
Quantitative 1H Nuclear Magnetic Resonance Method for Assessing the Purity of Dipotassium Glycyrrhizinate

 , , and
, , and
Abstract
:
1. Introduction
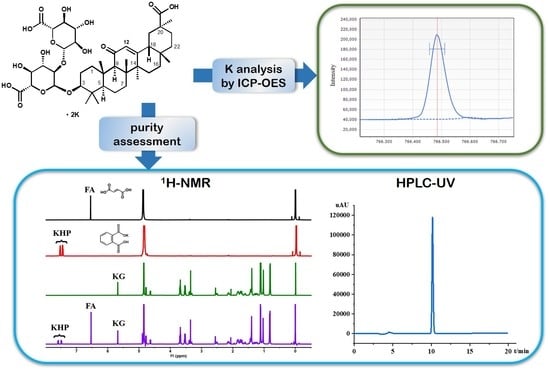
2. Results and Discussion
2.1. Selection of Quantification Signal from Analytes
2.2. Selection of Solvent and Internal Standard
2.3. Optimization of Experiment Parameters
2.4. Method Validation
2.4.1. Specificity
2.4.2. Linearity and Range
2.4.3. Limit of Quantification (LOQ)
2.4.4. Precision and Stability
2.4.5. Accuracy
2.4.6. Robustness
2.5. Analysis Results by qNMR and HPLC
3. Experimental Section
3.1. Materials
3.2. Instrument
3.3. Sample Preparations and Calculations
3.3.1. qNMR Analysis of Analytes
3.3.2. qNMR Method Validation
3.3.3. HPLC Analysis of Analytes
3.3.4. Potassium Analysis by ICP-OES
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability: Samples of the compounds are available from the authors. |
References
- Pauli, G.F.; Gödecke, T.; Jaki, B.U.; Lankin, D.C. Quantitative 1H NMR. Development and potential of an analytical method: An update. J. Nat. Prod. 2012, 75, 834–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadape, H.; Parikh, K. Quantitative determination and validation of Carvedilol in pharmaceuticals using quantitative nuclear magnetic resonance spectroscopy. Anal. Methods 2011, 3, 2341–2347. [Google Scholar] [CrossRef]
- Loópez-Rituerto, E.; Cabredo, S.; López, M.; Avenoza, A.; Busto, J.H.; Peregrina, J.M. A thorough study on the use of quantitative 1H NMR in Rioja red wine fermentation processes. J. Agric. Food. Chem. 2009, 57, 2112–2118. [Google Scholar] [CrossRef]
- Pauli, G.F.; Jaki, B.U.; Lankin, D.C. Quantitative 1H NMR: Development and potential of a method for natural products analysis. J. Nat. Prod. 2005, 68, 133–149. [Google Scholar] [CrossRef]
- Chauthe, S.K.; Sharma, R.J.; Aqil, F.; Gupta, R.C.; Singh, I.P. Quantitative NMR: An applicable method for quantitative analysis of medicinal plant extracts and herbal products. Phytochem. Anal. 2012, 23, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, L.; Irving, A.M. Assay by nuclear magnetic resonance spectroscopy: Quantification limits. Analyst 1998, 123, 1061–1068. [Google Scholar] [CrossRef]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR spectroscopy. TRAC Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Holzgrabe, U.; Deubner, R.; Schollmayer, C.; Waibel, B. Quantitative NMR spectroscopy-Applications in drug analysis. J. Pharm. Biomed. Anal. 2005, 38, 806–812. [Google Scholar] [CrossRef]
- Webster, G.K.; Kumar, S. Expanding the analytical toolbox: Pharmaceutical application of quantitative NMR. Anal. Chem. 2014, 86, 11474–11480. [Google Scholar] [CrossRef]
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef]
- Huo, H.Z.; Wang, B.; Liang, Y.K.; Bao, Y.Y.; Gu, Y. Hepatoprotective and antioxidant effects of licorice extract against CCl4-induced oxidative damage in rats. Int. J. Mol. Sci. 2011, 12, 6529–6543. [Google Scholar] [CrossRef]
- Abe, K.; Ikeda, T.; Wake, K.; Sato, T.; Sato, T.; Inoue, H. Glycyrrhizin prevents of lipopolysaccharide/D-galactosamine-induced liver injury through down-regulation of matrix metalloproteinase-9 in mice. J. Pharm. Pharmacol. 2008, 60, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wang, L.Q.; Yuan, B.C.; Liu, Y. The pharmacological activities of licorice. Planta Med. 2015, 81, 1654–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M.; Inoue, H.; Hirabayashi, K.; Natsume, H.; Ogihara, M. Glycyrrhizin and some analogues induce growth of primary cultured adult rat hepatocytes via epidermal growth factor receptors. Eur. J. Pharmacol. 2001, 431, 151–161. [Google Scholar] [CrossRef]
