
Chirality and Relativistic Effects in Os3(CO)12


Abstract
:1. Introduction
2. Results and Discussion
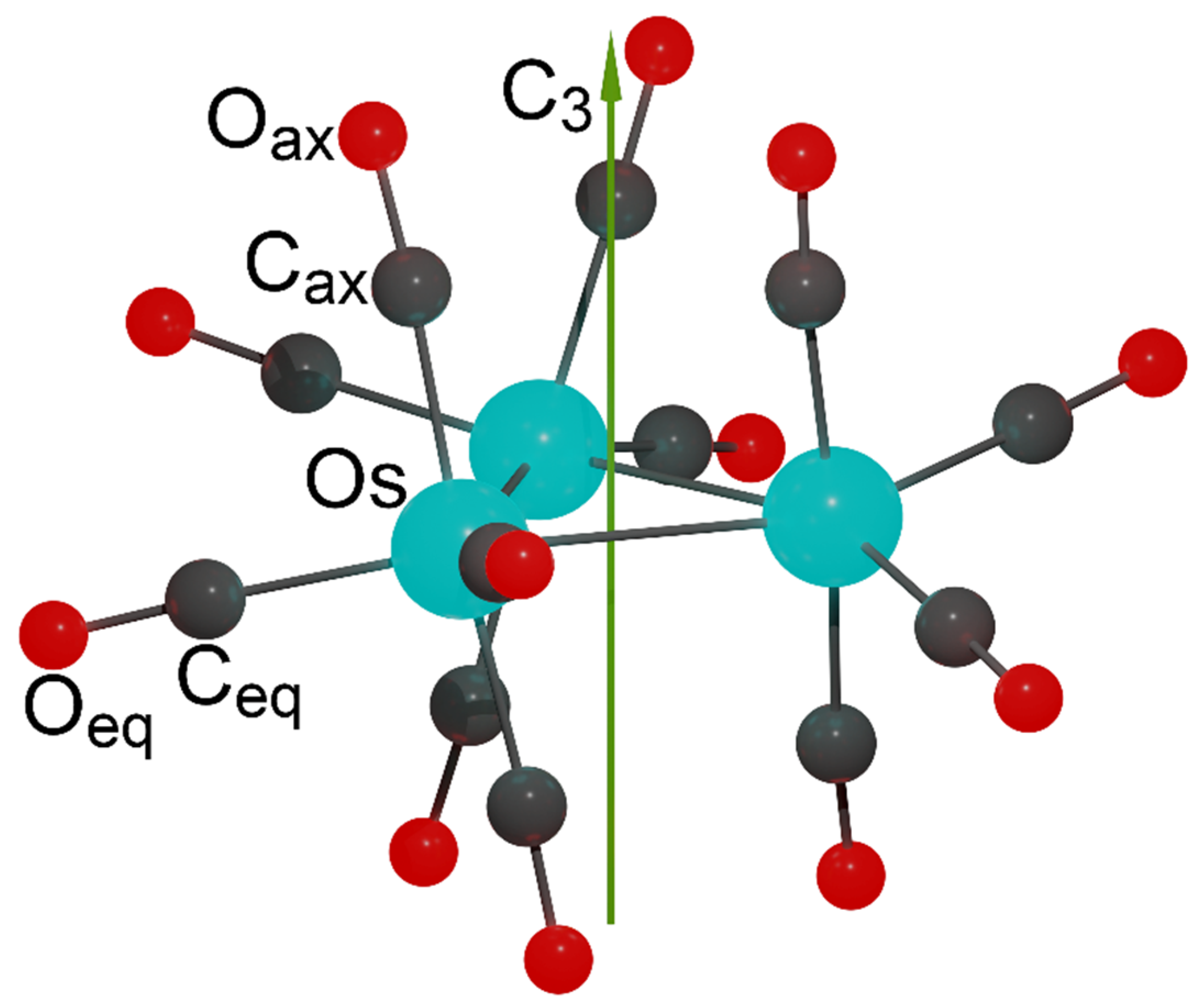
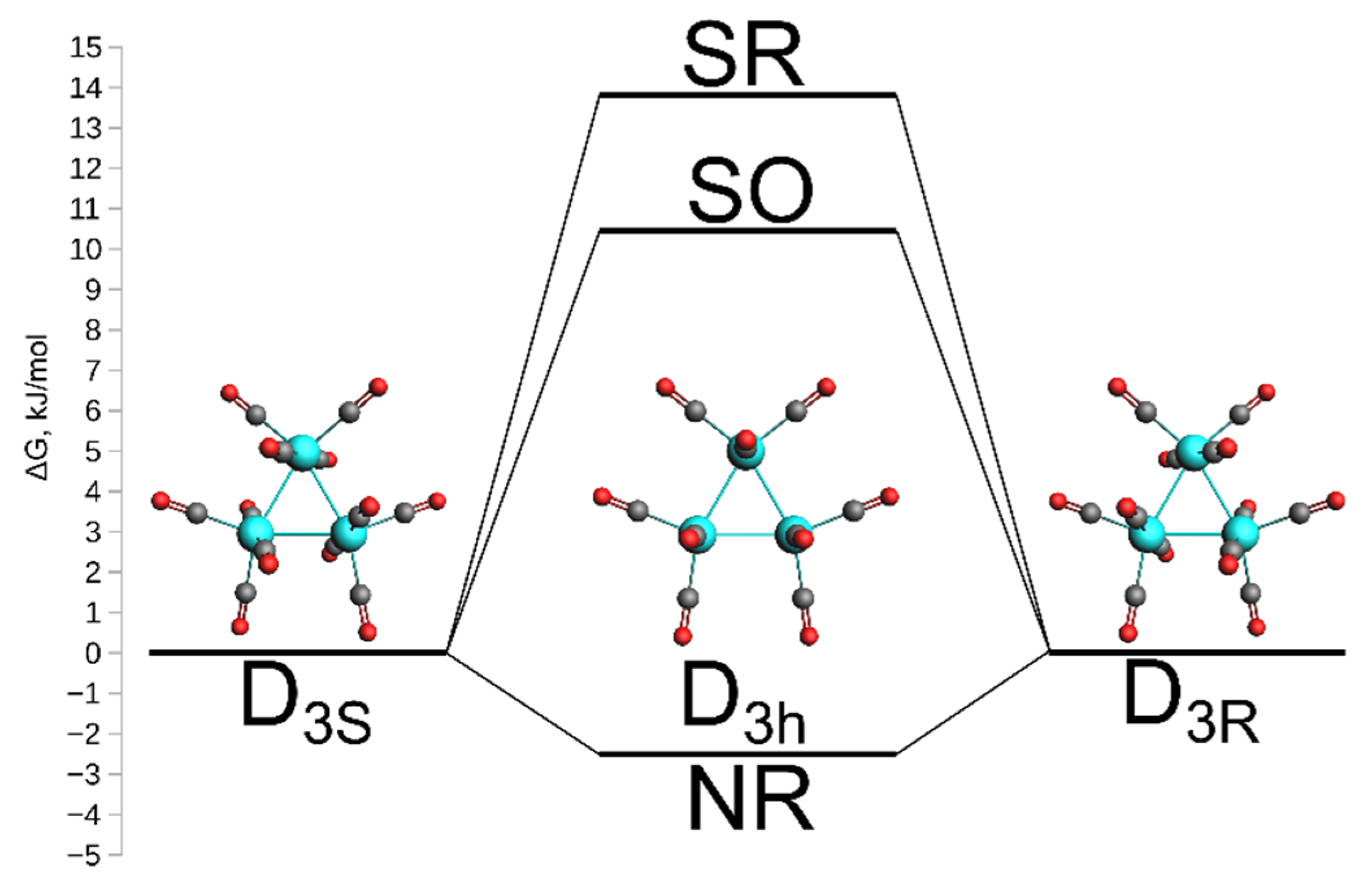
2.1. Structure and Energetics of Os3(CO)12
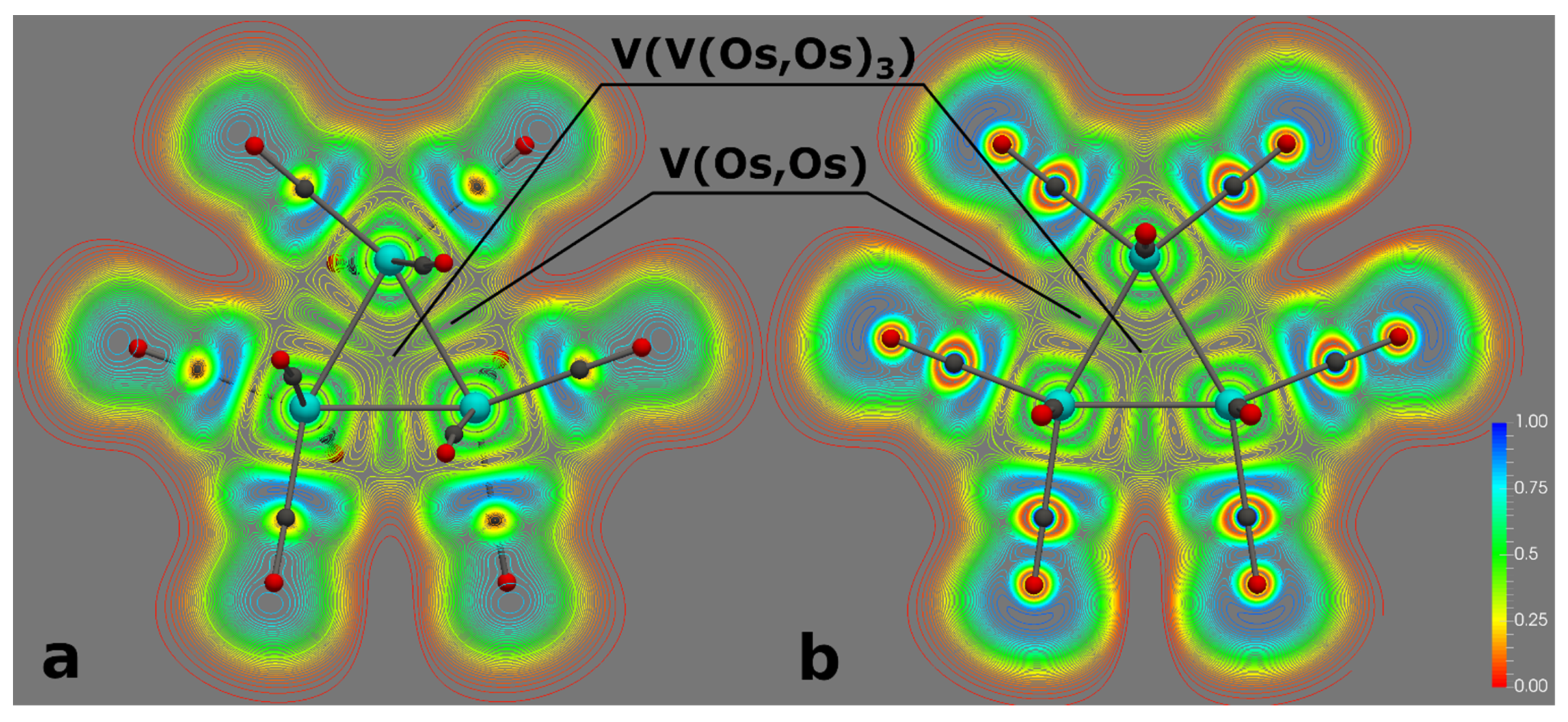
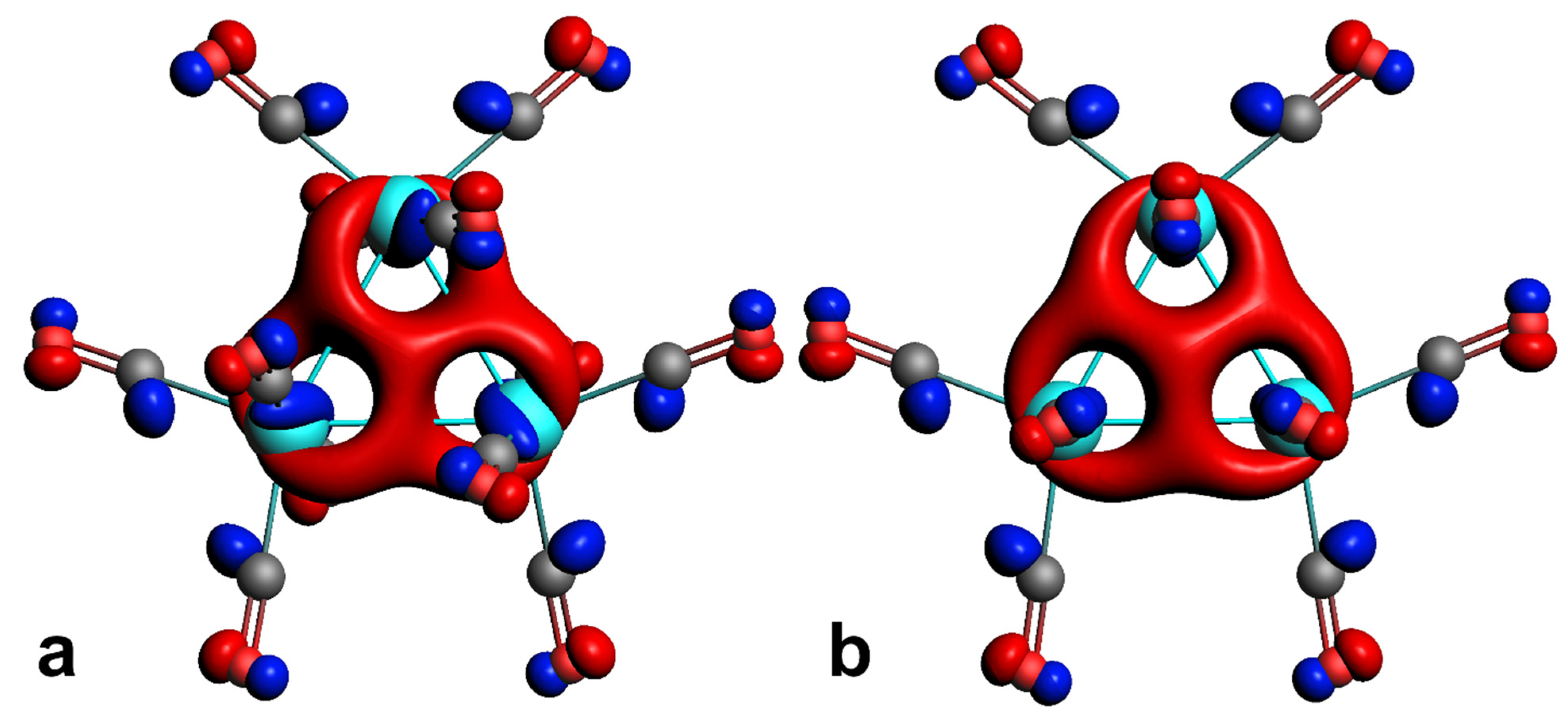
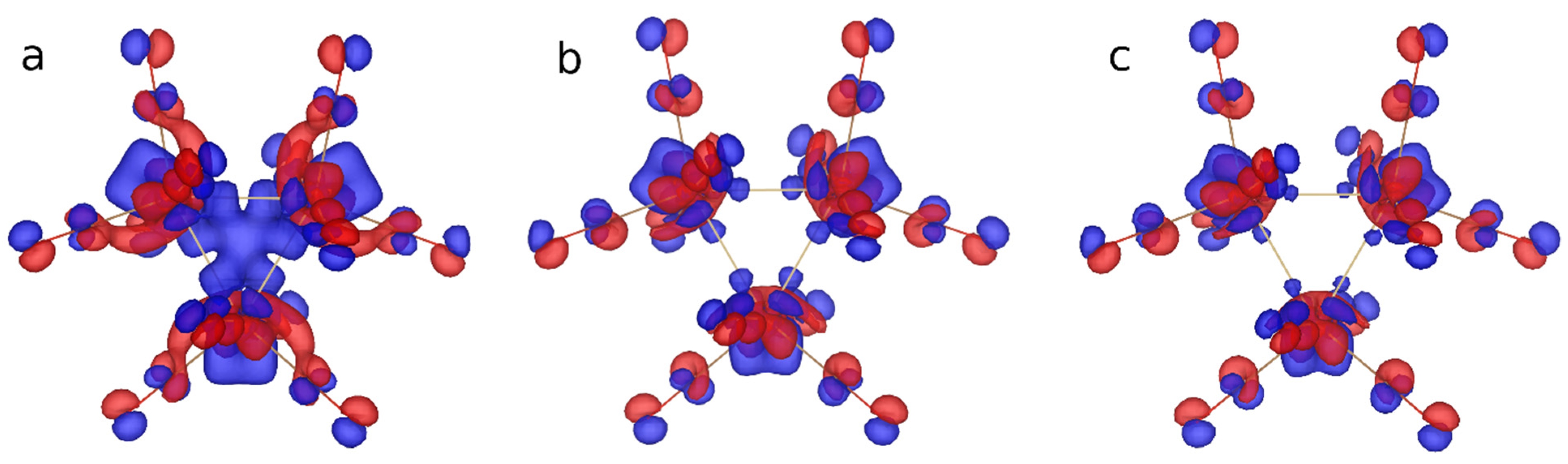
2.2. Characterization of the Interactions in the Os3(CO)12 Cluster
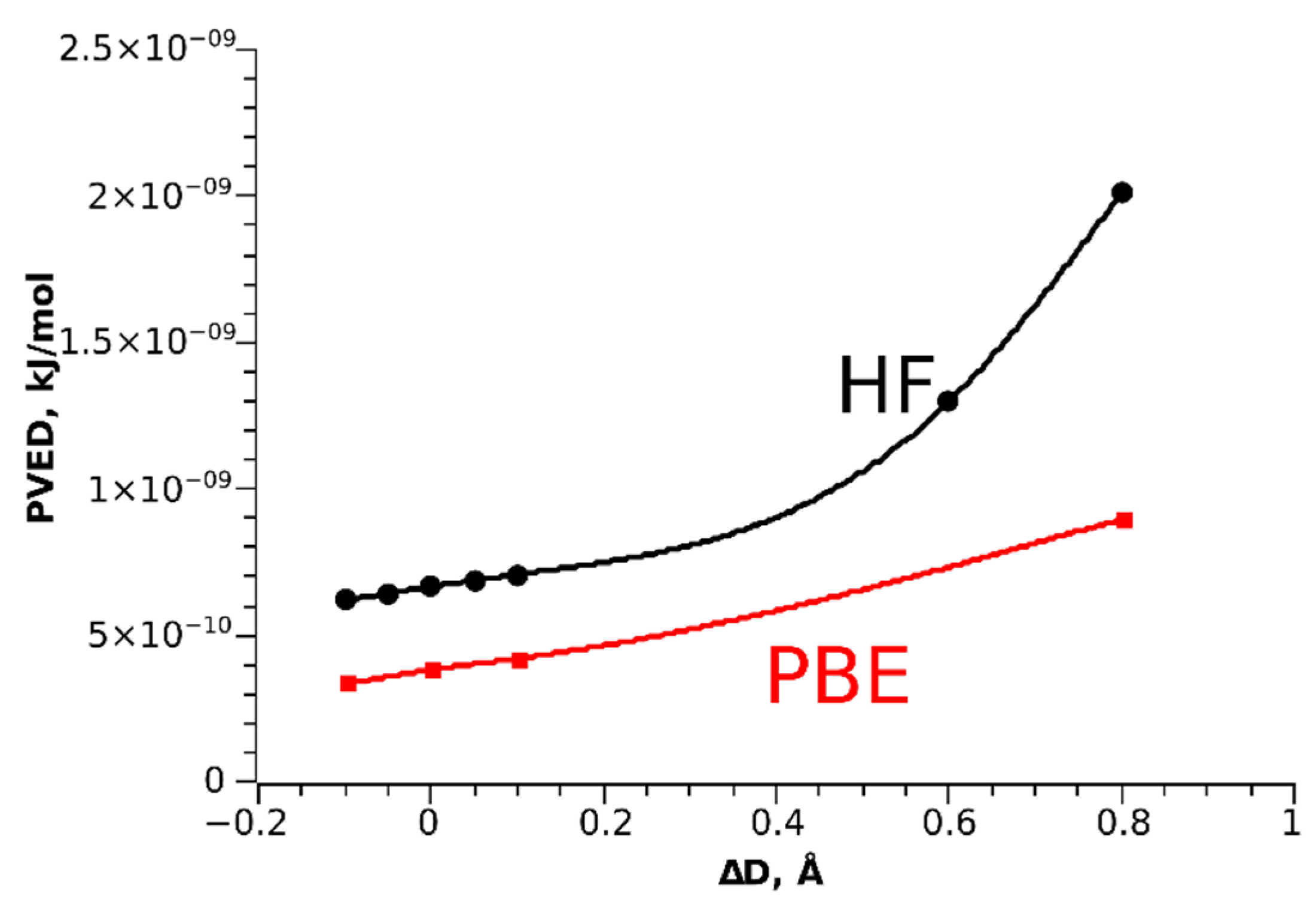
2.3. Chirality and Parity Violation in Os3(CO)12
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kozlova, S.G.; Mirzaeva, I.V.; Ryzhikov, M.R. DABCO molecule in the M2(C8H4O4)2·C6H12N2 (M = Co, Ni, Cu, Zn) metal-organic frameworks. Coord. Chem. Rev. 2018, 376, 62–74. [Google Scholar] [CrossRef]
- Kozlova, S.; Ryzhikov, M.; Pishchur, D.; Mirzaeva, I. Overview of Low-Temperature Heat Capacity Data for Zn2(C8H4O4)2 C6H12N2 and the Salam Hypothesis. Symmetry 2019, 11, 657. [Google Scholar] [CrossRef] [Green Version]
- Puneet, P.; Singh, S.; Fujiki, M.; Nandan, B. Handed Mirror Symmetry Breaking at the Photo-Excited State of π-Conjugated Rotamers in Solutions. Symmetry 2021, 13, 272. [Google Scholar] [CrossRef]
- Salam, A. The role of chirality in the origin of life. J. Mol. Evol. 1991, 33, 105–113. [Google Scholar] [CrossRef]
