
Combining Molecular Dynamic Information and an Aspherical-Atom Data Bank in the Evaluation of the Electrostatic Interaction Energy in Multimeric Protein-Ligand Complex: A Case Study for HIV-1 Protease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
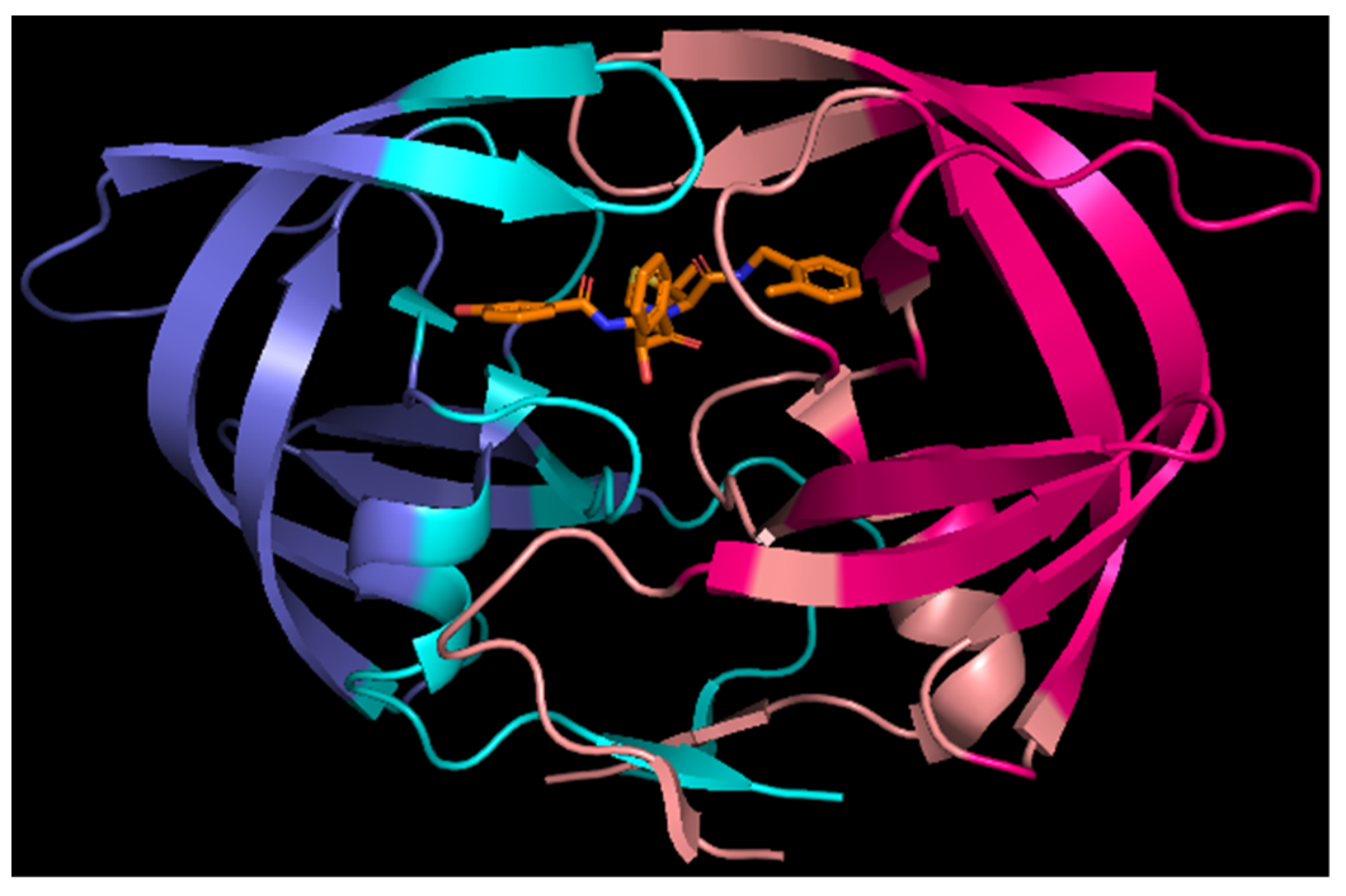
Abstract
:1. Background
2. Theory and Computation Details
2.1. Molecular Dynamics (MD) Simulation
2.2. Electrostatic Interaction Energy
3. Results and Discussion
3.1. HIV-1 Protease Interactions with Ligand
3.2. Monomer–Monomer Interactions in HIV-1 Protease
3.3. Mutants Disrupting Monomer–Monomer Interactions in HIV-1 Protease
3.4. Information Gained When Compared to Conventional Atomic Point-Charge Model of Electrostatics
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Appadurai, R.; Senapati, S. Dynamical Network of HIV-1 Protease Mutants Reveals the Mechanism of Drug Resistance and Unhindered Activity. Biochemistry 2016, 55, 1529–1540. [Google Scholar] [CrossRef]
- Shafer, R.W. Genotypic Testing for Human Immunodeficiency Virus Type 1 Drug Resistance. Clin. Microbiol. Rev. 2002, 15, 247–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Li, M.; Nguyen, J.T.; Kiso, Y.; Gustchina, A.; Wlodawer, A. Crystal structures of inhibitor complexes of human T-cell leukemia virus (HTLV-1) protease. J. Mol. Biol. 2010, 401, 626–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinaga, M.; Cherney, M.M.; Tarasova, N.I.; Bartlett, P.A.; Hanson, J.E.; James, M.N.G. Structural study of the complex between human pepsin and a phosphorus-containing peptidic transition-state analog. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 272–279. [Google Scholar] [CrossRef]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the Protease Domain of Memapsin 2 (β-Secretase) Complexed with Inhibitor. Science 2000, 290, 150–153. [Google Scholar] [CrossRef]
- Jaskólski, M.; Tomasselli, A.G.; Sawyer, T.K.; Staples, D.G.; Heinrikson, R.L.; Schneider, J.; Kent, S.B.H.; Wlodawer, A. Structure at 2.5-.ANG. resolution of chemically synthesized Human Immunodeficiency Virus Type 1 protease complexed with a hydroxyethylene-based inhibitor. Biochemistry 1991, 30, 1600–1609. [Google Scholar] [CrossRef]
- Gulnik, S.; Erickson, J.W.; Xie, D. HIV protease: Enzyme function and drug resistance. Vitam. Horm. 2000, 58, 213–256. [Google Scholar] [CrossRef]
- Kozal, M.J.; Shah, N.; Shen, N.; Yang, R.; Fucini, R.; Merigan, T.C.; Richman, D.D.; Morris, D.; Hubbell, E.; Chee, M.; et al. Extensive polymorphisms observed in HIV–1 clade B protease gene using high–density oligonucleotide arrays. Nat. Med. 1996, 2, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Shafer, R.W.; Chuang, T.K.; Hsu, P.; White, C.B.; Katzenstein, D. Sequence and Drug Susceptibility of Subtype C Protease from Human Immunodeficiency Virus Type 1 Seroconverters in Zimbabwe. AIDS Res. Hum. Retrovir. 1999, 15, 65–69. [Google Scholar] [CrossRef]
- Boden, D.; Markowitz, M. Resistance to Human Immunodeficiency Virus Type 1 Protease Inhibitors. Antimicrob. Agents Chemother. 1998, 42, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Hertogs, K.; Bloor, S.; Kemp, S.D.; Eynde, C.V.D.; Alcorn, T.M.; Pauwels, R.; Van Houtte, M.; Staszewski, S.; Miller, V.; Larder, B.A. Phenotypic and genotypic analysis of clinical HIV-1 isolates reveals extensive protease inhibitor cross-resistance: A survey of over 6000 samples. AIDS 2000, 14, 1203–1210. [Google Scholar] [CrossRef]
