
Pharmacological Activities of Aminophenoxazinones

 , , , and
, , , and 
Abstract
:1. Introduction
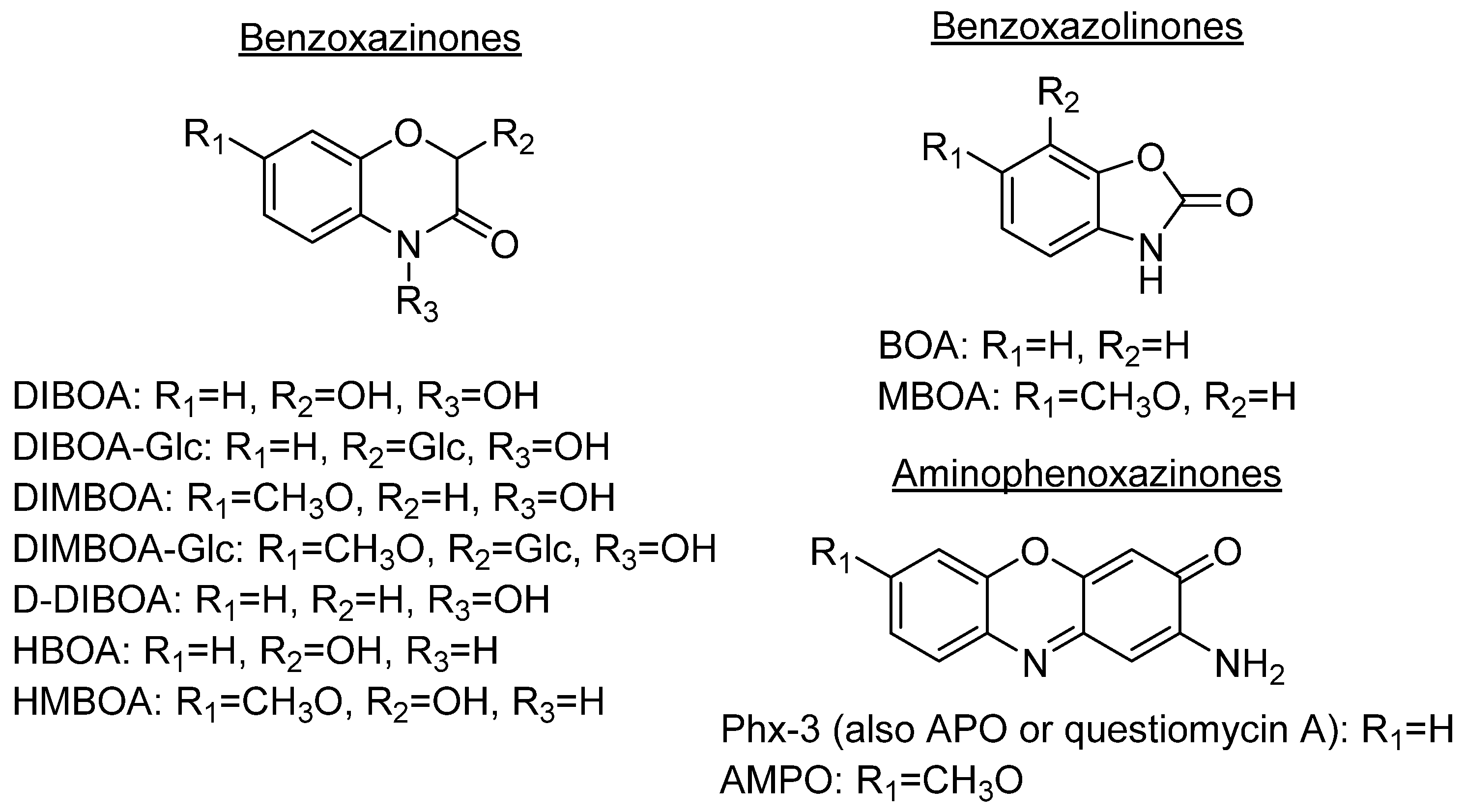
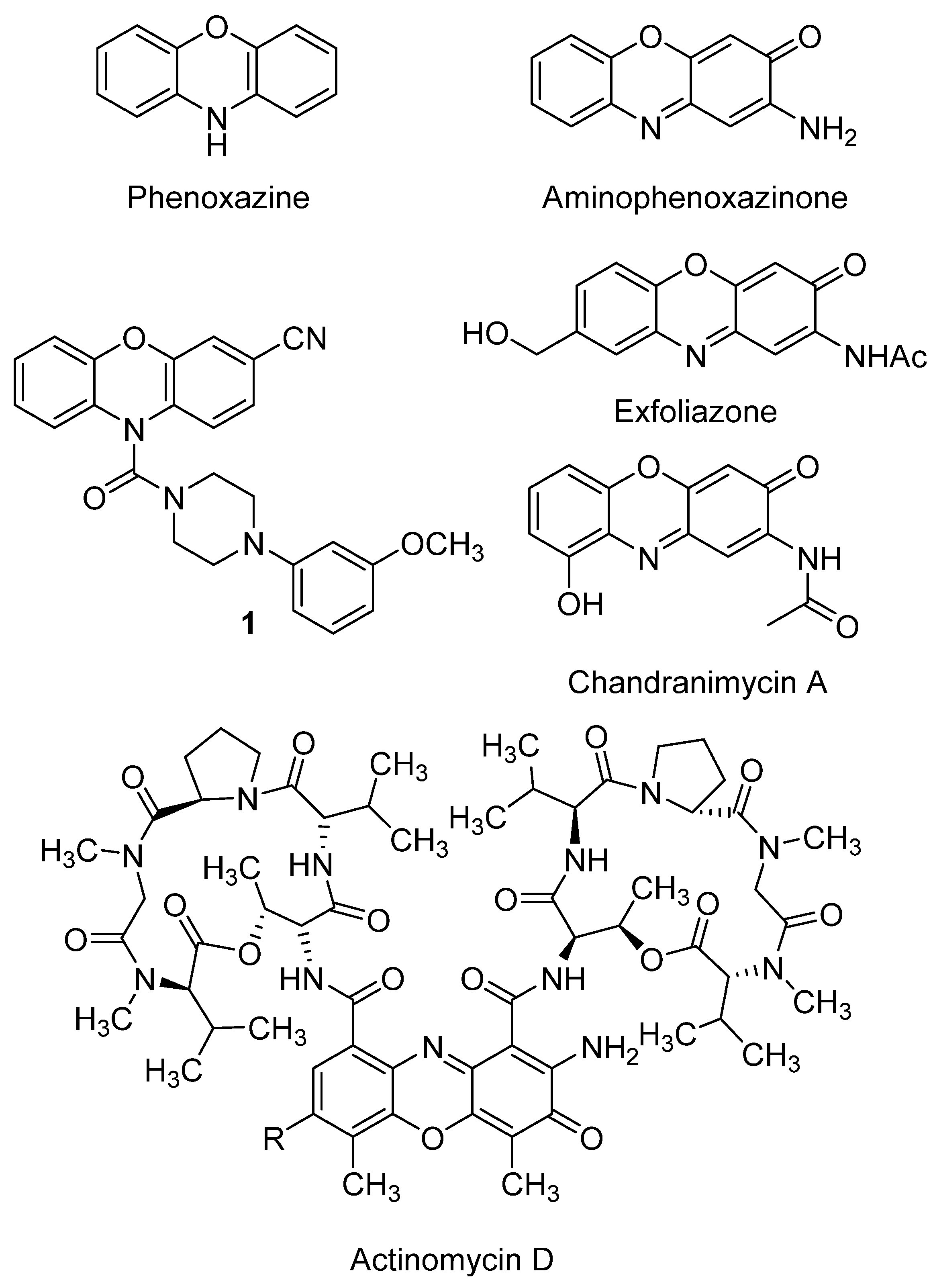
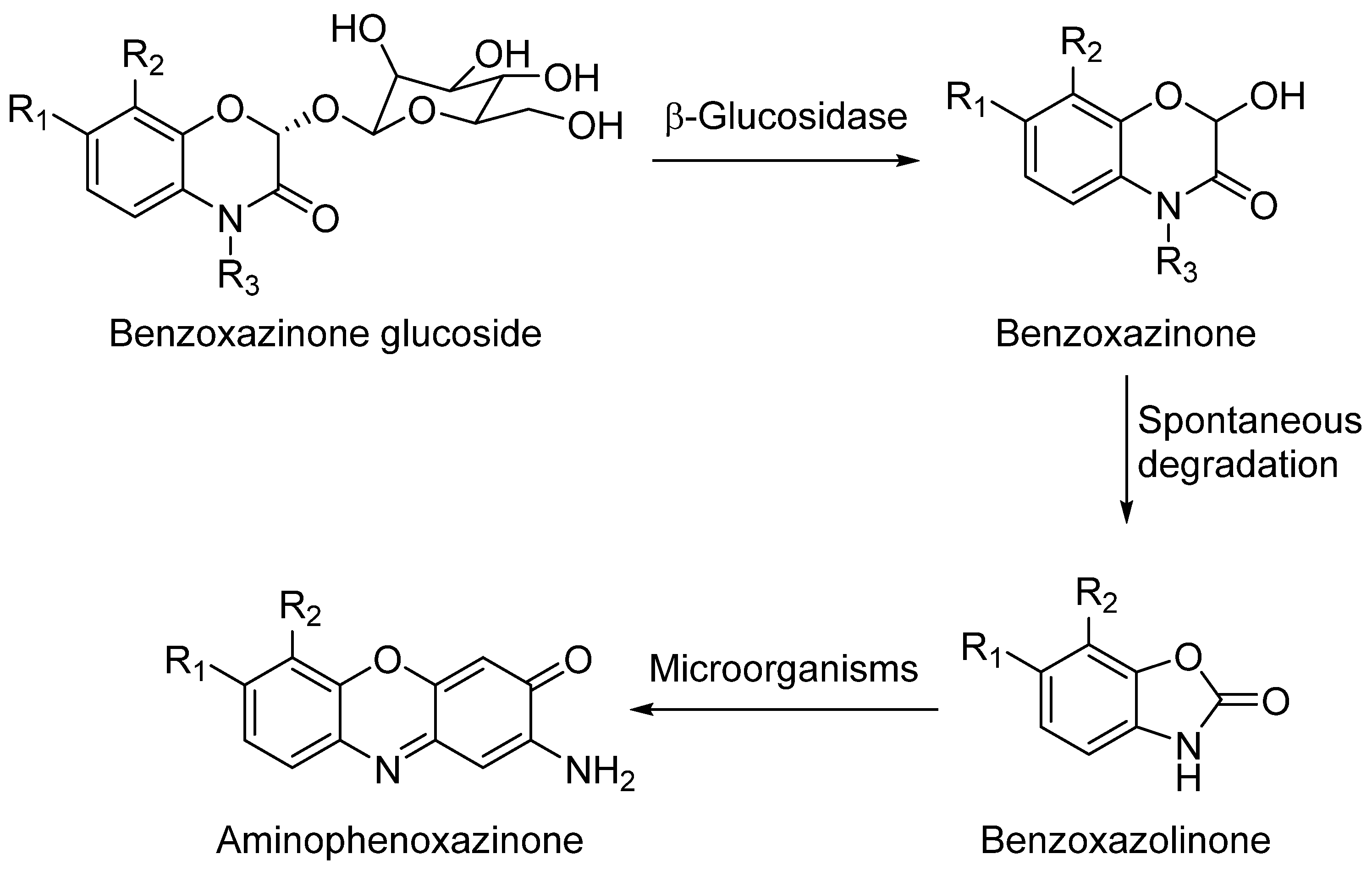
2. Aminophenoxazinones as Degradation Products of Benzoxazinones
3. Anticancer Activity
3.1. Gastric and Colon Cancer
3.2. Glioblastoma
3.3. Melanoma
4. Activity of Aminophenoxazinones on Other Cell Lines
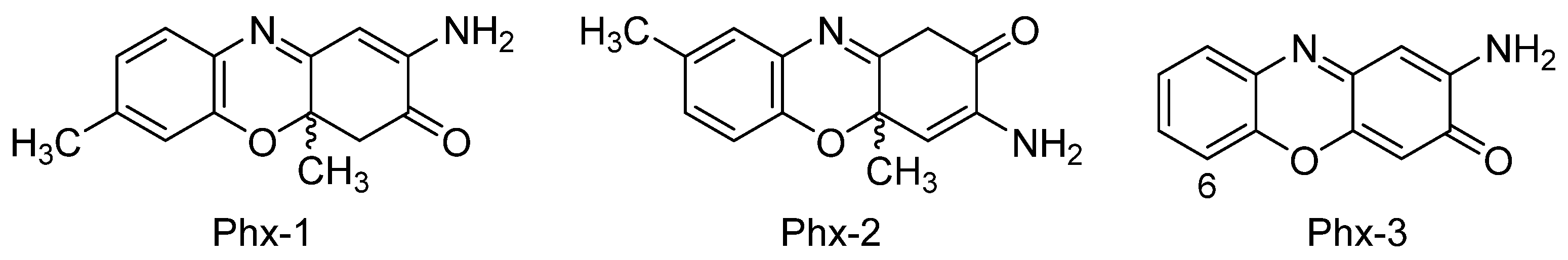
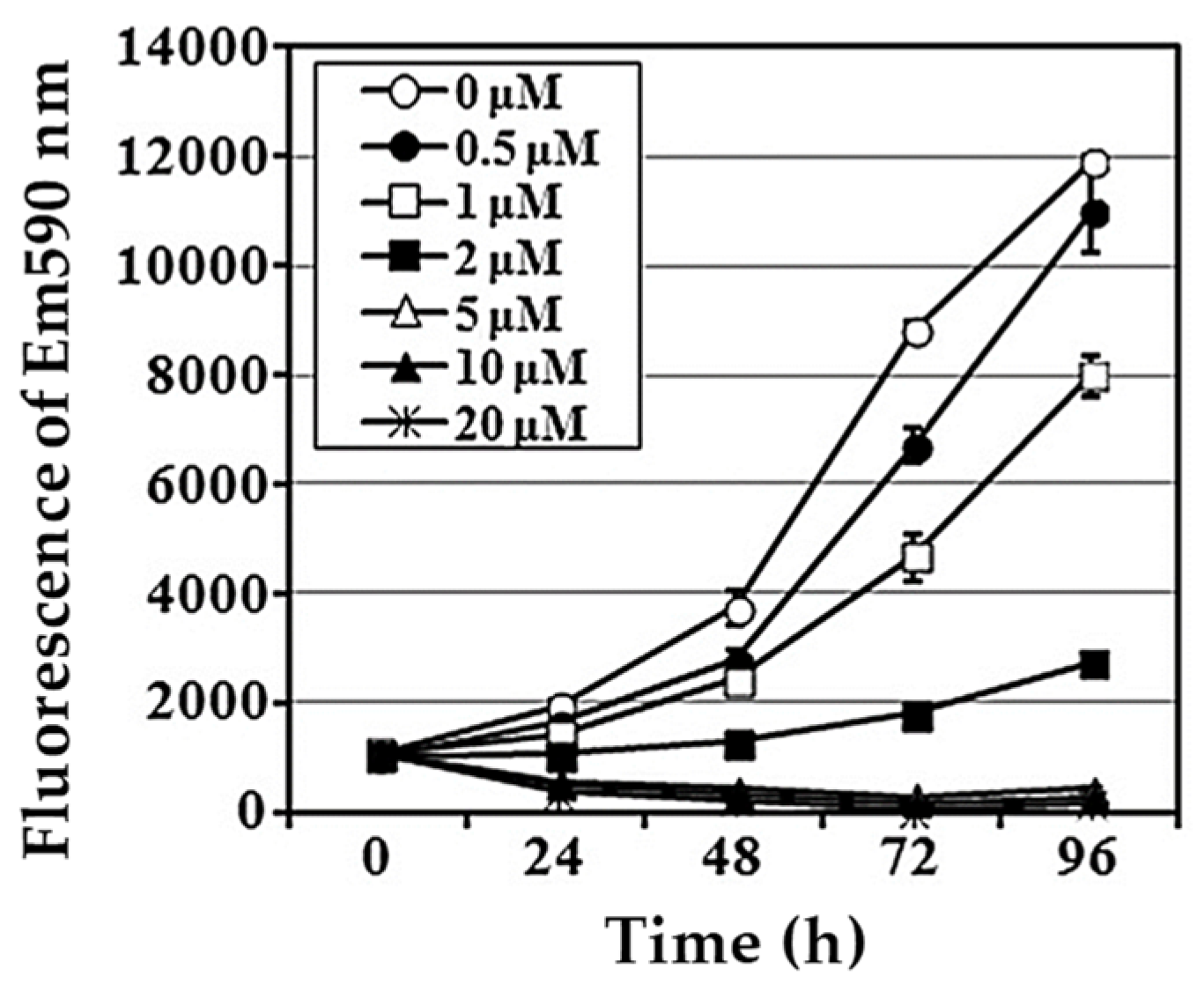
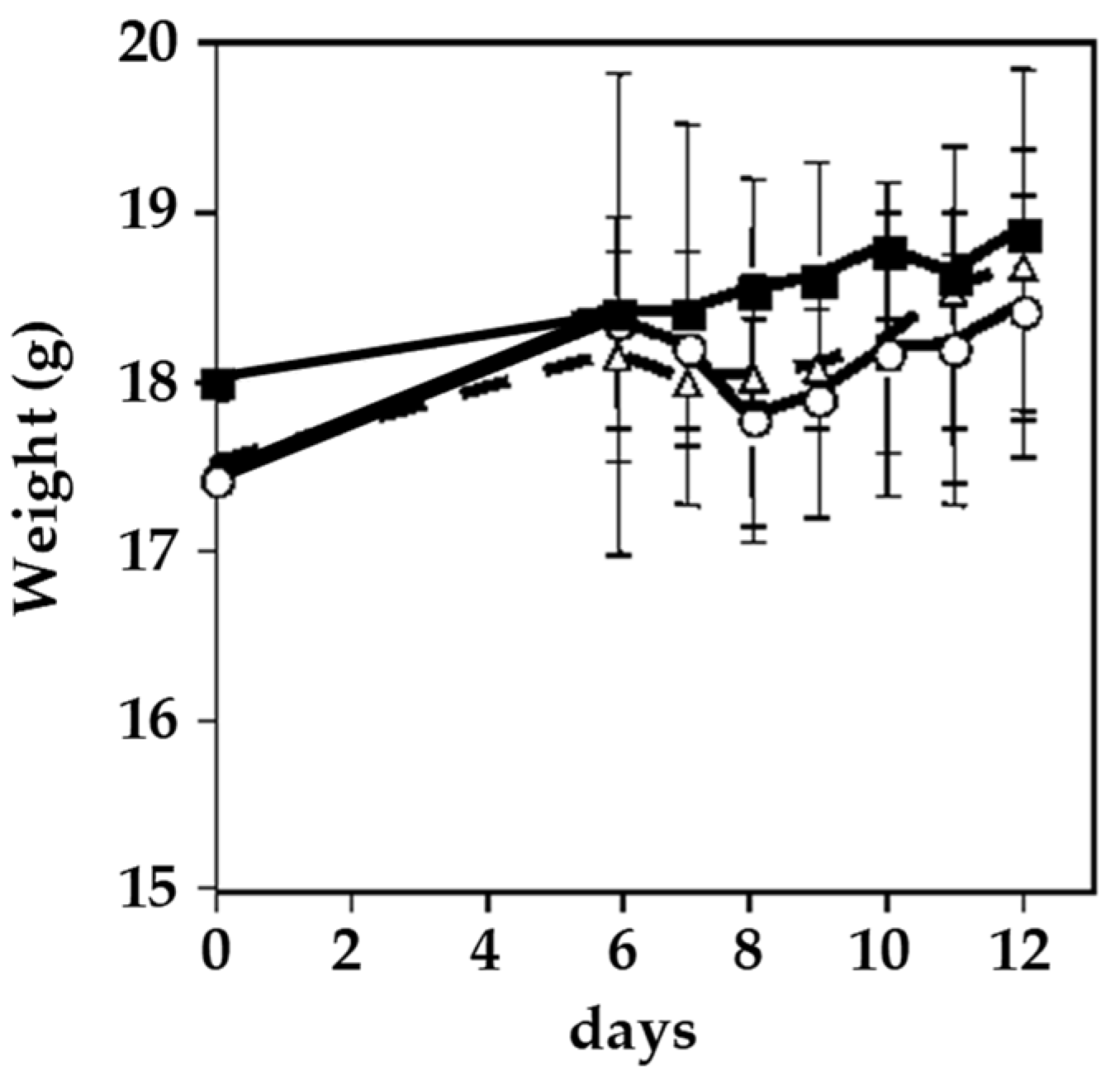
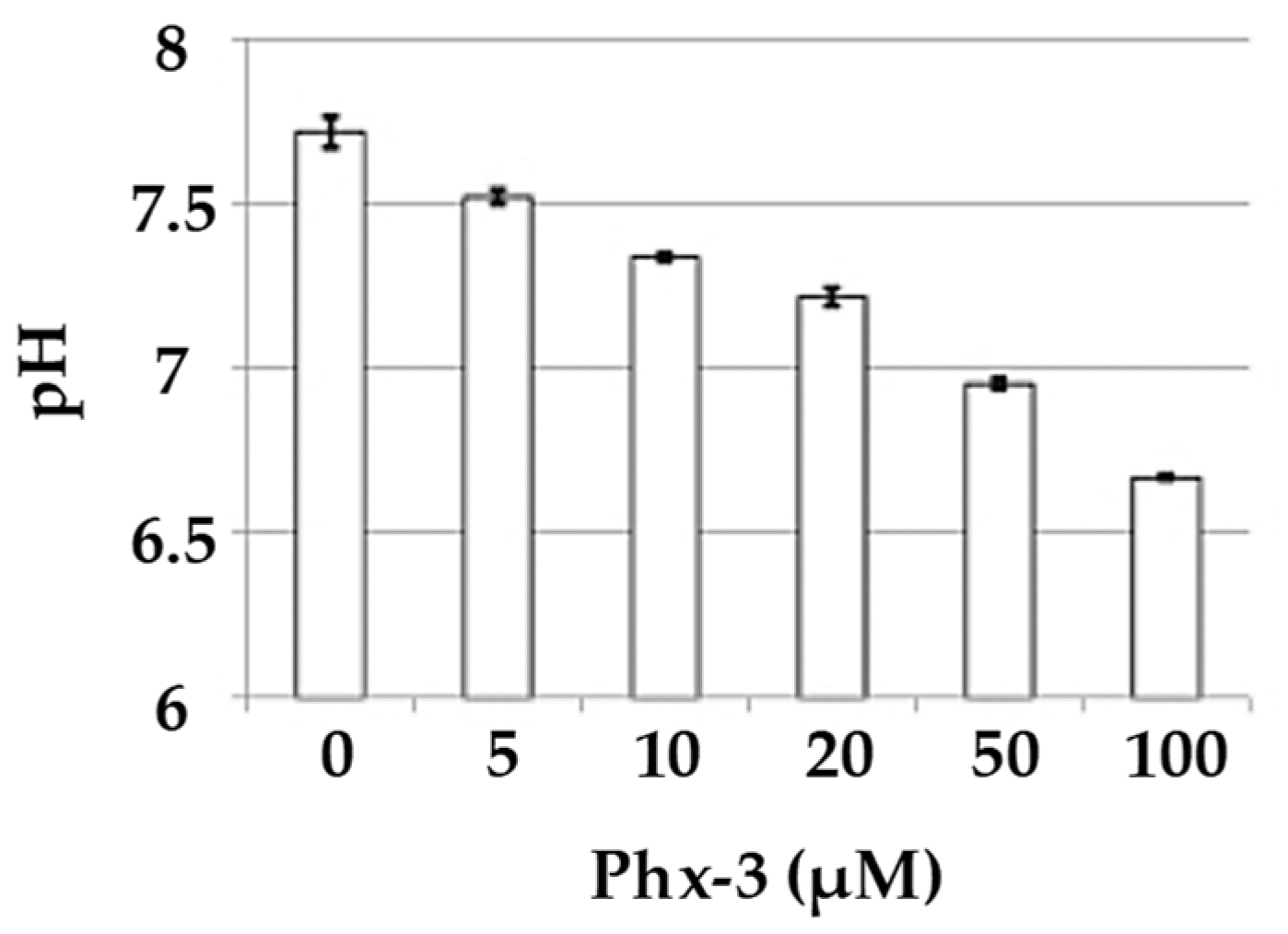
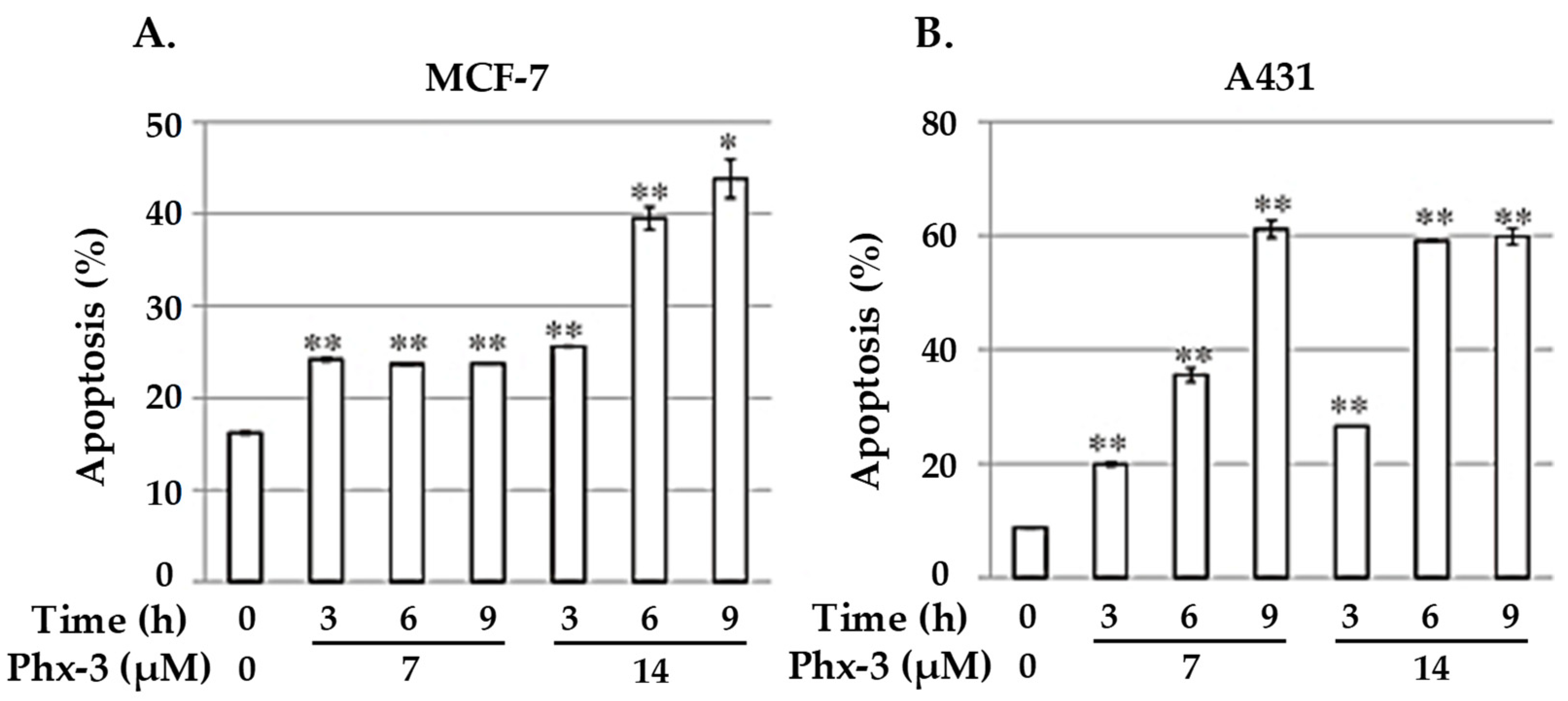
4.1. 2-Aminophenoxazine-3-one (Phx-3)
4.2. Amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazin-3-one (Phx-1)
4.3. Phenoxazine-Indole Conjugates
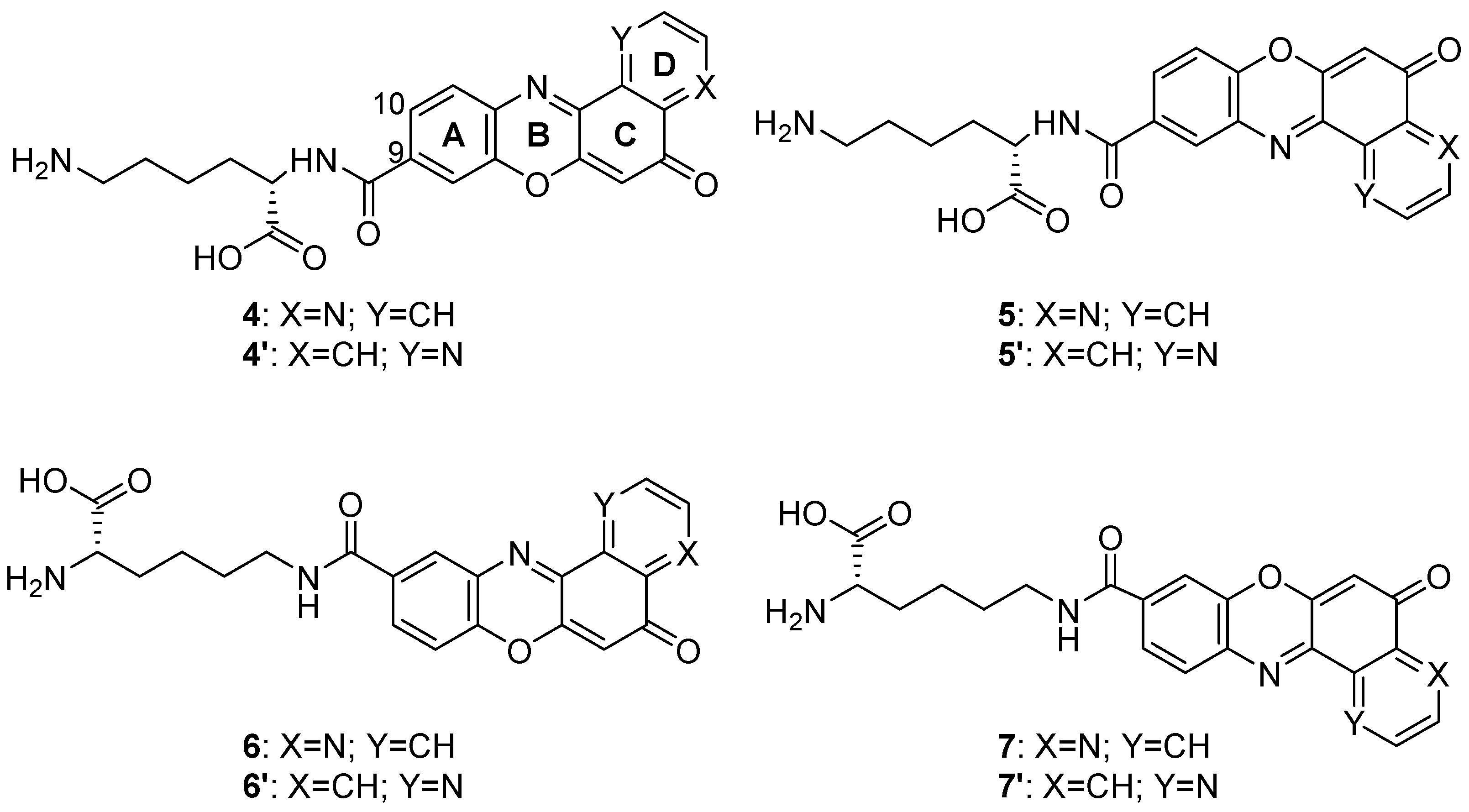
4.4. Pyridophenoxazinone Derivatives Conjugated to L-Lysine
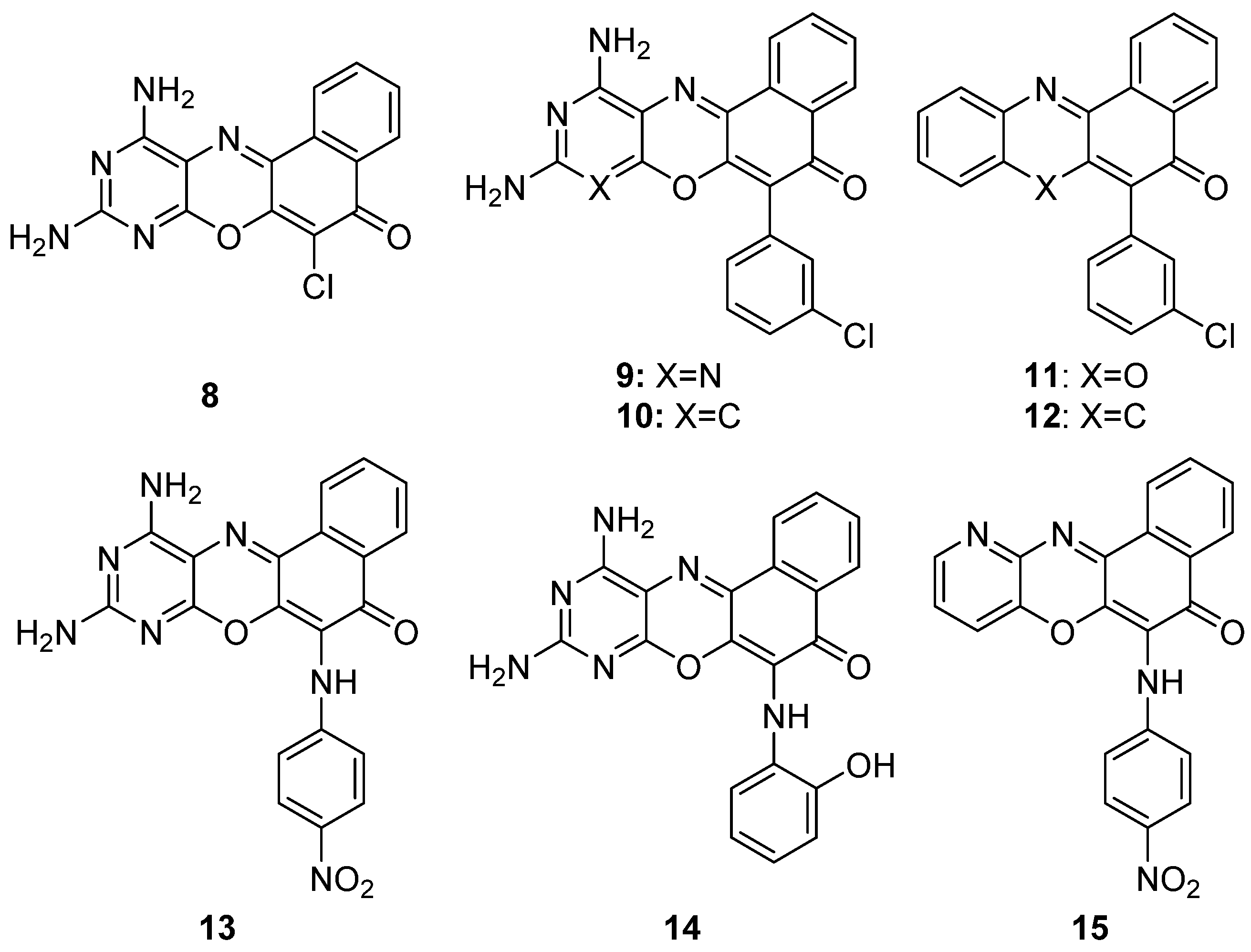
5. Antibacterial and Antifungal Activities
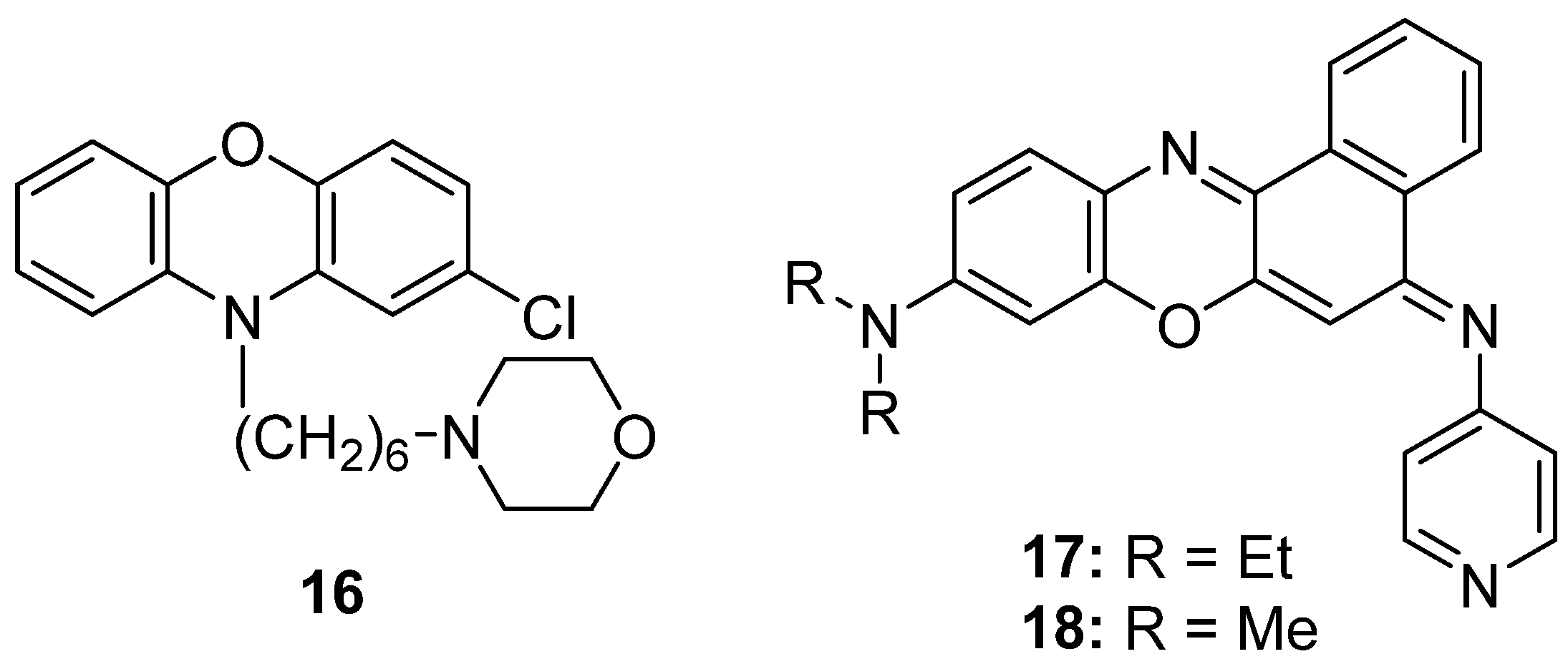
5.1. Antibacterial Activity on Genera Helicobacter
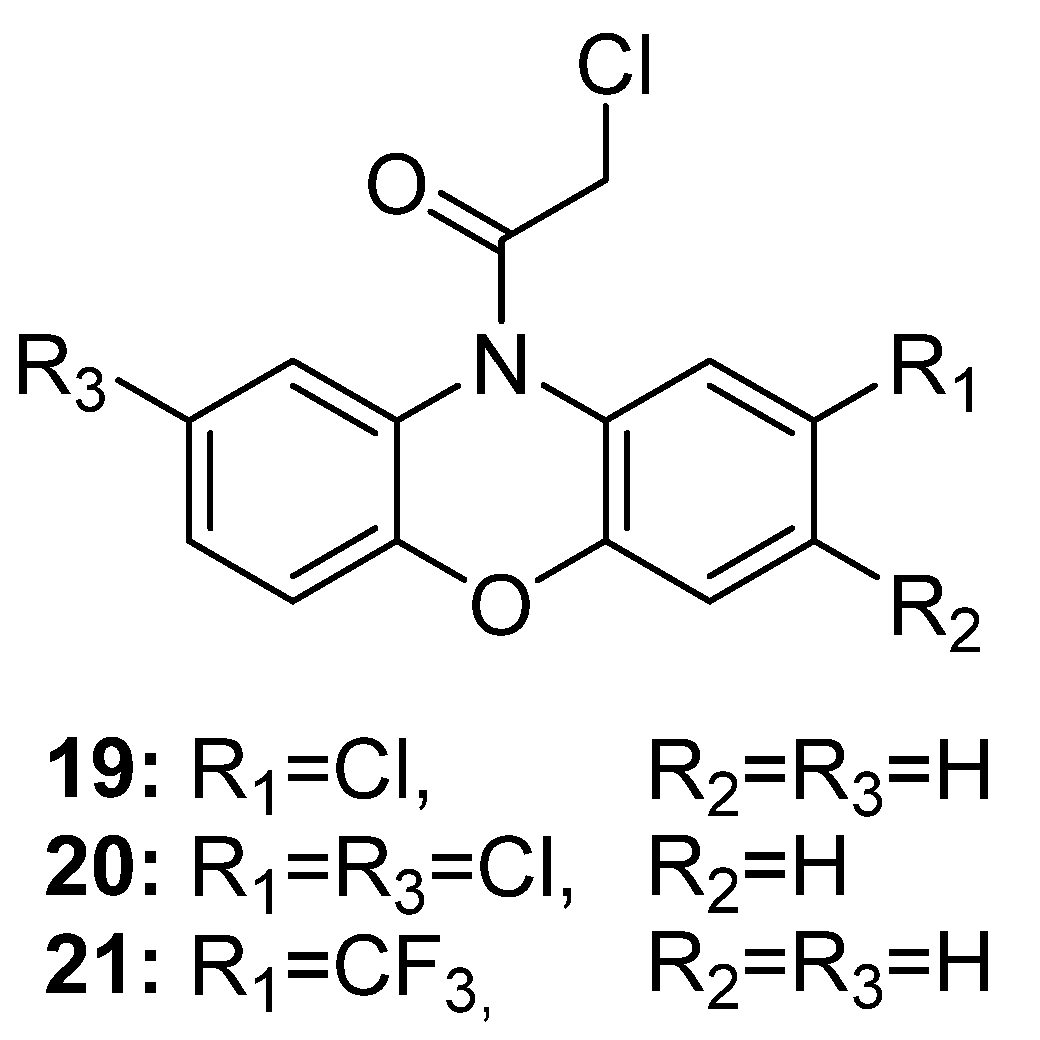
5.2. Other Antibacterial and Antifungal Activities
6. Other Activities
6.1. Antiviral Activity
6.2. Antiparasitic Activity
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, A.; Miyazawa, K.; Tabuchi, T. Prevention of carcinogenesis and development of gastric and colon cancers by 2-Aminophenoxazine-3-one (Phx-3): Direct and indirect Anti-Cancer activity of Phx-3. Int. J. Mol. Sci. 2013, 14, 17573–17583. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Moreiras, A.M.; Coba de la Peña, T.; Martínez, A.; González, L.; Pellisier, F.; Reigosa, M.J. Mode of action of the hydroxamic acid BOA and other related compounds. Allelopathy 2004, 239–252. [Google Scholar]
- Bravo, H.R.; Lazo, W. Antialgal and antifungal activity of natural hydroxamic acids and related compounds. J. Agric. Food Chem. 1996, 44, 1569–1571. [Google Scholar] [CrossRef]
- Jensen, B.M.; Adhikari, K.B.; Schnoor, H.J.; Juel-Berg, N.; Fomsgaard, I.S.; Poulsen, L.K. Quantitative analysis of absorption, metabolism, and excretion of benzoxazinoids in humans after the consumption of high- and low-benzoxazinoid diets with similar contents of cereal dietary fibres: A crossover study. Eur. J. Nutr. 2017, 56, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Scott Chilton, W. Biosynthesis of DIMBOA in maize using deuterium oxide as a tracer. Phytochemistry 1994, 37, 167–171. [Google Scholar] [CrossRef]
- Kumar, P.; Moreland, D.E.; Chilton, W.S. 2H-1,4-benzoxazin-3(4H)-one, an intermediate in the biosynthesis of cyclic hydroxamic acids in maize. Phytochemistry 1994, 36, 893–898. [Google Scholar] [CrossRef]
- Adhikari, K.B.; Lærke, H.N.; Mortensen, A.G.; Fomsgaard, I.S. Plasma and Urine Concentrations of Bioactive Dietary Benzoxazinoids and Their Glucuronidated Conjugates in Rats Fed a Rye Bread-Based Diet. J. Agric. Food Chem. 2012, 60, 11518–11524. [Google Scholar] [CrossRef]
- Steffensen, S.K.; Pedersen, H.A.; Adhikari, K.B.; Laursen, B.B.; Jensen, C.; Høyer, S.; Borre, M.; Pedersen, H.H.; Borre, M.; Edwards, D.; et al. Benzoxazinoids in Prostate Cancer Patients after a Rye-Intensive Diet: Methods and Initial Results. J. Agric. Food Chem. 2016, 64, 8235–8245. [Google Scholar] [CrossRef]
- Kumar, P.; Gagliardo, R.W.; Chilton, W.S. Soil transformation of wheat and corn metabolites mboa and DIM2BOA into aminophenoxazinones. J. Chem. Ecol. 1993, 19, 2453–2461. [Google Scholar] [CrossRef]