- Berger, R. Parity-Violation Effects in Molecules. In Relativistic Electronic Structure Theory, Part 2: Applications; Schwerdtfeger, P., Ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2004; pp. 188–288. [Google Scholar]
- MacDermott, A.J.; Fu, T.; Nakatsuka, R.; Coleman, A.P.; Hyde, G.O. Parity-Violating Energy Shifts of Murchison L-Amino Acids are Consistent with an Electroweak Origin of Meteorite L-Enantiomeric Excesses Orig. Life Evol. Biosph. 2009, 39, 459–478. [Google Scholar] [CrossRef]
- Schwerdtfeger, P. The Search for Parity Violation in Chiral Molecules. In Computational Spectroscopy: Methods, Experiments and Applications; Grunenberg, J., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 201–221. [Google Scholar]
- Nizovtsev, A.S.; Ryzhikov, M.R.; Kozlova, S.G. Structural flexibility of DABCO. Ab initio and DFT benchmark study. Chem. Phys. Lett. 2017, 667, 87–90. [Google Scholar] [CrossRef]
- Mirzaeva, I.V.; Kozlova, S.G. Parity violating energy difference for mirror conformers of DABCO linker between two M2+ cations (M = Zn, Cd, and Hg). J. Chem. Phys. 2018, 149, 214302. [Google Scholar] [CrossRef] [PubMed]
- Gabuda, S.P.; Kozlova, S.G. Abnormal difference between the mobilities of left- and right-twisted conformations of C6H12N2 roto-symmetrical molecules at very low temperatures. J. Chem. Phys. 2015, 142, 234302. [Google Scholar] [CrossRef]