- Yang, R.; Yuan, B.C.; Ma, Y.S.; Zhou, S.; Liu, Y. The anti-inflammatory activity of licorice, a widely used Chinese herb. Pharm. Biol. 2017, 55, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Trotta, M.; Peira, E.; Debernardi, F.; Gallarate, M. Elastic liposomes for skin delivery of dipotassium glycyrrhizinate. Int. J. Pharm. 2002, 241, 319–327. [Google Scholar] [CrossRef]
- Michaelis, M.; Geiler, J.; Naczk, P.; Sithisarn, P.; Leutz, A.; Doerr, H.W.; Cinatl, J., Jr. Glycyrrhizin exerts antioxidative effects in H5N1 influenza A virus-infected cells and inhibits virus replication and pro-inflammatory gene expression. PLoS ONE 2011, 6, e19705. [Google Scholar] [CrossRef] [Green Version]
- Hardy, M.E.; Hendricks, J.M.; Paulson, J.M.; Faunce, N.R. 18 β-glycyrrhetinic acid inhibits rotavirus replication in culture. Virol. J. 2012, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosmetic Ingredient Review Expert Panel. Final report on the safety assessment of glycyrrhetinic acid, potassium glycyrrhetinate, disodium succinoyl glycyrrhetinate, glyceryl glycyrrhetinate, glycyrrhetinyl stearate, stearyl glycyrrhetinate, glycyrrhizic acid, ammonium glycyrrhizate, dipotassium glycyrrhizate, disodium glycyrrhizate, trisodium glycyrrhizate, methyl glycyrrhizate, and potassium glycyrrhizinate. Int. J. Toxicol. 2007, 26, 79–112. [Google Scholar]
- Deng, L.; Yao, D.; Li, T.; Ju, W. Study on the Content of Glycyrhiza Acid in Glycyrhiza uralensis Fisch by Thin-Layer Colorimetry. Chem. Eng. 2001, 1, 35–36. [Google Scholar]
- Shen, Y.; Shen, J.; Zhuang, Z.; Wang, X. Comparison between HPCE and HPLC for the analysis of main active components in licorice. Chin. J. Pharm. Anal. 2006, 26, 421–425. [Google Scholar]
- Li, P.; Wang, Y.; Gao, H.; Wang, Q. The detection of dipotassium glycyrrhizinate based on polarograph and its application. Chin. J. Health Lab. Technol. 2005, 15, 1435–1436. [Google Scholar]
- Li, Z.Y.; Welbeck, E.; Yang, L.; He, C.Y.; Hu, H.J.; Song, M.; Bi, K.S.; Wang, Z.T. A quantitative 1H nuclear magnetic resonance (qHNMR) method for assessing the purity of iridoids and secoiridoids. Fitoterapia 2015, 100, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Malz, F.; Jancke, H. Validation of quantitative NMR. J. Pharm. Biomed. Anal. 2005, 38, 813–823. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| No. | mFA (mg) | mKHP (mg) | mKG (mg) | mKG/mFA | mKG/mKHP | AKG/AFA | AKG/AKHP |
|---|---|---|---|---|---|---|---|
| 1 | 0.51 | 0.52 | 2.04 | 4.02 | 3.96 | 0.24 | 0.21 |
| 2 | 0.51 | 0.52 | 3.06 | 6.02 | 5.94 | 0.36 | 0.32 |
| 3 | 0.51 | 0.52 | 5.22 | 10.27 | 10.12 | 0.62 | 0.54 |
| 4 | 0.51 | 0.52 | 10.55 | 20.77 | 20.47 | 1.22 | 1.06 |
| 5 | 0.51 | 0.52 | 12.56 | 24.72 | 24.36 | 1.44 | 1.25 |
| 6 | 0.51 | 0.52 | 15.43 | 30.37 | 29.93 | 1.74 | 1.52 |
| Linear equation | y = 0.0571x + 0.0237 | y = 0.0503x + 0.0207 | |||||
| R2 | 0.9996 | 0.9996 | |||||
| No. | mFA (mg) | mKHP (mg) | mKG (mg) | Px (%) * | ||
|---|---|---|---|---|---|---|
| FA as IS | KHP as IS | |||||
| Precision (n = 6) | 1 | 0.51 | 0.51 | 10.44 | 96.67 | 98.09 |
| 2 | 0.51 | 0.51 | 10.44 | 97.20 | 97.63 | |
| 3 | 0.51 | 0.51 | 10.44 | 97.20 | 98.09 | |
| 4 | 0.51 | 0.51 | 10.44 | 97.63 | 98.06 | |
| 5 | 0.51 | 0.51 | 10.44 | 97.20 | 98.09 | |