- Macías, F.A.; Oliveros-Bastidas, A.; Marín, D.; Castellano, D.; Simonet, A.M.; Molinillo, J.M.G. Degradation studies on benzoxazinoids. Soil degradation dynamics of (2R)-2-O-β-D-glucopyranosyl-4-hydroxy-(2H)-1,4-benzoxazin-3(4H)-one (DIBOA-Glc) and its degradation products, phytotoxic allelochemicals from gramineae. J. Agric. Food Chem. 2005, 53, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fomsgaard, I.S.; Mortensen, A.G.; Carlsen, S.C.K. Microbial transformation products of benzoxazolinone and benzoxazinone allelochemicals—A review. Chemosphere 2004, 54, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Macías, F.A.; Marín, D.; Oliveros-Bastidas, A.; Molinillo, J.M.G. Rediscovering the bioactivity and ecological role of 1,4-benzoxazinones. Nat. Prod. Rep. 2009, 26, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Macías, F.A.; Oliveros-Bastidas, A.; Marín, D.; Castellano, D.; Simonet, A.M.; Molinillo, J.M.G. Degradation studies on benzoxazinoids. Soil degradation dynamics of 2,4-dihydroxy-7-methoxy-(2H)-1,4-benzoxazin-3(4H)-one (DIMBOA) and its degradation products, phytotoxic allelochemicals from gramineae. J. Agric. Food Chem. 2004, 52, 6402–6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturelli, S.; Belz, R.G.; Kämper, A.; Berger, A.; von Horn, K.; Wegner, A.; Böcker, A.; Zabulon, G.; Langenecker, T.; Kohlbacher, O.; et al. Plants release precursors of histone deacetylase inhibitors to suppress growth of competitors. Plant Cell 2015, 27, 3175–3189. [Google Scholar] [CrossRef] [Green Version]
- Macías, F.A.; Oliveros-Bastidas, A.; Marín, D.; Chinchilla, N.; Castellano, D.; Molinillo, J.M.G. Evidence for an allelopathic interaction between rye and wild oats. J. Agric. Food Chem. 2014, 62, 9450–9457. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, N.; Marín, D.; Oliveros-Bastidas, A.; Molinillo, J.M.G.; Macías, F.A. Soil biodegradation of a benzoxazinone analog proposed as a natural products-based herbicide. Plant Soil 2015, 393, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Gierl, A.; Gruen, S.; Genschel, U.; Huettl, R.; Frey, M. Evolution of indole and benzoxazinone biosynthesis in Zea mays. Recent Adv. Phytochem. 2004, 69–83. [Google Scholar]
- Farrés, M.; Villagrasa, M.; Eljarrat, E.; Barceló, D.; Tauler, R. Chemometric evaluation of different experimental conditions on wheat (Triticum aestivum L.) development using liquid chromatography mass spectrometry (LC–MS) profiles of benzoxazinone derivatives. Anal. Chim. Acta 2012, 731, 24–31. [Google Scholar] [CrossRef]
- Schulz, M.; Marocco, A.; Tabaglio, V.; Macias, F.A.; Molinillo, J.M.G. Benzoxazinoids in rye allelopathy—From discovery to application in sustainable weed control and organic farming. J. Chem. Ecol. 2013, 39, 154–174. [Google Scholar] [CrossRef]
- Tomoda, A.; Yamaguchi, T.; Sato, K.; Iwata, A. Antiviral agents containing aminophenoxazines. Tohoku J. Exp. Med. 2004, 203, 47–52. [Google Scholar]
- Bitzer, M.; Lauer, U.M.; Venturelli, S.; Armeanu, S. Aminophenoxazinone compounds as antitumor and antiinflammatory agents. Interdiscip. Int. J. Am. Cancer Soc. 2009, 109, 2132–2141. [Google Scholar]
- Pandurangan, K.; Gallagher, S.; Morgan, G.G.; Müller-Bunz, H.; Paradisi, F. Structure and antibacterial activity of the silver(I) complex of 2-aminophenoxazine-3-one. Metallomics 2010, 2, 530. [Google Scholar] [CrossRef]
- Kehrmann, F. Ueber Oxydationsproducte vono-Aminophenolen. Berichte Dtsch. Chem. Gesellschaft 1906, 39, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Szeverényi, Z.; Milaeva, E.R.; Simándi, L.I. Kinetics of the oxidation of 2-aminophenol by dioxygen in the presence of tetrakis(3,5-di-t-butyl-4-hydroxyphenyl)-dodecachlorophthalocyaninatocobalt(II). J. Mol. Catal. 1991, 67, 251–258. [Google Scholar] [CrossRef]
- Gabriel, S. Ueber eine Darstellungsweise primärer Amine aus den entsprechenden Halogenverbindungen. Berichte der Dtsch. Chem. Gesellschaft 1887, 20, 2224–2236. [Google Scholar] [CrossRef] [Green Version]