- Erickson, J.W.; Burt, S.K. Structural mechanisms of HIV drug resistance. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 545–571. [Google Scholar] [CrossRef] [PubMed]
- Muzammil, S.; Ross, P.; Freire, E. A Major Role for a Set of Non-Active Site Mutations in the Development of HIV-1 Protease Drug Resistance. Biochemistry 2003, 42, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Olsen, D.B.; Stahlhut, M.W.; Rutkowski, C.A.; Schock, H.B.; Vanolden, A.L.; Kuo, L.C. Non-active Site Changes Elicit Broad-based Cross-resistance of the HIV-1 Protease to Inhibitors. J. Biol. Chem. 1999, 274, 23699–23701. [Google Scholar] [CrossRef] [Green Version]
- Bojarowski, S.A.; Kumar, P.; Dominiak, P.M. A Universal and Straightforward Approach to Include Penetration Effects in Electrostatic Interaction Energy Estimation. ChemPhysChem 2016, 17, 2455–2460. [Google Scholar] [CrossRef]
- Kumar, P.; Bojarowski, S.; Jarzembska, K.N.; Domagała, S.; Vanommeslaeghe, K.; MacKerell, A.D.; Dominiak, P. A Comparative Study of Transferable Aspherical Pseudoatom Databank and Classical Force Fields for Predicting Electrostatic Interactions in Molecular Dimers. J. Chem. Theory Comput. 2014, 10, 1652–1664. [Google Scholar] [CrossRef]
- Bojarowski, S.A.; Kumar, P.; Wandtke, C.M.; Dittrich, B.; Dominiak, P.M. Universal Method for Electrostatic Interaction Energies Estimation with Charge Penetration and Easily Attainable Point Charges. J. Chem. Theory Comput. 2018, 14, 6336–6345. [Google Scholar] [CrossRef]
- Kramer, C.; Spinn, A.; Liedl, K. Charge Anisotropy: Where Atomic Multipoles Matter Most. J. Chem. Theory Comput. 2014, 10, 4488–4496. [Google Scholar] [CrossRef]
- Shi, Y.; Xia, Z.; Zhang, J.; Best, R.; Wu, C.; Ponder, J.W.; Ren, P. Polarizable Atomic Multipole-Based AMOEBA Force Field for Proteins. J. Chem. Theory Comput. 2013, 9, 4046–4063. [Google Scholar] [CrossRef] [Green Version]
- Söderhjelm, P.; Ryde, U. How Accurate Can a Force Field Become? A Polarizable Multipole Model Combined with Fragment-wise Quantum-Mechanical Calculations. J. Phys. Chem. A 2009, 113, 617–627. [Google Scholar] [CrossRef]
- Stone, A.J. Electrostatic Damping Functions and the Penetration Energy. J. Phys. Chem. A 2011, 115, 7017–7027. [Google Scholar] [CrossRef]
- Wang, B.; Truhlar, D.G. Screened Electrostatic Interactions in Molecular Mechanics. J. Chem. Theory Comput. 2014, 10, 4480–4487. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and Efficient Model Energies for Exploring Intermolecular Interactions in Molecular Crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojarowski, S.A.; Kumar, P.; Dominiak, P.M. Interplay of point multipole moments and charge penetration for intermolecular electrostatic interaction energies from the University at Buffalo pseudoatom databank model of electron density. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A. The use of the promolecular charge density to approximate the penetration contribution to intermolecular electrostatic energies. Chem. Phys. Lett. 2006, 418, 158–162. [Google Scholar] [CrossRef]
