
Modeling Adsorption and Optical Properties for the Design of CO2 Photocatalytic Metal-Organic Frameworks


Abstract
:1. Introduction
2. Methods
3. Results and Discussion
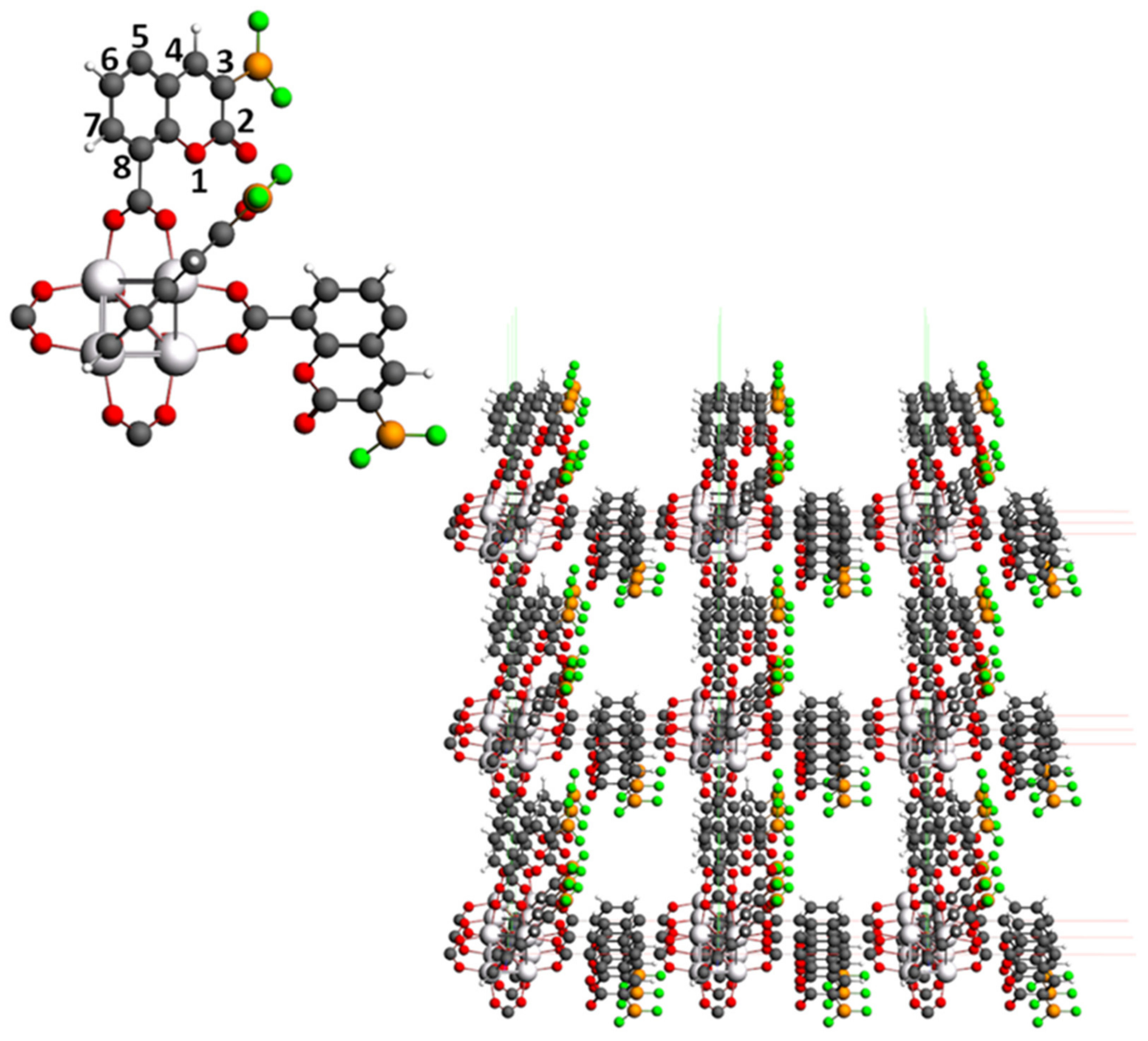
3.1. IRMOF-C-BF2
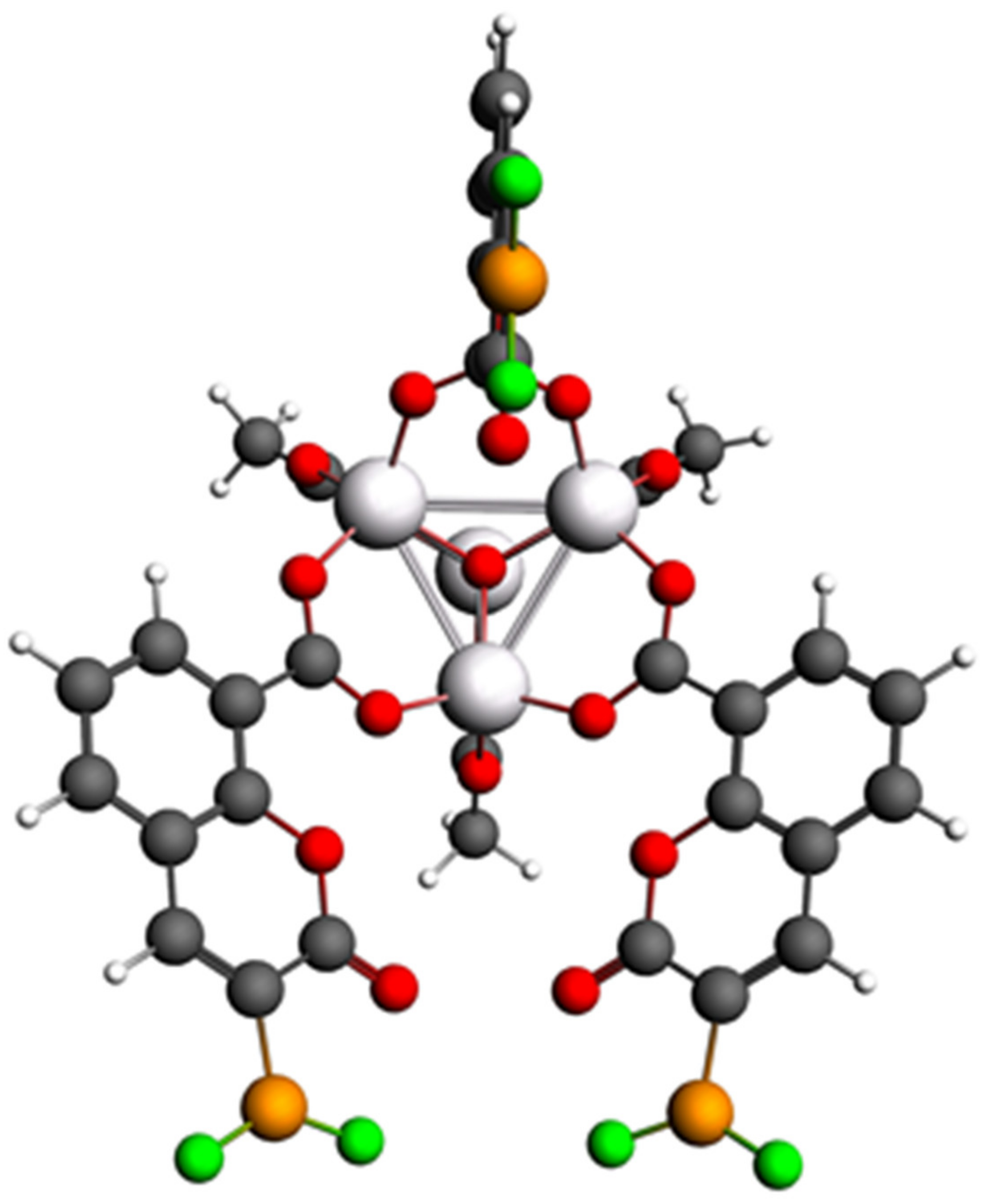
3.1.1. Primitive Cell of IRMOF-C-BF2
Optical Properties of Primitive Cell of IRMOF-C-BF2 during Adsorption Process
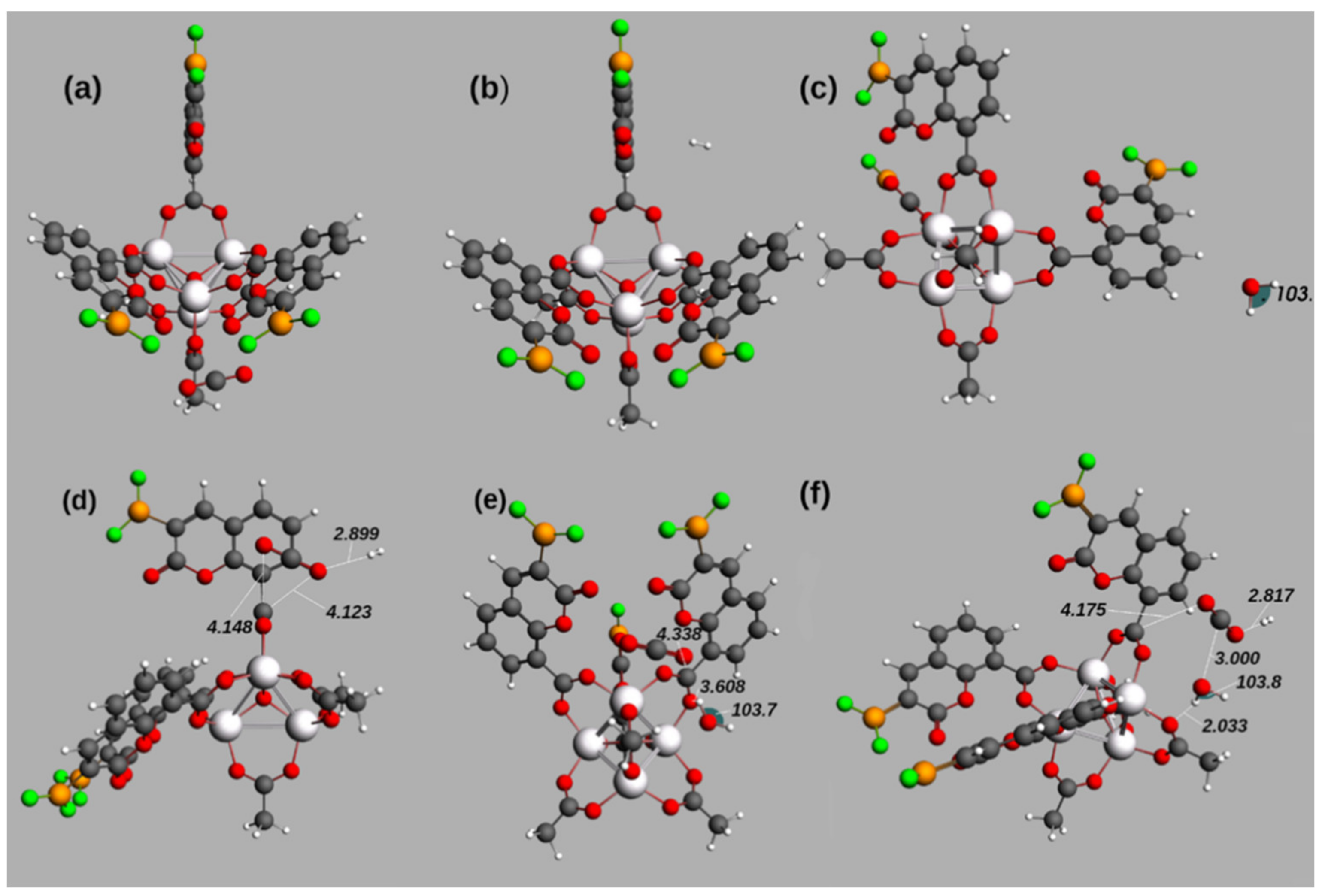
Change in Order of the Guests’ Adsorption for Geometry Optimization in IRMOF-C-BF2
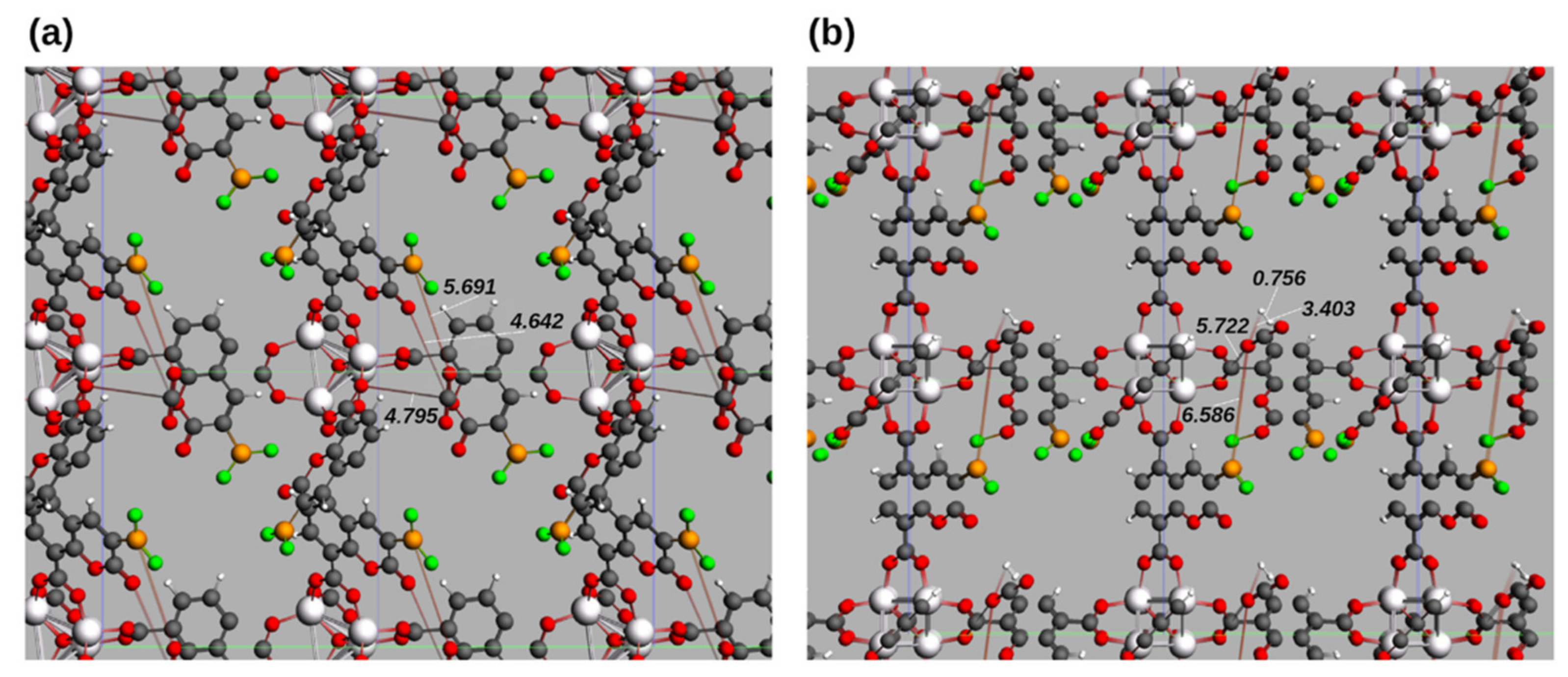