- Prinz, H.; Ridder, A.K.; Vogel, K.; Böhm, K.J.; Ivanov, I.; Ghasemi, J.B.; Aghaee, E.; Müller, K. N-heterocyclic (4-phenylpiperazin-1-yl)methanones derived from phenoxazine and phenothiazine as highly potent inhibitors of tubulin polymerization. J. Med. Chem. 2017, 60, 749–766. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cheng, T.; Yin, C.; Zhou, S.; Fan, Q.; Wu, W.; Jiang, X. Phenothiazine versus Phenoxazine: Structural effects on the photophysical properties of NIR-II AIE fluorophores. ACS Appl. Mater. Interfaces 2020, 12, 43466–43473. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Yang, J.; Shen, Y.; Yao, Y.; Lv, G.; Hao, S.; Li, C. The near-infrared fluorescent probes based on phenoxazine for the rapid detection of hypochlorous acid. Dye. Pigment. 2020, 179, 108404. [Google Scholar] [CrossRef]
- Farmer, L.A.; Haidasz, E.A.; Griesser, M.; Pratt, D.A. Phenoxazine: A Privileged Scaffold for Radical-Trapping Antioxidants. J. Org. Chem. 2017, 82, 10523–10536. [Google Scholar] [CrossRef]
- Onoabedje, E.A.; Egu, S.A.; Ezeokonkwo, M.A.; Okoro, U.C. Highlights of molecular structures and applications of phenothiazine & phenoxazine polycycles. J. Mol. Struct. 2019, 1175, 956–962. [Google Scholar] [CrossRef]
- Alves, C.M.A.; Naik, S.; Coutinho, P.J.G.; Gonçalves, M.S.T. Novel DNA fluorescence probes based on N-[5-(11-functionalised-undecylamino)-9H-benzo[a]phenoxazin-9-ylidene]propan-1-aminium chlorides: Synthesis and photophysical studies. Tetrahedron Lett. 2011, 52, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.-J.; Niu, J.-Y.; He, D.-D.; Sun, R.; Xu, Y.-J.; Ge, J.-F. Near-infrared pH probes based on phenoxazinium connecting with nitrophenyl and pyridinyl groups. Dye. Pigment. 2018, 149, 481–490. [Google Scholar] [CrossRef]
- Jaszczyszyn, A.; Gąsiorowski, K.; Świątek, P.; Malinka, W.; Cieślik-Boczula, K.; Petrus, J.; Czarnik-Matusewicz, B. Chemical structure of phenothiazines and their biological activity. Pharmacol. Rep. 2012, 64, 16–23. [Google Scholar] [CrossRef]
- Peng, M.; Ding, Y.; Yu, L.; Deng, Y.; Lai, W.; Hu, Y.; Zhang, H.; Wu, X.; Fan, H.; Ding, H.; et al. Tegafur Substitution for 5-Fu in Combination with Actinomycin D to Treat Gestational Trophoblastic Neoplasm. PLoS ONE 2015, 10, e0143531. [Google Scholar] [CrossRef] [PubMed]
- Hadi, L.M.; Yaghini, E.; Macrobert, A.J.; Loizidou, M. Synergy between photodynamic therapy and dactinomycin chemotherapy in 2D and 3D ovarian cancer cell cultures. Int. J. Mol. Sci. 2020, 21, 3203. [Google Scholar] [CrossRef]
- Kühlborn, J.; Konhäuser, M.; Groß, J.; Wich, P.R.; Opatz, T. Xylochemical Synthesis of Cytotoxic 2-Aminophenoxazinone-Type Natural Products Through Oxidative Cross Coupling. ACS Sustain. Chem. Eng. 2019, 7, 4414–4419. [Google Scholar] [CrossRef]
- Pasceri, R.; Siegel, D.; Ross, D.; Moody, C.J. Aminophenoxazinones as inhibitors of indoleamine 2,3-dioxygenase (IDO). Synthesis of exfoliazone and chandranimycin A. J. Med. Chem. 2013, 56, 3310–3317. [Google Scholar] [CrossRef]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, A.; Lyndin, M.; Sikora, V.; Lyndina, Y.; Romaniuk, S.; Sikora, K. Heavy metals effect on breast cancer progression. J. Occup. Med. Toxicol. 2017, 12, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [Green Version]
- JGCA Japanese gastric cancer treatment guidelines 2018 (5th edition). Gastric Cancer 2021, 24, 1–21. [CrossRef] [Green Version]
- Zhao, Y.; Guo, Q.; Chen, J.; Hu, J.; Wang, S.; Sun, Y. Role of long non-coding RNA HULC in cell proliferation, apoptosis and tumor metastasis of gastric cancer: A clinical and in vitro investigation. Oncol. Rep. 2014, 31, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Waddingham, W.; Nieuwenburg, S.A.V.; Carlson, S.; Rodriguez-Justo, M.; Spaander, M.; Kuipers, E.J.; Jansen, M.; Graham, D.G.; Banks, M. Recent advances in the detection and management of early gastric cancer and its precursors. Frontline Gastroenterol. 2020. [Google Scholar] [CrossRef]