- Mirzaeva, I.V.; Kozlova, S.G. Computational estimation of parity violation effects in a metal-organic framework containing DABCO. Chem. Phys. Lett. 2017, 687, 110–115. [Google Scholar] [CrossRef]
- Vergeer, F.W.; Kleverlaan, C.J.; Matousek, P.; Towrie, M.; Stufkens, D.J.; Hartl, F. Redox Control of Light-Induced Charge Separation in a Transition Metal Cluster: Photochemistry of a Methyl Viologen-Substituted [Os3(CO)10(α-diimine)] Cluster. Inorg. Chem. 2005, 44, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Nijhoff, J.; Bakker, M.J.; Hartl, F.; Stufkens, D.J.; Fu; van Eldik, R. Photochemistry of the Triangular Clusters Os3(CO)10(α-diimine): Homolysis of an Os−Os Bond and Solvent-Dependent Formation of Biradicals and Zwitterions. Inorg. Chem. 1998, 37, 661–668. [Google Scholar] [CrossRef]
- Adams, R.D.; Captain, B.; Zhu, L. A New Tris(diphenylstannylene)triosmium Carbonyl Cluster Complex and Its Reactions with Pt(PBut3)2 and Pt(PPh3)4. Organometallics 2006, 25, 2049–2054. [Google Scholar] [CrossRef]
- Kuznetsov, B.N.; Kovalchuk, V.I.; Chesnokov, N.V.; Mikova, N.M.; Naimushina, L.V. Cluster carbonyls of Os, Fe and Fe-Rh on oxide supports: Synthesis and properties. Macromol. Symp. 1998, 136, 41–46. [Google Scholar] [CrossRef]
