
Synthesis, Structure and Electrochemical Properties of Acetamide- and Caprolactam-Containing Silicon Catecholates

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
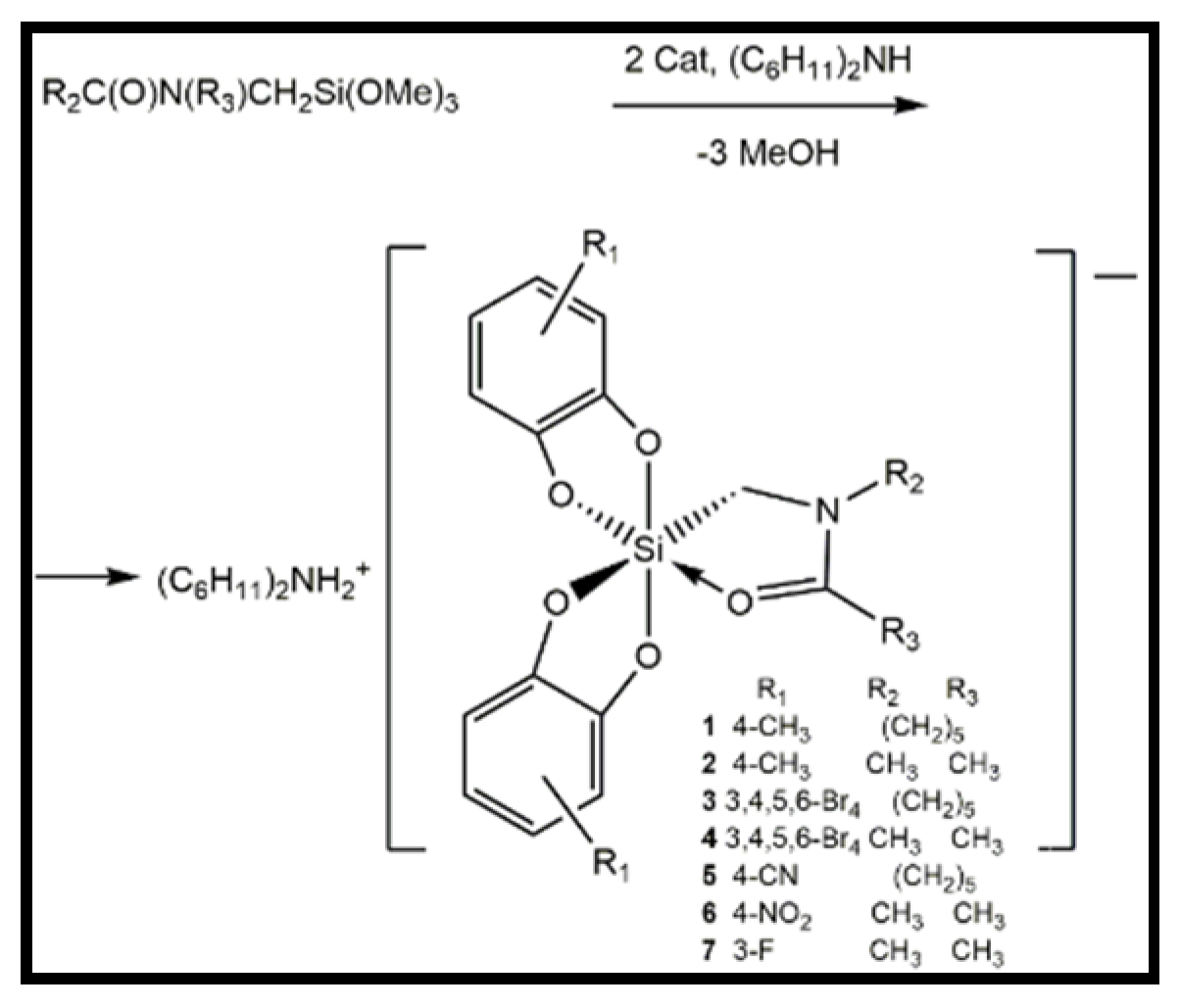
2.1. Synthesis
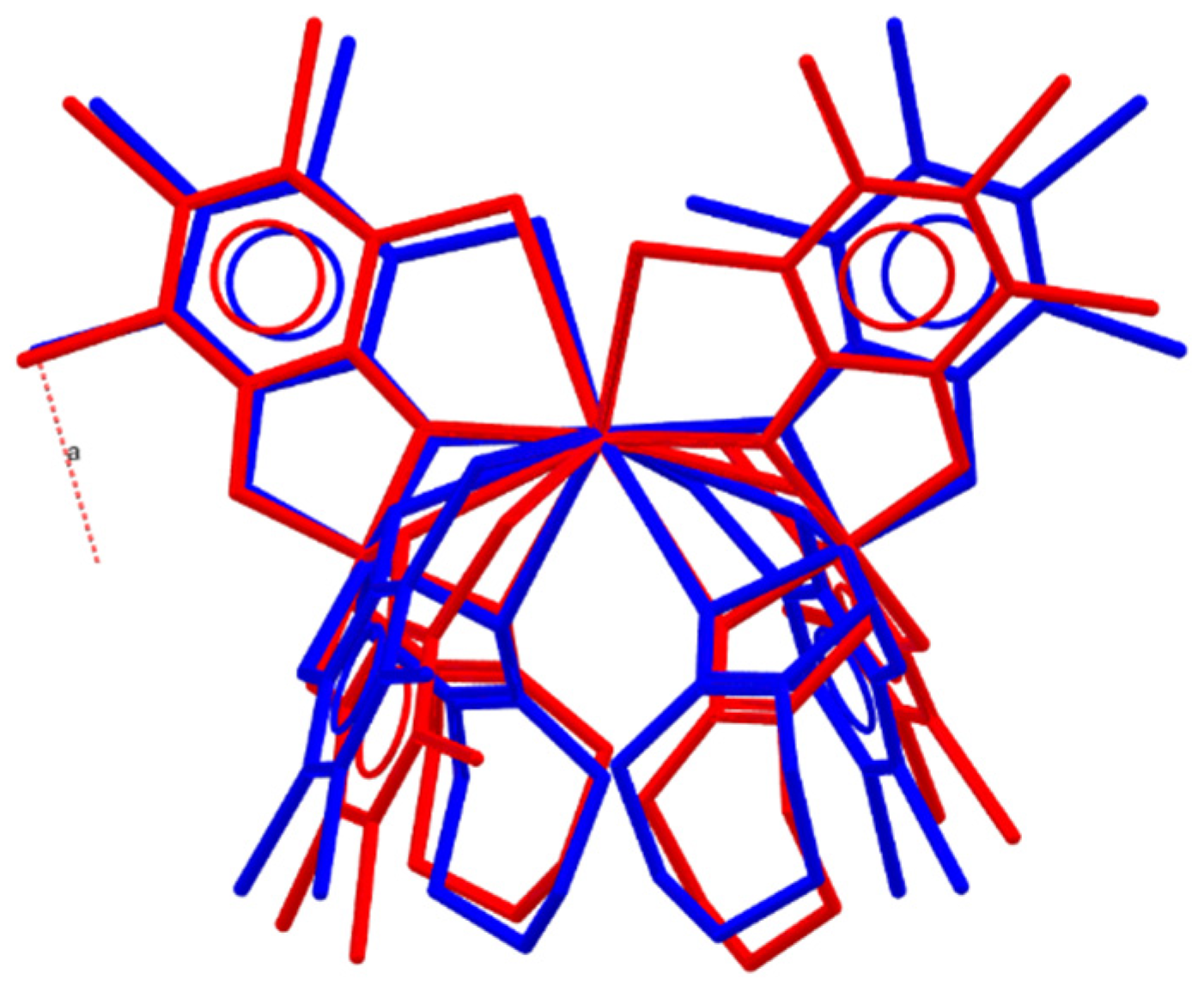
2.2. Crystal Structures
2.3. Electrochemical Characterization
3. Materials and Methods
3.1. General
3.2. Synthesis
3.3. X-ray Diffraction Studies
3.4. Electrochemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Baramov, T.; Keijzer, K.; Irran, E.; Mösker, E.; Baik, M.-H.; Süssmuth, R. Synthesis and Structural Characterization of Hexacoordinate Silicon, Germanium, and Titanium Complexes of the E. Coli Siderophore Enterobactin. Chem. Eur. J. 2013, 19, 10536–10542. [Google Scholar] [CrossRef] [PubMed]
- Kenla, T.J.N.; Tatong, M.D.K.; Talontsi, F.M.; Dittrich, B.; Frauendorf, H.; Laatsch, H. Si-Enterobactin from the Endophytic Streptomyces Sp. KT-S1-B5–a Potential Silicon Transporter in Nature? Chem. Commun. 2013, 49, 7641–7643. [Google Scholar] [CrossRef] [Green Version]
- Liberman-Martin, A.L.; Bergman, R.G.; Tilley, T.D. Lewis Acidity of Bis(Perfluorocatecholato)Silane: Aldehyde Hydrosilylation Catalyzed by a Neutral Silicon Compound. J. Am. Chem. Soc. 2015, 137, 5328–5331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maskey, R.; Schädler, M.; Legler, C.; Greb, L. Bis(Perchlorocatecholato)Silane-A Neutral Silicon Lewis Super Acid. Angew. Chem. Int. Ed. 2018, 57, 1717–1720. [Google Scholar] [CrossRef]
- Hartmann, D.; Schädler, M.; Greb, L. Bis(Catecholato)Silanes: Assessing, Rationalizing and Increasing Silicon’s Lewis Superacidity. Chem. Sci. 2019, 10, 7379–7388. [Google Scholar] [CrossRef] [Green Version]