- Stock, C.; Pedersen, S.F. Roles of pH and the Na+/H+ exchanger NHE1 in cancer: From cell biology and animal models to an emerging translational perspective? Semin. Cancer Biol. 2017, 43, 5–16. [Google Scholar] [CrossRef]
- Pérez-Sala, D.; Collado-Escobar, D.; Mollinedo, F. Intracellular alkalinization suppresses lovastatin-induced apoptosis in HL-60 cells through the inactivation of a pH-dependent endonuclease. J. Biol. Chem. 1995, 270, 6235–6242. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Wang, Z.; Hao, Z.; Li, L.; Lu, J.; Kang, H.; Lu, Y.; You, Y.; Li, L.; Chen, Q.; et al. Requirement for etoposide in the treatment of pregnancy related hemophagocytic lymphohistiocytosis: A multicenter retrospective study. Orphanet J. Rare Dis. 2019, 14, 1–9. [Google Scholar] [CrossRef]
- Kaufmann, S.H. Induction of Endonucleolytic DNA Cleavage in Human Acute Myelogenous Leukemia Cells by Etoposide, Camptothecin, and Other Cytotoxic Anticancer Drugs: A Cautionary Note. Cancer Res. 1989, 49, 5870–5878. [Google Scholar]
- Che, X.-F.; Akiyama, S.-I.; Tomoda, A. Suppression of the proliferation of cancer cell lines, KB-3-1 and K562 cells preceded by a decrease in intracellular pH caused by phenoxazine derivatives. Oncol. Rep. 2008, 19, 1253–1258. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Honda, K.; Ishida, R.; Nohira, T.; Tomoda, A. Antitumor activity of 2-amino-4,4α-dihydro-4α,7-dimethyl-3Hphenoxazine-3-one against Meth A tumor transplanted into BALB/c mice. Anticancer Drugs 2000, 11, 653–657. [Google Scholar] [CrossRef]
- Miyano-Kurosaki, N.; Kurosaki, K.; Hayashi, M.; Takaku, H.; Hayafune, M.; Shirato, K.; Kasuga, T.; Endo, T.; Tomoda, A. 2-Aminophenoxazine-3-one suppresses the growth of mouse malignant melanoma B16 cells transplanted into C57BL/6Cr Slc mice. Biol. Pharm. Bull. 2006, 29, 2197–2201. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, T.; Tabuchi, T.; Shirato, K.; Imaizumi, K.; Tomoda, A. Caspase-independent cell death revealed in human gastric cancer cell lines, MKN45 and KATO III treated with phenoxazine derivatives. Oncol. Rep. 2007, 17, 409–415. [Google Scholar] [CrossRef]
- Nagata, H.; Che, X.F.; Miyazawa, K.; Tomoda, A.; Konishi, M.; Ubukata, H.; Tabuchi, T. Rapid decrease of intracellular pH associated with inhibition of Na+/H+ exchanger precedes apoptotic events in the MNK45 and MNK74 gastric cancer cell lines treated with 2-aminophenoxazine-3-one. Oncol. Rep. 2011, 25, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Pedersen, S.F.; Kramhøft, B.; Skovsgaard, T.; Hoffmann, E.K. pH regulation in sensitive and multidrug resistant Ehrlich ascites tumor cells. Cell. Physiol. Biochem. 1998, 8, 138–150. [Google Scholar] [CrossRef]
- Nakachi, T.; Tabuchi, T.; Takasaki, A.; Arai, S.; Miyazawa, K.; Tomoda, A. Anticancer activity of phenoxazines produced by bovine erythrocytes on colon cancer cells. Oncol. Rep. 2010, 1517–1522. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; He, W.; Qin, X.; Wei, X.; Tian, X.; Liao, L.; Liao, S.; Yang, B.; Tu, Z.; Chen, B.; et al. Three new indolyl diketopiperazine metabolites from the antarctic soil-derived fungus Penicillium sp. SCSIO 05705. RSC Adv. 2015, 5, 68736–68742. [Google Scholar] [CrossRef]