- Bentsen, J.G.; Wrighton, M.S. Wavelength-, medium-, and temperature-dependent competition between photosubstitution and photofragmentation in triruthenium dodecacarbonyl and triiron dodecacarbonyl: Detection and characterization of coordinatively unsaturated trimetal undecacarbonyl complexes. J. Am. Chem. Soc. 1987, 109, 4530–4544. [Google Scholar]
- Leadbeater, N.E. Control of the photochemistry of Ru3(CO)12 and Os3(CO)12 by variation of the solvent. J. Organomet. Chem. 1999, 573, 211–216. [Google Scholar] [CrossRef]
- Schalk, O.; Josefsson, I.; Richter, R.; Prince, K.C.; Odelius, M.; Mucke, M. Ionization and photofragmentation of Ru3(CO)12 and Os3(CO)12. J. Chem. Phys. 2015, 143, 154305. [Google Scholar] [CrossRef] [Green Version]
- Huggins, D.K.; Flitcroft, N.; Kaesz, H.D. Infrared Spectrum of Osmium Tetracarbonyl Trimer, Os3(CO)12; Assignment of CO and MC Stretching Absorptions. Inorg. Chem. 1965, 4, 166–169. [Google Scholar] [CrossRef]
- Yan, S.; Seidel, M.T.; Zhang, Z.; Leong, W.K.; Tan, H.-S. Ultrafast vibrational relaxation dynamics of carbonyl stretching modes in Os3(CO)12. J. Chem. Phys. 2011, 135, 024501. [Google Scholar] [CrossRef]