3.1.2. Unit Cell of IRMOF-C-BF2
3.2. IRMOF-C’-BF2
3.2.1. Primitive Cells of IRMOF-C-(2)-BF2 and IRMOF-C’-BF2
3.2.2. Unit Cells of IRMOF-C’-BF2
3.3. IRMOF-C-CH2BF2
Primitive Cells of IRMOF-C-CH2BF2
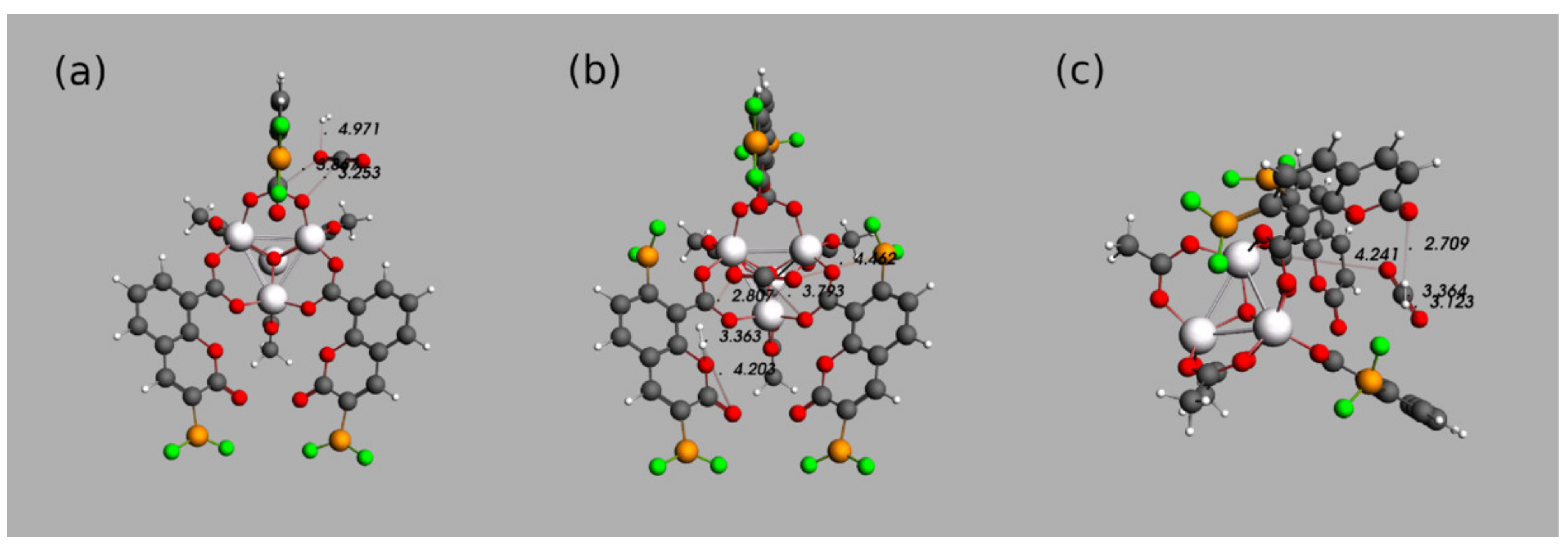
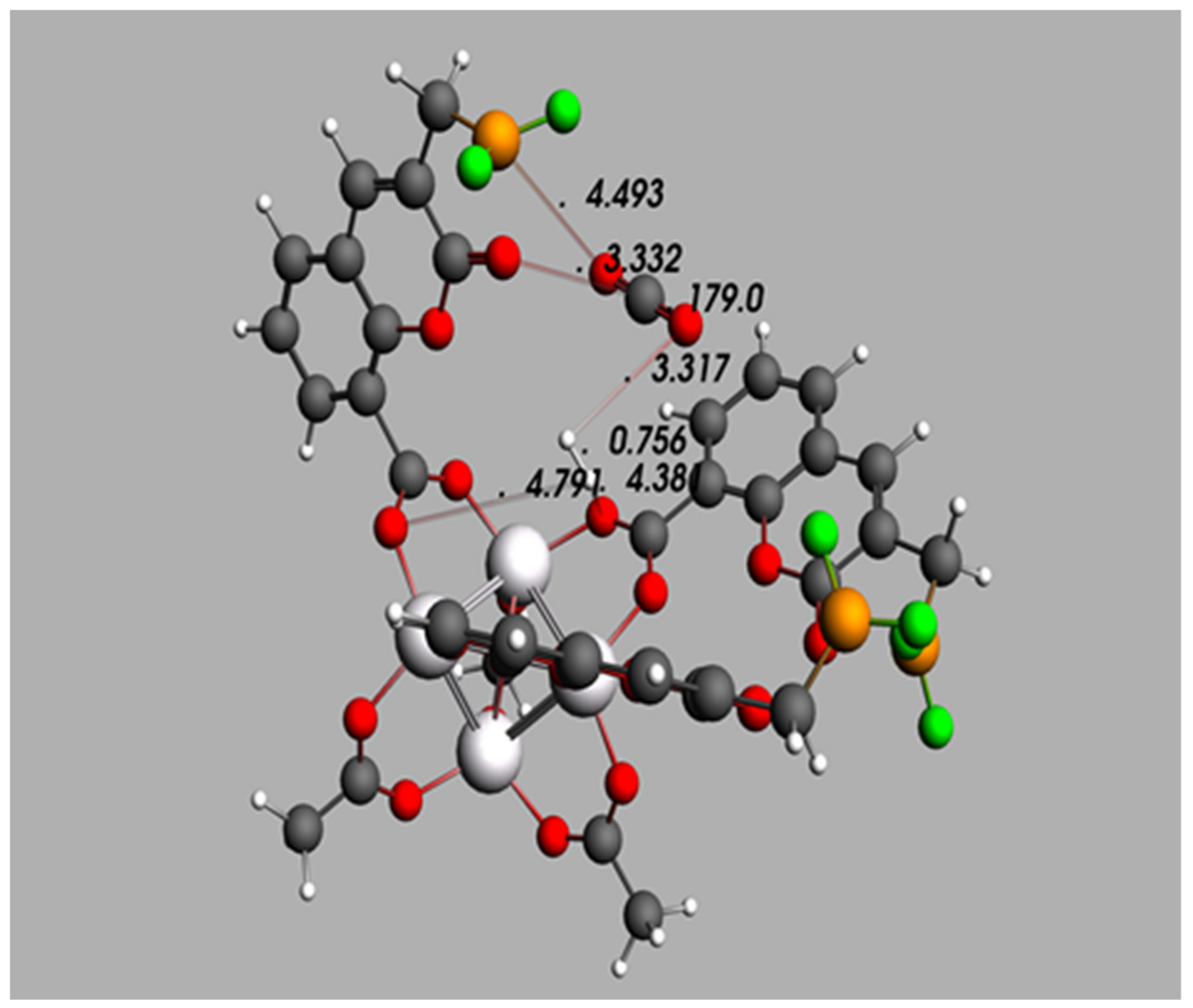
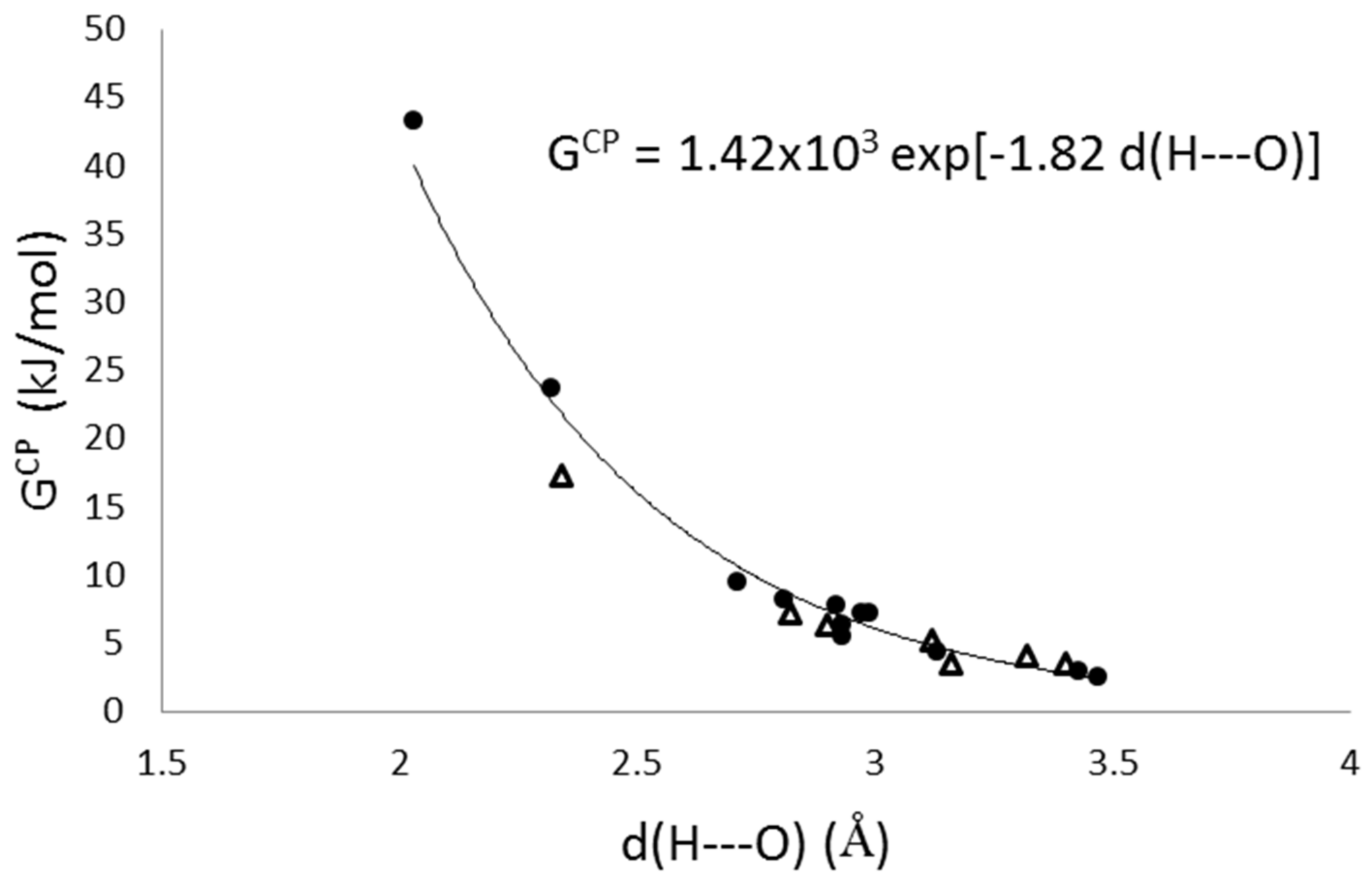
3.4. Further Topological Analysis of All Possible Intermolecular Hydrogen Bond
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, Q.; Astruc, D. State of the Art and Prospects in Metal–Organic Framework (MOF)-Based and MOF-Derived Nanocatalysis. Chem. Rev. 2020, 120, 1438–1511. [Google Scholar] [CrossRef]