- Che, X.-F.; Zheng, C.-L.; Akiyama, S.-I.; Tomoda, A. 2-Aminophenoxazine-3-one and 2-amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazine-3-one cause cellular apoptosis by reducing higher intracellular pH in cancer cells. Proc. Jpn. Acad. Ser. B 2011, 87, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabuchi, T.; Che, X.-F.; Hiraishi, K.; Adachi, M.; Miyano, K.; Sumimoto, H.; Tabuchi, T.; Miyazawa, K.; Tomoda, A. Selectively Induced Apoptosis in Human Neutrophils in the Presence of Oxidative Phenoxazines, 2-Amino-4,4α-dihydryo-4α-7H-phenoxazine-3-one and 2-Aminophenoxazine-3-one, Preceded by Decrease of Intracellular pH, Depolarization of the Mitochondria, and Inh. J. Pharmacol. Sci. 2011, 117, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Kohno, K.; Miyake, M.; Sano, O.; Tanaka-Kataoka, M.; Yamamoto, S.; Koya-Miyata, S.; Arai, N.; Fujii, M.; Watanabe, H.; Ushio, S.; et al. Anti-inflammatory and immunomodulatory properties of 2-amino-3H-phenoxazin-3-one. Biol. Pharm. Bull. 2008, 31, 1938–1945. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Lu, Y.; Xing, Y.; Ma, Y.; Lu, J.; Bao, W.; Wang, Y.; Xi, T. A novel anticancer and antifungus phenazine derivative from a marine actinomycete BM-17. Microbiol. Res. 2012, 167, 616–622. [Google Scholar] [CrossRef]
- Takemura, A.; Che, X.F.; Tabuchi, T.; Moriya, S.; Miyazawa, K.; Tomoda, A. Enhancement of cytotoxic and pro-apoptotic effects of 2-aminophenoxazine-3- one on the rat hepatocellular carcinoma cell line dRLh-84, the human hepatocellular carcinoma cell line HepG2, and the rat normal hepatocellular cell line RLN-10 in combination wi. Oncol. Rep. 2012, 27, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidros, D.S.; Vogelbaum, M.A. Novel Drug Delivery Strategies in Neuro-Oncology. Neurotherapeutics 2009, 6, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuine, M.A.; Tokuda, H.; Takayasu, J.; Enjyo, F.; Mukainaka, T.; Konoshima, T.; Nishino, H.; Kapadia, G.J. Cancer chemopreventive effect of phenothiazines and related tri-heterocyclic analogues in the 12-O-tetradecanoylphorbol-13-acetate promoted Epstein-Barr virus early antigen activation and the mouse skin two-stage carcinogenesis models. Pharmacol. Res. 2004, 49, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kenia, H.; Shivkumar, B.; Kotnal, R.B.; Ramesha, A.; Devadiga, P.; Simpi, C.C.; Chandrashekar, V.M. Synthesis and Evaluation of Phenothiazine Derivatives. IOSR J. Pharm. 2020, 10, 54–62. [Google Scholar]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J.-I. Aberrant signaling pathways in glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [Green Version]
- Soni, D.; King, J.A.J.; Kaye, A.H.; Hovens, C.M. Genetics of glioblastoma multiforme: Mitogenic signaling and cell cycle pathways converge. J. Clin. Neurosci. 2005, 12, 1–5. [Google Scholar] [CrossRef]
- Los, M.; Maddika, S.; Erb, B.; Schulze-Osthoff, K. Switching Akt: From survival signaling to deadly response. BioEssays 2009, 31, 492–495. [Google Scholar] [CrossRef] [Green Version]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. In Inflammatory Processes: Molecular Mechanisms and Therapeutic Opportunities; Letts, L.G., Morgan, D.W., Eds.; Birkhäuser Basel: Basel, Switzerland, 2000; pp. 13–21. [Google Scholar]
- Che, X.-F.; Moriya, S.; Zheng, C.-L.; Abe, A.; Tomoda, A.; Miyazawa, K. 2-Aminophenoxazine-3-one-induced apoptosis via generation of reactive oxygen species followed by c-jun N-terminal kinase activation in the human glioblastoma cell line LN229. Int. J. Oncol. 2013, 43, 1456–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, S.; Miyazawa, K.; Kawaguchi, T.; Che, X.F.; Tomoda, A. Involvement of endoplasmic reticulum stress-mediated CHOP (GADD153) induction in the cytotoxicity of 2-aminophenoxazine-3-one in cancer cells. Int. J. Oncol. 2011, 39, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.-L.; Che, Z.-F.; Akiyama, S.-I.; Miyazawa, K.; Tomoda, A. 2-Aminophenoxazine-3-one induces cellular apoptosis by causing rapid intracellular acidification and generating reactive oxygen species in human lung adenocarcinoma cells. Int. J. Oncol. 2010, 36, 641–650. [Google Scholar] [CrossRef]