- Koridze, A.A.; Kizas, O.A.; Astakhova, N.M.; Petrovskii, P.V.; Grishin, Y.K. Internuclear exchange of carbonyl groups in Os3(CO)12: Coupling constants J(187Os–13C) in trinuclear osmium carbonyls. J. Chem. Soc. Chem. Commun. 1981, 853–855. [Google Scholar] [CrossRef]
- Churchill, M.R.; DeBoer, B.G. Structural studies on polynuclear osmium carbonyl hydrides. 1. Crystal structures of the isomorphous species H2Os3(CO)11 and Os3(CO)12. Role of an equatorial μ2-bridging hydride ligand in perturbing the arrangement of carbonyl ligands in a triangular cluster. Inorg. Chem. 1977, 16, 878–884. [Google Scholar]
- Van der Maelen, J.F.; García-Granda, S.; Cabeza, J.A. Theoretical topological analysis of the electron density in a series of triosmium carbonyl clusters: [Os3(CO)12], [Os3(μ-H)2(CO)10], [Os3(μ-H)(μ-OH)(CO)10], and [Os3(μ-H)(μ-Cl)(CO)10]. Comput. Theor. Chem. 2011, 968, 55–63. [Google Scholar] [CrossRef]
- Hunstock, E.; Mealli, C.; Calhorda, M.J.; Reinhold, J. Molecular Structures of M2(CO)9 and M3(CO)12 (M = Fe, Ru, Os): New Theoretical Insights. Inorg. Chem. 1999, 38, 5053–5060. [Google Scholar] [CrossRef]
- Li, Q.-S.; Xu, B.; Xie, Y.; King, R.B.; Schaefer, H.F. Unsaturated trinuclear osmium carbonyls: Comparison with their iron analogues. Dalt. Trans. 2007, 4312–4322. [Google Scholar] [CrossRef]