- Zhao, X.; Feng, J.; Liu, J.; Lu, J.; Shi, W.; Yang, G.; Wang, G.; Feng, P.; Cheng, P. Metal-Organic Framework-Derived ZnO/ZnS Heteronanostructures for Efficient Visible-Light-Driven Photocatalytic Hydrogen Production. Adv. Sci. 2018, 5, 1700590. [Google Scholar] [CrossRef]
- Chen, J.; Shen, Z.; Lv, S.; Shen, K.; Wu, R.; Jiang, X.-F.; Fan, T.; Chen, J.; Li, Y. Fabricating sandwich-shelled ZnCdS/ZnO/ZnCdS dodecahedral cages with “one stone” as Z-scheme photocatalysts for highly efficient hydrogen production. J. Mater. Chem. A 2018, 6, 19631–19642. [Google Scholar] [CrossRef]
- Wenlong, Z.; Jiiantai, M.; Gongxuan, L. Small-sized Ni(1 1 1) particles in metal-organic frameworks with low over-potential for visible photocatalytic hydrogen generation. Appl. Catal. B Environ. 2016, 190, 12–25. [Google Scholar]
- Shunning, X.; Peijue, L.; Wei, Z.; Guisheng, L.; Dieqing, Z.; Hexing, L. Hierarchical Nanostructured WO3 with Biomimetic Proton Channels and Mixed Ionic-Electronic Conductivity for Electrochemical Energy Storage. Nano Lett. 2015, 15, 4853–4858. [Google Scholar]