- Goddard, L.; Yorozuya, L.; Hirokane, J. Art of prevention: The importance of melanoma surveillance. Int. J. Women’s Dermatol. 2020, 6, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Čelakovská, J.; Bukač, J.; Čáková, L.; Šimková, M.; Jandová, E. Epidemiology of melanoma in the Czech Republic in east bohemia in the period 2002–2017 and the effect of the annual sunshine exposure. Acta Med. 2020, 63, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Yamamoto, S.; Sano, O.; Fujii, M.; Kohno, K.; Ushio, S.; Iwaki, K.; Fukuda, S. Inhibitory effects of 2-amino-3H-phenoxazin-3-one on the melanogenesis of murine B16 melanoma cell line. Biosci. Biotechnol. Biochem. 2010, 74, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-H.; Li, M.-G.; Li, Y.-Q.; Zhao, J.-Y.; Ding, Z.-G.; Yang, P.-W.; Cui, X.-L.; Wen, M.-L. Cytotoxic metabolites of Streptimonospora salina. Chem. Nat. Compd. 2008, 44, 503–505. [Google Scholar] [CrossRef]
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384. [Google Scholar]
- Machihara, K.; Tanaka, H.; Hayashi, Y.; Murakami, I.; Namba, T. Questiomycin A stimulates sorafenib-induced cell death via suppression of glucose-regulated protein 78. Biochem. Biophys. Res. Commun. 2017, 492, 33–40. [Google Scholar] [CrossRef]
- Tomoda, A.; Arai, S.; Ishida, R.; Shimamoto, T.; Ohyashiki, K. An improved method for the rapid preparation of 2-Amino-4,4a-dihydro-4a,7-dimethyl-3H-phenoxazine-3-one, a Novel Antitumor Agent. Bioorg. Med. Chem. Lett. 2001, 11, 1057–1058. [Google Scholar] [CrossRef]
- Kimura, K.; Usui, Y.; Hattori, T.; Yamakawa, N.; Goto, H.; Usui, M.; Okada, S.; Shirato, K.; Tomoda, A. Phenoxazine derivative, 2-amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazine-3-one suppresses growth of human retinoblastoma cell line Y79 in vitro and in vivo. Oncol. Rep. 2008, 19, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Shimamoto, T.; Tomoda, A.; Ishida, R.; Ohyashiki, K. Antitumor effects of a novel phenoxazine derivative on human leukemia cell lines through activation of caspase-3 and telomerase. Clin. Cancer Res. 2001, 7, 704–708. [Google Scholar] [PubMed]
- Shirato, K.; Imaizumi, K.; Abe, A.; Tomoda, A. Phenoxazine derivatives 2-amino-4,4α-dihydro-4α-phenoxazine-3- one and 2-aminophenoxazine-3-one-induced apoptosis through a caspase-independent mechanism in human neuroblastoma cell line NB-1 cells. Biol. Pharm. Bull. 2007, 30, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejías, F.J.R.; Durán, A.G.; Zorrilla, J.G.; Varela, R.M.; Molinillo, J.M.G.; Valdivia, M.M.; Macías, F.A. Acyl Derivatives of Eudesmanolides To Boost their Bioactivity: An Explanation of Behavior in the Cell Membrane Using a Molecular Dynamics Approach. ChemMedChem 2020, 1–12. [Google Scholar] [CrossRef]
- Chauhan, M.; Saxena, A.; Saha, B. An insight in anti-malarial potential of indole scaffold: A review. Eur. J. Med. Chem. 2021, 218, 113400. [Google Scholar] [CrossRef]
- Chadha, N.; Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef]
- Paul, K.; Bindal, S.; Luxami, V. Synthesis of new conjugated coumarin–benzimidazole hybrids and their anticancer activity. Bioorg. Med. Chem. Lett. 2013, 23, 3667–3672. [Google Scholar] [CrossRef]
- Nunewar, S.N.; Kotla, N.; Lakshmi Uppu, J.; Dixit, A.; Pooladanda, V.; Godugu, C.; Tangellamudi, N.D. Synthesis of 1-(Indol-2-yl)-phenoxazine hybrids from quinacetophenone precursors and their biological evaluation as DNA intercalating agents. J. Mol. Struct. 2020, 1217, 128311. [Google Scholar] [CrossRef]