- Corey, E.R.; Dahl, L.F. The Molecular and Crystal Structure of Os3(CO)12. Inorg. Chem. 1962, 1, 521–526. [Google Scholar] [CrossRef]
- Roberts, B.M.; Dzuba, V.A.; Flambaum, V.V. Parity and Time-Reversal Violation in Atomic Systems. Annu. Rev. Nucl. Part. Sci. 2015, 65, 63–86. [Google Scholar] [CrossRef] [Green Version]
- Bast, R.; Koers, A.; Gomes, A.S.P.; Iliaš, M.; Visscher, L.; Schwerdtfeger, P.; Saue, T. Analysis of parity violation in chiral molecules. Phys. Chem. Chem. Phys. 2011, 13, 864–876. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Bast, R. Large Parity Violation Effects in the Vibrational Spectrum of Organometallic Compounds. J. Am. Chem. Soc. 2004, 126, 1652–1653. [Google Scholar] [CrossRef]
- Faglioni, F.; Lazzeretti, P. Parity-violation effect on vibrational spectra. Phys. Rev. A 2003, 67, 032101. [Google Scholar] [CrossRef]
- Berger, R.; van Wüllen, C. Density functional calculations of molecular parity-violating effects within the zeroth-order regular approximation. J. Chem. Phys. 2005, 122, 134316. [Google Scholar] [CrossRef]
- Laerdahl, J.; Schwerdtfeger, P. Fully relativistic ab initio calculations of the energies of chiral molecules including parity-violating weak interactions. Phys. Rev. A. 1999, 60, 4439–4453. [Google Scholar] [CrossRef]
- Rein, D.W.; Hegstrom, R.A.; Sandars, P.G.H. Parity non-conserving energy difference between mirror image molecules. Phys. Lett. A 1979, 71, 499–502. [Google Scholar] [CrossRef]
- Harris, R.A.; Stodolsky, L. Quantum beats in optical activity and weak interactions. Phys. Lett. B 1978, 78, 313–317. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- ADF, Version 2020.102, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: https://www.scm.com/product/adf/ (accessed on 14 May 2021).
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.A.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- van Lenthe, E.; Snijders, J.G.; Baerends, E.J. The zero–order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules. J. Chem. Phys. 1996, 105, 6505–6516. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta. 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Savin, A.; Jepsen, O.; Flad, J.; Andersen, O.K.; Preuss, H.; von Schnering, H.G. Electron Localization in Solid-State Structures of the Elements: The Diamond Structure. Angew. Chem. Int. Ed. English. 1992, 31, 187–188. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Kohout, M. DGrid, Version 4.6, Radebeul, 2011. Available online: http://www.cpfs.mpg.de/~kohout/dgrid.html (accessed on 14 May 2021).