- Maskey, R.; Wadepohl, H.; Greb, L. Silicon Tris(Perchloro)Dioxolene: A Neutral Triplet Diradical. Angew. Chem. Int. Ed. 2019, 58, 3616–3619. [Google Scholar] [CrossRef]
- Holmes, J.L.; Abrahams, B.F.; Ahveninen, A.; Boughton, B.A.; Hudson, T.A.; Robson, R.; Thinagaran, D. Self-Assembly of a Si-Based Cage by the Formation of 24 Equivalent Covalent Bonds. Chem. Commun. 2018, 54, 11877–11880. [Google Scholar] [CrossRef] [PubMed]
- Wolff, B.; Weiss, A. Novel Octahedral Si and Ge Complexes with a Hexadentate Diphenol Ligand. Angew. Chem. Int. Ed. Engl. 1986, 25, 162–163. [Google Scholar] [CrossRef]
- Hong, C.M.; Morimoto, M.; Kapustin, E.A.; Alzakhem, N.; Bergman, R.G.; Raymond, K.N.; Toste, F.D. Deconvoluting the Role of Charge in a Supramolecular Catalyst. J. Am. Chem. Soc. 2018, 140, 6591–6595. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Ogishima, T.; Kawara, T.; Yamauchi, S.; Okamoto, K.; Nikaido, S.; Souma, D.; Jin, R.-H.; Kabe, Y. Silane Catecholates: Versatile Tools for Self-Assembled Dynamic Covalent Bond Chemistry. Chem. Commun. 2019, 55, 6066–6069. [Google Scholar] [CrossRef]
- Corcé, V.; Chamoreau, L.-M.; Derat, E.; Goddard, J.-P.; Ollivier, C.; Fensterbank, L. Silicates as Latent Alkyl Radical Precursors: Visible-Light Photocatalytic Oxidation of Hypervalent Bis-Catecholato Silicon Compounds. Angew. Chem. Int. Ed. 2015, 54, 11414–11418. [Google Scholar] [CrossRef]
- Do, T.H.; Brown, S.N. Mono- and Bimetallic Pentacoordinate Silicon Complexes of a Chelating Bis(Catecholimine) Ligand. Dalton Trans. 2019, 48, 11565–11574. [Google Scholar] [CrossRef]
- Maguylo, C.; Chukwu, C.; Aun, M.; Blake Monroe, T.; Ceccarelli, C.; Jones, D.S.; Merkert, J.W.; Donovan-Merkert, B.T.; Schmedake, T.A. Exploring the Structure and Redox Activity of Hexacoordinate Bis(Bipyridyl)Silicon(IV) Complexes. Polyhedron 2015, 94, 52–58. [Google Scholar] [CrossRef]