| 6 | 0.51 | 0.51 | 10.44 | 97.20 | 97.63 | |
| Average value | / | / | / | 97.18 | 97.93 | |
| RSD% | / | / | / | 0.31 | 0.24 | |
| Reproducibility | 1 | 0.52 | 0.51 | 10.47 | 95.88 | 96.45 |
| 2 | 0.52 | 0.51 | 10.13 | 96.14 | 95.81 | |
| 3 | 0.52 | 0.51 | 10.40 | 96.65 | 96.49 | |
| 4 | 0.52 | 0.51 | 10.54 | 96.62 | 96.84 | |
| 5 | 0.52 | 0.51 | 10.12 | 95.48 | 96.04 | |
| 6 | 0.52 | 0.51 | 10.14 | 97.30 | 97.35 | |
| Average value | / | / | / | 96.34 | 96.50 | |
| RSD% | / | / | / | 0.67 | 0.57 | |
| Stability | 0 # | 0.51 | 0.51 | 10.44 | 97.30 | 97.66 |
| 1 | 0.51 | 0.51 | 10.44 | 97.30 | 97.66 | |
| 2 | 0.51 | 0.51 | 10.44 | 96.86 | 97.23 | |
| 4 | 0.51 | 0.51 | 10.44 | 98.16 | 98.06 | |
| 8 | 0.51 | 0.51 | 10.44 | 97.73 | 97.63 | |
| 12 | 0.51 | 0.51 | 10.44 | 97.73 | 97.63 | |
| 24 | 0.51 | 0.51 | 10.44 | 98.16 | 98.06 | |
| Average value | / | / | / | 97.60 | 97.70 | |
| RSD% | / | / | / | 0.49 | 0.29 | |
| No. | Amount Added (mg) | FA as IS | KHP as IS | ||
|---|---|---|---|---|---|
| Amount Found (mg) | Recovery (%) | Amount Found (mg) | Recovery (%) | ||
| 1 | 7.93 | 7.89 | 99.49 | 8.00 | 100.83 |
| 2 | 7.93 | 7.88 | 99.31 | 8.00 | 100.83 |
| 3 | 7.93 | 7.92 | 99.80 | 8.05 | 101.51 |
| 4 | 10.34 | 10.21 | 98.79 | 10.39 | 100.75 |
| 5 | 10.34 | 10.22 | 98.80 | 10.37 | 100.49 |
| 6 | 10.34 | 10.18 | 98.46 | 10.32 | 99.96 |
| 7 | 12.02 | 12.06 | 100.33 | 11.80 | 98.23 |
| 8 | 12.02 | 11.89 | 98.94 | 11.74 | 98.23 |
| 9 | 12.02 | 11.98 | 99.71 | 11.83 | 98.23 |
| Average value (%) | / | / | 99.29 | / | 99.90 |
| RSD (%) | / | / | 0.60 | / | 1.31 |
| Parameters | Variation | FA as IS | KHP as IS | ||
|---|---|---|---|---|---|
| Px (%) | Diff (%) | Px (%) | Diff (%) | ||
| Relaxation delay (D1) | 1 s | 106.06 | 9.04 | 100.30 | 2.36 |
| 30 s # | 97.02 | / | 97.94 | / | |
| 70 s | 97.67 | 0.65 | 98.05 | 0.11 | |
| Time domain (TD) | 32 k | 97.58 | 0.56 | 97.64 | 0.30 |
| 64 k | 97.02 | / | 97.94 | / | |
| Number of scans (NS) | 8 | 97.30 | 0.28 | 97.66 | 0.28 |
| 16 | 97.02 | / | 97.94 | / | |
| 32 | 97.69 | 0.67 | 98.08 | 0.14 | |
| Pulse length (Pl) | 2.2 μs | 97.62 | 0.60 | 97.80 | 0.14 |
| 2.4 μs | 97.02 | / | 97.94 | / | |
| 2.6 μs | 97.47 | 0.45 | 97.50 | 0.44 | |
| Batch No. | qNMR Method (n = 6) | HPLC Method | K% * | Water% # | |||
|---|---|---|---|---|---|---|---|
| IS | % | RSD% | % | RSD% | |||
| 8060633 | FA | 99.27 | 0.30 | 99.61 | 0.23 | 9.48 | 2.25 |
| KHP | 99.42 | 0.27 | |||||
| 8070201 | FA | 99.59 | 0.29 | 99.38 | 0.50 | 8.19 | 2.38 |
| KHP | 99.20 | 0.27 | |||||
| 9010291 | FA | 97.18 | 0.31 | 97.22 | 0.34 | 9.47 | 3.59 |
| KHP | 97.93 | 0.24 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.-Y.; Zhang, J.; Zhang, W.-X.; Wang, Y.; Wang, Y.-H.; Yang, Q.-Y.; Wu, S. Quantitative 1H Nuclear Magnetic Resonance Method for Assessing the Purity of Dipotassium Glycyrrhizinate. Molecules 2021, 26, 3549. https://doi.org/10.3390/molecules26123549
Zhang Y-Y, Zhang J, Zhang W-X, Wang Y, Wang Y-H, Yang Q-Y, Wu S. Quantitative 1H Nuclear Magnetic Resonance Method for Assessing the Purity of Dipotassium Glycyrrhizinate. Molecules. 2021; 26(12):3549. https://doi.org/10.3390/molecules26123549
Chicago/Turabian StyleZhang, Yuan-Yuan, Jie Zhang, Wen-Xuan Zhang, Yue Wang, Ying-Hong Wang, Qing-Yun Yang, and Song Wu. 2021. "Quantitative 1H Nuclear Magnetic Resonance Method for Assessing the Purity of Dipotassium Glycyrrhizinate" Molecules 26, no. 12: 3549. https://doi.org/10.3390/molecules26123549