- Llabrés i Xamena, F.X.; Corma, A.; Garcia, H. Applications for Metal−Organic Frameworks (MOFs) as Quantum Dot Semiconductors. J. Phys. Chem. C 2007, 111, 80–85. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nat. Cell Biol. 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nat. Cell Biol. 1999, 402, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Wei, J.; Hu, Z.; Zheng, R. Preparation and photocatalytic properties of ZnO/C/TiO2 nanoparticles. Guocheng Gongcheng Xuebao 2018, 18, 1068–1074. [Google Scholar]
- Hussain, M.Z.; Pawar, G.S.; Huang, Z.; Tahir, A.; Fischer, R.A.; Zhu, Y.; Xia, Y. Porous ZnO/Carbon nanocomposites derived from metal organic frameworks for highly efficient photocatalytic applications: A correlational study. Carbon 2019, 146, 348–363. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhou, J.; Chen, X.; Feng, Q.; Cai, W. MOF-derived C-doped ZnO composites for enhanced photocatalytic performance under visible light. J. Alloy. Compd. 2019, 777, 109–118. [Google Scholar] [CrossRef]
- Thakare, S.R.; Ramteke, S.M. Fast and regenerative photocatalyst material for the disinfection of E. coli from water: Silver nano particle anchor on MOF-5. Catal. Commun. 