- Doddi, A.; Kingston, J.V.; Ramkumar, V.; Suzuki, M.; Hojo, M.; Rao, M.N.S. Synthesis and Characterization of Dianionic Hexacoordinate Silicon(IV) Complexes of Substituted Catechols, Flavones, and Fluorone: X-Ray Crystal Structures of [(n-C3H7)2NH2]2[(Cl4C6O2)3Si] · 3 CH3CN and [(n-C3H7)2NH2]2[(Br4C6O2)3Si] · 2 (CH3)2SO. Phosph. Sulf. Sil. Rel. Elem. 2012, 187, 343–356. [Google Scholar] [CrossRef]
- Bindu, P.; Varghese, B.; Rao, M.N.S. Six Coordinate Tris(Catecholato)Silicates of Primary Amine Residues—Synthesis, Characterization, and Thermolysis Studies. X-Ray Structures of [n-C3H7NH3]2[Si(C6H4O2)3]·1/2(C6H14N2) and of a Bulky Secondary Ammonium Ion, [(i-C4H9)2NH2]2[Si(C6H4O2)3]·H2O. Phosph. Sulf. Sil. Rel. El. 2003, 178, 2373–2386. [Google Scholar] [CrossRef]
- Hahn, F.E.; Keck, M.; Raymond, K.N. Catecholate Complexes of Silicon: Synthesis and Molecular and Crystal Structures of [Si(Cat)2]...2THF and Li2[Si(Cat)3]...3.5dme (Cat = Catecholate Dianion). Inorg. Chem. 1995, 34, 1402–1407. [Google Scholar] [CrossRef]
- Carre, F.; Chuit, C.; Corriu, R.J.P.; Fanta, A.; Mehdi, A.; Reye, C. Use of the 2,6-Bis[(Dimethylamino)Methyl]Phenyl Ligand for the Study of Nucleophilic Substitution at Hexacoordinate Silicon Centers. Evidence Suggestive of a Heptacoordinate Silicon Transition State. Organometallics 1995, 14, 194–198. [Google Scholar] [CrossRef]
- Carré, F.; Chuit, C.; Corriu, R.J.P.; Mehdi, A.; Reyé, C. Unexpected Behaviour of a Hexacoordinate Silicon Compound. J. Organomet. Chem. 1993, 446, C6–C8. [Google Scholar] [CrossRef]