- Pedatella, S.; Cerchia, C.; Manfra, M.; Cioce, A.; Bolognese, A.; Lavecchia, A. Antitumor agents 7. Synthesis, antiproliferative activity and molecular modeling of new l-lysine-conjugated pyridophenoxazinones as potent DNA-binding ligands and topoisomerase IIα inhibitors. Eur. J. Med. Chem. 2020, 187, 111960. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Suzuki, M.; Tomoda, A.; Arai, S.; Taguchi, H.; Hanawa, T.; Kamiya, S. Phenoxazine compounds produced by the reactions with bovine hemoglobin show antimicrobial activity against non-tuberculosis mycobacteria. Tohoku J. Exp. Med. 2004, 203, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Vissa, V.D.; Brennan, P.J. The genome of Mycobacterium leprae: A minimal myocobacterial gene set. Genome Biol. 2001, 2, 1–8. [Google Scholar] [CrossRef]
- Asaka, M.; Sugiyama, T.; Kato, M.; Satoh, K.; Kuwayama, H.; Fukuda, Y.; Fujioka, T.; Takemoto, T.; Kimura, K.; Shimoyama, T.; et al. A multicenter, double-blind study on triple therapy with lansoprazole, amoxicillin and clarithromycin for eradication of Helicobacter pylori in japanese peptic ulcer patients. Helicobacter 2001, 6, 254–262. [Google Scholar] [CrossRef]
- Soni, M.; Sharma, P.; Bhadauria, R.S.; Choudhary, M.L. A Review on Antibacterial Resistance. Pharm. Chem. J. 2020, 7, 68–71. [Google Scholar]
- Hanawa, T.; Osaki, T.; Manzoku, T.; Fukuda, M.; Kawakami, H.; Tomoda, A.; Kamiya, S. In vitro antibacterial activity of Phx-3 against Helicobacter pylori. Biol. Pharm. Bull. 2010, 33, 188–191. [Google Scholar] [CrossRef] [Green Version]
- Kamoda, O.; Anzai, K.; Mizoguchi, J.; Shiojiri, M.; Yanagi, T.; Nishino, T.; Kamiya, S. In vitro activity of a novel antimicrobial agent, TG44, for treatment of Helicobacter pylori infection. Antimicrob. Agents Chemother. 2006, 50, 3062–3069. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, A.R. Helicobacter, inflammation, and gastric cancer. Curr. Pathobiol. Rep. 2013, 1, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.; Panda, S.S.; Oliferenko, A.A.; Oliferenko, P.V.; Girgis, A.S.; Elagawany, M.; Küçükbay, F.Z.; Panda, C.S.; Pillai, G.G.; Samir, A.; et al. Macrocyclic peptidomimetics with antimicrobial activity: Synthesis, bioassay, and molecular modeling studies. Org. Biomol. Chem. 2015, 13, 9492–9503. [Google Scholar] [CrossRef] [Green Version]
- Kettle, A.J.; Carere, J.; Batley, J.; Benfield, A.H.; Manners, J.M.; Kazan, K.; Gardiner, D.M. A γ-lactamase from cereal infecting Fusarium spp. catalyses the first step in the degrdation of the benzoxazolinone class of phytoalexins. Fungal Genet. Biol. 2015, 83, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bacon, C.W.; Hinton, D.M.; Glenn, A.E.; Macías, F.A.; Marin, D. Interactions of Bacillus mojavensis and Fusarium verticillioides with a Benzoxazolinone (BOA) and its Transformation Product, APO. J. Chem. Ecol. 2007, 33, 1885–1897. [Google Scholar] [CrossRef] [Green Version]
- Bitzer, J.; Große, T.; Wang, L.; Lang, S.; Beil, W.; Zeeck, A. New aminophenoxazinones from a marine Halomonas sp.: Fermentation, structure elucidation, and biological activity. J. Antibiot. 2006, 59, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Tse, H.; Chan, E.; Lam, C.-W.; Leung, K.-F.; Chow, P.; Lee, K.-C.; Sze, K.-H.; Cheung, S.K.K.; Tse, M.-K.; Ho, P.-L.; et al. Production of 2-aminophenoxazin-3-one by Staphylococcus aureus causes false-positive results in β-galactosidase assays. J. Clin. Microbiol. 2012, 50, 3780–3782. [Google Scholar] [CrossRef] [Green Version]
- Maskey, R.P.; Li, F.C.; Qin, S.; Fiebig, H.H.; Laatsch, H. Chandrananimycins A-C: Production of Novel Anticancer Antibiotics from a Marine Actinomadura sp. Isolate M048 by Variation of Medium Composition and Growth Conditions. J. Antibiot. 2003, 56, 622–629. [Google Scholar] [CrossRef] [Green Version]