- Dominiak, P.; Volkov, A.; Li, X.; Messerschmidt, M.; Coppens, P. A Theoretical Databank of Transferable Aspherical Atoms and Its Application to Electrostatic Interaction Energy Calculations of Macromolecules. J. Chem. Theory Comput. 2006, 3, 232–247. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Dominiak, P.M. New version of the theoretical databank of transferable aspherical pseudoatoms, UBDB2011—Towards nucleic acid modelling. Acta Crystallogr. Sect. A Found. Crystallogr. 2012, 68, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Gruza, B.; Bojarowski, S.A.; Dominiak, P.M. Extension of the transferable aspherical pseudoatom data bank for the comparison of molecular electrostatic potentials in structure–activity studies. Acta Crystallogr. Sect. A Found. Adv. 2019, 75, 398–408. [Google Scholar] [CrossRef]
- Volkov, A.; Li, X.; Koritsanszky, A.T.; Coppens, P. Ab Initio Quality Electrostatic Atomic and Molecular Properties Including Intermolecular Energies from a Transferable Theoretical Pseudoatom Databank. J. Phys. Chem. A 2004, 108, 4283–4300. [Google Scholar] [CrossRef]
- Boggavarapu, R.; Jeckelmann, J.-M.; Harder, D.; Ucurum, Z.; Fotiadis, D.I. Role of electrostatic interactions for ligand recognition and specificity of peptide transporters. BMC Biol. 2015, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sartori, G.R.; Nascimento, A.S. Comparative Analysis of Electrostatic Models for Ligand Docking. Front. Mol. Biosci. 2019, 6, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitencourt-Ferreira, G.; Veit-Acosta, M.; De Azevedo, W.F. Electrostatic Energy in Protein–Ligand Complexes. Methods Mol. Biol. 2019, 2053, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Masukawa, K.M.; Kollman, A.P.A.; Kuntz, I.D. Investigation of Neuraminidase-Substrate Recognition Using Molecular Dynamics and Free Energy Calculations. J. Med. Chem. 2003, 46, 5628–5637. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.; Pindi, C.; Senapati, S. Electrostatics Plays a Crucial Role in HIV-1 Protease Substrate Binding, Drugs Fail to Take Advantage. Biochemistry 2020, 59, 3316–3331. [Google Scholar] [CrossRef] [PubMed]
- Langner, K.M.; Beker, W.; Sokalski, W.A. Robust Predictive Power of the Electrostatic Term at Shortened Intermolecular Distances. J. Phys. Chem. Lett. 2012, 3, 2785–2789. [Google Scholar] [CrossRef]
- Reiling, K.K.; Endres, N.F.; Dauber, D.S.; Craik, C.S.; Stroud, R.M. Anisotropic Dynamics of the JE-2147−HIV Protease Complex: Drug Resistance and Thermodynamic Binding Mode Examined in a 1.09 Å Structure. Biochemistry 2002, 41, 4582–4594. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Cai, Y.; Nalam, M.N.L.; Bolon, D.N.A.; Schiffer, C.A. Hydrophobic Core Flexibility Modulates Enzyme Activity in HIV-1 Protease. J. Am. Chem. Soc. 2012, 134, 4163–4168. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufrievl, A. H++: A server for estimating p Ka s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Case, D.A.; Walker, B.C.; Cheatham, T.E., III; Simmerling, C.; Roitberg, A.; Merz, K.M.; Luo, R.; Darden, T.; Wang, J.; Duke, R.E.; et al. Amber 2018; University of California: Auckland, CA, USA, 2018. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.; Brereton, I.M.; Chai, R.Y.; Kent, S.B. Ionization states of the catalytic residues in HIV-1 protease. Nat. Genet. 1996, 3, 946–950. [Google Scholar] [CrossRef]