- Korlyukov, A.; Shipov, A.; Kramarova, E.; Negrebetskii, V.V.; Baukov, Y. Synthesis and Specific Features of the Structure of the Mixed Anionic Six-Coordinate Silicon Complexes with the (O,O)-Dianionic and (C,O)-Monoanionic Chelate Ligands. Russ. Chem. Bull. 2008, 57, 2093–2100. [Google Scholar] [CrossRef]
- Shipov, A.G.; Kramarova, E.P.; Artamkina, O.B.; Baukov, Y.I. Monoreactor Synthesis of N-(trialkoxysilylmethyl)amides and N-(trialkoxysilylmethyl)lactams. Zh. Obshch. Khim. 1993, 63, 1434–1435. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3a 1 | 3b 1 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|
| Si1-O1 | 1.877(2) | 1.8851(15) | 1.880(12) | 1.896(6) | 1.867(3) | 1.882(3) | 1.860(17) | 1.8873(19) |
| Si1-O2 | 1.788(2) | 1.7935(14) | 1.792(12) | 1.777(6) | 1.784(3) | 1.809(3) | 1.778(2) | 1.8090(18) |
| Si1-O3 | 1.800(2) | 1.7638(17) | 1.795(11) | 1.795(6) | 1.830(3) | 1.782(3) | 1.7861(18) | 1.8052(19) |
| Si1-O4 | 1.761(2) | 1.7889(17) | 1.798(11) | 1.795(6) | 1.784(3) | 1.798(4) | 1.7868(18) | 1.7894(18) |
| Si1-O5 | 1.801(2) | 1.7934(16) | 1.801(11) | 1.807(6) | 1.815(3) | 1.815(3) | 1.8036(17) | 1.768(2) |
| Si1-C1 | 1.918(3) | 1.923(2) | 1.927(19) | 1.918(8) | 1.925(4) | 1.932(4) | 1.996(17) | 1.923(3) |
| O1Si1O2 | 174.71(11) | 174.95(8) | 176.5(6) | 177.5(3) | 175.38(14) | 175.97(14) | 174.0(5) | 173.13(9) |
| O3Si1O4 | 173.31(11) | 174.13(8) | 172.8(6) | 173.7(3) | 175.10(14) | 173.59(14) | 176.33(9) | 175.12(10) |
| O5Si1C1 | 171.91(12) | 170.34(8) | 167.2(7) | 168.4(3) | 169.15(16) | 172.13(15) | 169.6(5) | 172.07(10) |
| Compound | Fc | E1ox (V) | E2ox (V) | E3ox (V) |
|---|---|---|---|---|
| 1 | 0.487 | 0.006 | 0.346 | 0.497 |
| 2 | 0.447 | 0.649 | ||
| 3 | 0.464 | 0.869 | ||
| 4 | 0.459 | 0.654 | 0.928 | |
| 5 | 0.485 | 0.047 | 0.511 | 0.845 |