- BAND, Version 2020.102, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: https://www.scm.com/product/band_periodicdft/ (accessed on 14 May 2021).
- te Velde, G.; Baerends, E.J. Precise density-functional method for periodic structures. Phys. Rev. B 1991, 44, 7888–7903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef] [Green Version]
- Saue, T.; Bast, R.; Gomes, A.S.P.; Jensen, H.J.A.; Visscher, L.; Aucar, I.A.; Di Remigio, R.; Dyall, K.G.; Eliav, E.; Fasshauer, E.; et al. The DIRAC code for relativistic molecular calculations. J. Chem. Phys. 2020, 152, 204104. [Google Scholar] [CrossRef]
- Dyall, K.G. Relativistic double-zeta, triple-zeta, and quadruple-zeta basis sets for the 6d elements Rf–Cn. Theor. Chem. Acc. 2011, 129, 603–613. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPSSh + D4(EEQ)/ TZ2P | TPSSh + D4(EEQ)/ TZ2P | XRD [22] | XRD [26] | MPW1PW91 /SDD [25] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Symmetry | D3 | D3h | Pseudo-D3h | Pseudo-D3h | D3 | D3h | ||||
| Relativity level | NR | SR | SO | NR | SR | SO | ECP | ECP | ||
| d(Os-Os) | 2.808 | 2.869 | 2.869 | 2.801 | 2.883 | 2.884 | <2.877>(3) | <2.881>(4) | 2.895 | 2.907 |
| d(Os-Ceq) | 1.968 | 1.916 | 1.914 | 1.966 | 1.915 | 1.914 | <1.912>(7) | <1.919>(36) | 1.917 | 1.917 |
| d(Os-Cax) | 2.001 | 2.11 | 1.955 | 1.992 | 1.954 | 1.953 | <1.946>(6) | <1.973>(12) | 1.953 | 1.95 |
| ∠C-Os-Os-C | 37.2 | 29.3 | 29.2 | 0 | 0 | 0 | <2.1>(5) | <1.3>(1.3) | 31.4 | 0 |
| ∠C3axes-Os-C | 21.2 | 16.8 | 16.8 | 0 | 0 | 0 | <1.2>(5) | <0.7>(1.0) | 18 | 0 |
| ΔE | 0 | 0 | 0 | −3.0 | 7.5 | 7.7 | - | - | −2.3 | 0 |
| ΔG | 0 | 0 | 0 | −2.5 | 13.8 | 10.5 | - | - | - | - |
| ni | 2 | 0 | 0 | 0 | 1 | 1 | - | - | 0 | 1 |
| Efrag(Os(CO)4) | ESteric | EOrbital | EDisp | EInt | |
|---|---|---|---|---|---|
| D3 | −7838.3 | 520.1 | −1191.6 | −118.4 | −789.9 |
| D3h | −7839.6 | 489.5 | −1153.1 | −114.2 | −778.2 |
| ∆(D3−D3h) | 1.3 | 30.5 | −38.5 | −4.2 | −11.7 |
| HF | PBE | PBE0 | BLYP | B3LYP | CAMB3LYP | |
|---|---|---|---|---|---|---|
| Os3(CO)12 | 6.66 × 10−10 | 3.87 × 10−10 | 4.61 × 10−10 | 4.14 × 10−10 | 4.77 × 10−10 | 5.51 × 10−10 |
| OsOSSeTe | 4.08 × 10−11 | 5.11 × 10−14 | 2.23 × 10−13 | 3.40 × 10−14 | 1.17 × 10−13 | 1.35 × 10−12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryzhikov, M.R.; Mirzaeva, I.V.; Kozlova, S.G.; Mironov, Y.V. Chirality and Relativistic Effects in Os3(CO)12. Molecules 2021, 26, 3333. https://doi.org/10.3390/molecules26113333
Ryzhikov MR, Mirzaeva IV, Kozlova SG, Mironov YV. Chirality and Relativistic Effects in Os3(CO)12. Molecules. 2021; 26(11):3333. https://doi.org/10.3390/molecules26113333
Chicago/Turabian StyleRyzhikov, Maxim R., Irina V. Mirzaeva, Svetlana G. Kozlova, and Yuri V. Mironov. 2021. "Chirality and Relativistic Effects in Os3(CO)12" Molecules 26, no. 11: 3333. https://doi.org/10.3390/molecules26113333