- Torbeev, V.Y.; Kent, S.B.H. Ionization state of the catalytic dyad Asp25/25’ in the HIV-1 protease: NMR studies of site-specifically 13C labelled HIV-1 protease prepared by total chemical synthesis. Org. Biomol. Chem. 2012, 10, 5887–5891. [Google Scholar] [CrossRef] [Green Version]
- Adachi, M.; Ohhara, T.; Kurihara, K.; Tamada, T.; Honjo, E.; Okazaki, N.; Arai, S.; Shoyama, Y.; Kimura, K.; Matsumura, H.; et al. Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography. Proc. Natl. Acad. Sci. USA 2009, 106, 4641–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-X.; Freedberg, D.I.; Yamazaki, T.; Wingfield, P.T.; Stahl, S.J.; Kaufman, J.D.; Kiso, A.Y.; Torchia, D.A. Solution NMR Evidence That the HIV-1 Protease Catalytic Aspartyl Groups Have Different Ionization States in the Complex Formed with the Asymmetric Drug KNI-272†. Biochemistry 1996, 35, 9945–9950. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Volkov, A.; Macchi, P.; Farrugia, L.J.; Gatti, C.; Mallinson, P.; Richter, T.; Koritsanszky, T. XD2006—A Computer Program for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental or Theoretical Structure Factors; University at Buffalo: New York, NY, USA, 2006. [Google Scholar]
- Volkov, A.; Li, X.; Koritsanszky, T.; Coppens, P. Combination of the exact potential and multipole methods (EP/MM) for evaluation of intermolecular electrostatic interaction energies with pseudoatom representations of molecular electron densities. Chem. Phys. Lett. 2004, 391, 170–175. [Google Scholar] [CrossRef]
- Jedwabny, W.; Dyguda-Kazimierowicz, E.; Pernal, K.; Szalewicz, K.; Patkowski, K. Extension of an Atom–Atom Dispersion Function to Halogen Bonds and Its Use for Rational Design of Drugs and Biocatalysts. J. Phys. Chem. A 2021, 125, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Leduc, T.; Aubert, E.; Espinosa, E.; Jelsch, C.; Iordache, C.; Guillot, B. Polarization of Electron Density Databases of Transferable Multipolar Atoms. J. Phys Chem. A 2019, 123, 7156–7170. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Dominiak, P.M. Combining Molecular Dynamic Information and an Aspherical-Atom Data Bank in the Evaluation of the Electrostatic Interaction Energy in Multimeric Protein-Ligand Complex: A Case Study for HIV-1 Protease. Molecules 2021, 26, 3872. https://doi.org/10.3390/molecules26133872
Kumar P, Dominiak PM. Combining Molecular Dynamic Information and an Aspherical-Atom Data Bank in the Evaluation of the Electrostatic Interaction Energy in Multimeric Protein-Ligand Complex: A Case Study for HIV-1 Protease. Molecules. 2021; 26(13):3872. https://doi.org/10.3390/molecules26133872
Chicago/Turabian StyleKumar, Prashant, and Paulina Maria Dominiak. 2021. "Combining Molecular Dynamic Information and an Aspherical-Atom Data Bank in the Evaluation of the Electrostatic Interaction Energy in Multimeric Protein-Ligand Complex: A Case Study for HIV-1 Protease" Molecules 26, no. 13: 3872. https://doi.org/10.3390/molecules26133872