| 6 | 0.486 | 0.593 | 0.837 | |
| 7 | 0.489 | 0.312 | 0.756 |
| 1∙C7H8O2∙2C7H8 | 2∙C7H8O2∙2.5C6H6 | 3a∙H2O∙0.5C2H3N | 3b∙2C2H3N | 4 | 5∙2H2O | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|
| Formula | C54H72N2O7Si | C52H67N2O7Si | C51H51·50Br16KN3·50O11Si2 | C54H54Br16KN5O10Si2 | C28H32Br8N2O5Si | C33H46N4O7Si | C28H38N4O9Si | C28H38F2N2O5Si |
| Fw | 889.22 | 860.16 | 2263.29 | 2306.86 | 1143.92 | 638.83 | 602.71 | 548.69 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | Triclinic | Monoclinic |
| Space group | P21/n | P21/c | Cc | Cc | P21 | Pbca | P1̅ | P21/c |
| a, Å | 16.231(3) | 12.302(3) | 20.160(4) | 20.754(4) | 9.3422(5) | 15.651(16) | 11.2726(17) | 9.192(2) |
| b, Å | 15.185(3) | 15.613(3) | 20.620(4) | 20.697(4) | 20.4930(11) | 19.49(2) | 11.7569(17) | 20.522(6) |
| c, Å | 19.933(4) | 25.066(5) | 17.620(4) | 18.740(4) | 9.4596(5) | 21.76(3) | 13.4405(19) | 14.798(3) |
| α, ° | 90 | 90 | 90 | 90 | 90 | 90 | 73.242(3) | 90 |
| β, ° | 92.22(3) | 100.08(3) | 113.78(3) | 115.59(3) | 98.6190(10) | 90 | 80.444(4) | 105.325(10) |
| γ, ° | 90 | 90 | 90 | 90 | 90 | 90 | 77.905(4) | 90 |
| V, Å | 4909.2(17) | 4740.1(17) | 6703(3) | 7260(3) | 1790.58(17) | 6637(13) | 1657.1(4) | 2692.2(12) |
| Z | 4 | 4 | 4 | 4 | 2 | 8 | 2 | 4 |
| dcalc, g cm−1 | 1.203 | 1.205 | 2.243 | 2.111 | 2.122 | 1.279 | 1.208 | 1.354 |
| μ, mm−1 | 0.132 | 0.134 | 12.912 | 11.924 | 9.031 | 0.123 | 0.124 | 0.143 |
| F(000) | 1920 | 1852 | 4308 | 4400 | 1096 | 2736 | 640 | 1168 |
| Ref. coll. | 29,344 | 43,014 | 46,282 | 40,319 | 40,981 | 23,440 | 16,813 | 18,206 |
| Ref. ind. Rint | 10,704 0.086 | 10,824 0.052 | 14,922 0.039 | 16,243 0.046 | 10,879 0.038 | 7593 0.145 | 9841 0.062 | 8030 (0.099) |