- Ocholi, S.S.; Onoabedje, E.A.; Egu, S.A. Synthesis and Antimicrobial Studies of 6-Aryl and 6-Anilino Benzo[a]phenoxazinones. Eur. J. Adv. Chem. Res. 2020, 1, 1–6. [Google Scholar] [CrossRef]
- Sridhar, B.T.; Girish, K.; Channu, B.C.; Thimmaiah, K.N.; Kumara, M.N. Antibacterial activity of phenoxazine derivatives. J. Chem. Pharm. Res. 2015, 7, 1074–1079. [Google Scholar]
- Bedernjak, A.F.; Zaytsev, A.V.; Babolat, M.; Cellier, M.; James, A.L.; Orenga, S.; Perry, J.D.; Groundwater, P.W.; Anderson, R.J. Synthesis and Evaluation of Novel 7- and 8-Aminophenoxazinones for the Detection of β-Alanine Aminopeptidase Activity and the Reliable Identification of Pseudomonas aeruginosa in Clinical Samples. J. Med. Chem. 2016, 59, 4476–4487. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Hayashi, T.; Miyazawa, K.; Tomoda, A. Phenoxazine Derivatives Suppress the Infections Caused by Herpes Simplex Virus Type-1 and Herpes Simplex Virus Type-2 Intravaginally Inoculated Into Mice. J. Pharmacol. Sci. 2010, 114, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Hayashi, T.; Tomoda, A. Phenoxazine Derivatives Inactivate Human Cytomegalovirus, Herpes Simplex Virus-1, and Herpes Simplex Virus-2 In Vitro. J. Pharmacol. Sci. 2008, 106, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Miyano-Kurosaki, N.; Ikegami, K.; Kurosaki, K.; Endo, T.; Aoyagi, H.; Hanami, M.; Yasumoto, J.; Tomoda, A. Anticancer effects of phenoxazine derivatives revealed by inhibition of cell growth and viability, disregulation of cell cycle, and apoptosis induction in HTLV-1-positive leukemia cells. J. Pharmacol. Sci. 2009, 110, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Divya, M.; Aparna, C.; Mayank, R.; Singh, M.P. In-silico insights to identify the bioactive compounds of edible mushrooms as potential MMP9 inhibitor for Hepatitis-B. Res. J. Biotechnol. 2021, 16, 116–126. [Google Scholar]
- Ansong, D.; Seydel, K.B.; Taylor, T.E. Malaria. In Hunter’s Tropical Medicine and Infectious Disease; Ryan, E.T., Hill, D.R., Solomon, T., Aronson, N.E., Endy, T.P., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 734–754. [Google Scholar]
- Ge, J.-F.; Arai, C.; Yang, M.; Bakar Md., A.; Lu, J.; Ismail, N.S.M.; Wittlin, S.; Kaiser, M.; Brun, R.; Charman, S.A.; et al. Discovery of novel benzo[a]phenoxazine SSJ-183 as a drug candidate for malaria. ACS Med. Chem. Lett. 2010, 1, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Marcu, A.; Schurigt, U.; Müller, K.; Moll, H.; Krauth-Siegel, R.L.; Prinz, H. Inhibitory effect of phenothiazine- and phenoxazine-derived chloroacetamides on Leishmania major growth and Trypanosoma brucei trypanothione reductase. Eur. J. Med. Chem. 2016, 108, 436–443. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Phx-1 | Phx-2 | Phx-3 |
|---|---|---|---|
| M. scrofulaceum | 2.8 | 1.4 | 2.8 |
| M. marinum | >45 | >45 | 11.3 |
| M. intracellulare | >45 | >45 | 5.6 |
| M. kansasii | 22.5 | 11.3 | >45 |
| M. tuberculosis, M. fortuitum and M. smegmatis | >45 | >45 | >45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zorrilla, J.G.; Rial, C.; Cabrera, D.; Molinillo, J.M.G.; Varela, R.M.; Macías, F.A. Pharmacological Activities of Aminophenoxazinones. Molecules 2021, 26, 3453. https://doi.org/10.3390/molecules26113453
Zorrilla JG, Rial C, Cabrera D, Molinillo JMG, Varela RM, Macías FA. Pharmacological Activities of Aminophenoxazinones. Molecules. 2021; 26(11):3453. https://doi.org/10.3390/molecules26113453
Chicago/Turabian StyleZorrilla, Jesús G., Carlos Rial, Daniel Cabrera, José M. G. Molinillo, Rosa M. Varela, and Francisco A. Macías. 2021. "Pharmacological Activities of Aminophenoxazinones" Molecules 26, no. 11: 3453. https://doi.org/10.3390/molecules26113453