphonium salts. J. Chem. Soc. 1961, 3208–3211. [Google Scholar] [CrossRef]
- Porter, K.A.; Willis, A.C.; Zank, J.; Wild, S.B. Phosphine-stabilized arsenium salts: Water-stable, labile, coordination complexes. Inorg. Chem. 2002, 41, 6380–6386. [Google Scholar] [CrossRef]
- Bernstein, H.J.; Pople, J.A.; Schneider, W.G. The Analysis of Nuclear Magnetic Resonance Spectra: I. Systems of two and three Nuclei. Can. J. Chem. 1957, 35, 65–81. [Google Scholar] [CrossRef]
- Adhikari, A.K.; Ziegler, C.G.P.; Schwedtmann, K.; Taube, C.; Weigand, J.J.; Wolf, R. Functionalization of Pentaphosphorus Cations by Complexation. Angew. Chem. Int. Ed. Engl. 2019, 58, 18584–18590. [Google Scholar] [CrossRef]
- Hennersdorf, F.; Weigand, J.J. A Tetracyclic Octaphosphane by Successive Addition, Inversion, and Condensation Reactions. Angew. Chem. Int. Ed. Engl. 2017, 56, 7858–7862. [Google Scholar] [CrossRef] [PubMed]
- Barr, M.E.; Adams, B.R.; Weller, R.R.; Dahl, L.F. Synthesis and structural-bonding analysis of [η5-C5H4Me)4Fe4(CO)6P8] and [η5-C5H4Me)4Fe6(CO)13P8]: Two unprecedented transition-metal complexes containing the cage-like P8 subunit of Hittorf’s monoclinic phosphorus allotrope. J. Am. Chem. Soc. 1991, 113, 3052–3060. [Google Scholar] [CrossRef]
- Deringer, V.L.; Pickard, C.J.; Proserpio, D.M. Hierarchically Structured Allotropes of Phosphorus from Data-Driven Exploration. Angew. Chem. Int. Ed. Engl. 2020, 59, 15880–15885. [Google Scholar] [CrossRef]
- Mädl, E.; Balázs, G.; Peresypkina, E.V.; Scheer, M. Unexpected Reactivity of (η5-1,2,4-tBuC5H2)Ni(η3-P3) towards Main Group Nucleophiles and by Reduction. Angew. Chem. Int. Ed. Engl. 2016, 55, 7702–7707. [Google Scholar] [CrossRef]
- Scheer, M.; Becker, U.; Matern, E. Ir-Komplexe mit P4-Bicyclotetraphosphan und P8-Cunean als Liganden—CO-Insertion in eine Ir–P-Bindung. Chem. Ber. 1996, 129, 721–724. [Google Scholar] [CrossRef]
- Hennersdorf, F.; Frötschel, J.; Weigand, J.J. Selective Derivatization of a Hexaphosphane from Functionalization of White Phosphorus. J. Am. Chem. Soc. 2017, 139, 14592–14604. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, F.; Graßl, C.; Balázs, G.; Zolnhofer, E.M.; Meyer, K.; Scheer, M. Influence of the nacnac Ligand in Iron(I)-Mediated P4 Transformations. Angew. Chem. Int. Ed. Engl. 2016, 55, 4340–4344. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Diaconescu, P.L. P4 activation by group 3 metal arene complexes. Chem. Commun. 2012, 48, 2216–2218. [Google Scholar] [CrossRef] [PubMed]
- Konchenko, S.N.; Pushkarevsky, N.A.; Gamer, M.T.; Köppe, R.; Schnöckel, H.; Roesky, P.W. {(η5-C5Me5)2Sm}4P8: A molecular polyphosphide of the rare-earth elements. J. Am. Chem. Soc. 2009, 131, 5740–5741. [Google Scholar] [CrossRef] [PubMed]
- Lerner, H.-W.; Margraf, G.; Kaufmann, L.; Bats, J.W.; Bolte, M.; Wagner, M. Syntheses, Reactivity, and X-ray Structure Analyses of the Dimeric Pentaphosphides [(tBu3Si)3P5M2]2 (M = Na, Ag) and the Bicyclo[2.1.0]pentaphosphane (tBu3Si)3P5. Eur. J. Inorg. Chem. 2005, 2005, 1932–1939. [Google Scholar] [CrossRef]
- Butovskiy, M.V.; Balázs, G.; Bodensteiner, M.; Peresypkina, E.V.; Virovets, A.V.; Sutter, J.; Scheer, M. Ferrocene and pentaphosphaferrocene: A comparative study regarding redox chemistry. Angew. Chem. Int. Ed. Engl. 2013, 52, 2972–2976. [Google Scholar] [CrossRef]
- Mädl, E.; Butovskii, M.V.; Balázs, G.; Peresypkina, E.V.; Virovets, A.V.; Seidl, M.; Scheer, M. Functionalization of a cyclo-P5 ligand by main-group element nucleophiles. Angew. Chem. Int. Ed. Engl. 2014, 53, 7643–7646. [Google Scholar] [CrossRef]
- Krossing, I. The Facile Preparation of Weakly Coordinating Anions: Structure and Characterisation of Silverpolyfluoroalkoxyaluminates AgAl(ORF)4, Calculation of the Alkoxide Ion Affinity. Chem. Eur. J. 2001, 7, 490–502. [Google Scholar] [CrossRef]
- Scherer, O.J.; Schwarz, G.; Wolmershäuser, G. Eisen-Zweikernkomplexe mit unterschiedlichen P4-Liganden. Z. Anorg. Allg. Chem. 1996, 622, 951–957. [Google Scholar] [CrossRef]
- Cullen, W.R.; Trotter, J. Crystal data for diphenylarsenicals. Can. J. Chem. 1961, 39, 2602–2603. [Google Scholar] [CrossRef]
- Burkhardt, A.; Pakendorf, T.; Reime, B.; Meyer, J.; Fischer, P.; Stübe, N.; Panneerselvam, S.; Lorbeer, O.; Stachnik, K.; Warmer, M.; et al. Status of the crystallography beamlines at PETRA III. Eur. Phys. J. Plus 2016, 131, 56. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, M.; Balázs, G.; Virovets, A.V.; Peresypkina, E.; Scheer, M. Insertion of Phosphenium Ions into a Bicyclo[1.1.0]Tetraphosphabutane Iron Complex. Molecules 2021, 26, 3920. https://doi.org/10.3390/molecules26133920
Weber M, Balázs G, Virovets AV, Peresypkina E, Scheer M. Insertion of Phosphenium Ions into a Bicyclo[1.1.0]Tetraphosphabutane Iron Complex. Molecules. 2021; 26(13):3920. https://doi.org/10.3390/molecules26133920
Chicago/Turabian StyleWeber, Martin, Gábor Balázs, Alexander V. Virovets, Eugenia Peresypkina, and Manfred Scheer. 2021. "Insertion of Phosphenium Ions into a Bicyclo[1.1.0]Tetraphosphabutane Iron Complex" Molecules 26, no. 13: 3920. https://doi.org/10.3390/molecules26133920