| Ref. obs. (I > 2σ(I)) | 5451 | 6465 | 13,780 | 11,537 | 9735 | 3465 | 4296 | 3840 |
| Parameters | 717 | 576 | 780 | 831 | 399 | 424 | 413 | 363 |
| R1 (I > 2σ(I)) | 0.067 | 0.057 | 0.033 | 0.066 | 0.025 | 0.073 | 0.068 | 0.066 |
| wR2 (all refls.) | 0.205 | 0.171 | 0.087 | 0.204 | 0.050 | 0.196 | 0.172 | 0.171 |
| GOF | 1.013 | 1.038 | 1.039 | 1.038 | 0.993 | 1.001 | 0.929 | 0.958 |
| ρₘᵢₙ/ρₘₐₓ, eÅ−3 | −0.276/0.253 | −0.322/0.242 | −1.028/0.946 | −0.717/1.565 | −0.494/0.776 | −0.298/0.463 | −0.288/0.303 | −0.616/0.623 |
| Flack | - | - | 0.095(8) | 0.368(15) | 0.006(4) | - | - | - |
| No. of restr. | 19 | 79 | 46 | 128 | 1 | 0 | 2 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kramarova, E.P.; Volodin, A.D.; Negrebetsky, V.V.; Shagina, A.D.; Aliev, T.M.; Dorovatovskii, P.V.; Novikov, R.A.; Vologzhanina, A.V.; Korlyukov, A.A. Synthesis, Structure and Electrochemical Properties of Acetamide- and Caprolactam-Containing Silicon Catecholates. Molecules 2021, 26, 3548. https://doi.org/10.3390/molecules26123548
Kramarova EP, Volodin AD, Negrebetsky VV, Shagina AD, Aliev TM, Dorovatovskii PV, Novikov RA, Vologzhanina AV, Korlyukov AA. Synthesis, Structure and Electrochemical Properties of Acetamide- and Caprolactam-Containing Silicon Catecholates. Molecules. 2021; 26(12):3548. https://doi.org/10.3390/molecules26123548
Chicago/Turabian StyleKramarova, Eugenia P., Alexander D. Volodin, Vadim V. Negrebetsky, Anastasia D. Shagina, Teimur M. Aliev, Pavel V. Dorovatovskii, Roman A. Novikov, Anna V. Vologzhanina, and Alexander A. Korlyukov. 2021. "Synthesis, Structure and Electrochemical Properties of Acetamide- and Caprolactam-Containing Silicon Catecholates" Molecules 26, no. 12: 3548. https://doi.org/10.3390/molecules26123548