2017, 102, 21–25. [Google Scholar] [CrossRef]
- Thakare, S.R.; Ramteke, S.M. Postmodification of MOF-5 using secondary complex formation using 8- hydroxyquinoline (HOQ) for the development of visible light active photocatalysts. J. Phys. Chem. Solids 2018, 116, 264–272. [Google Scholar] [CrossRef]
- Tachikawa, T.; Choi, J.R.; Fujitsuka, M.; Majima, T. Photoinduced Charge-Transfer Processes on MOF-5 Nanoparticles: Elucidating Differences between Metal-Organic Frameworks and Semiconductor Metal Oxides. J. Phys. Chem. C 2008, 112, 14090–14101. [Google Scholar] [CrossRef]
- Alvaro, M.; Carbonell, E.; Ferrer, B.; I Xamena, F.X.L.; Garcia, H. Semiconductor Behavior of a Metal-Organic Framework (MOF). Chem. A Eur. J. 2007, 13, 5106–5112. [Google Scholar] [CrossRef]
- Abu-Eittah, R.; Moustafa, H.; Al-Omar, A. The electronic absorption spectra of some N-sulfinylanilines. A molecular orbital treatment. Can. J. Chem. 1997, 75, 934–941. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Liu, T.; Sun, J.; Wang, X.-J. Synthesis and application of coumarin fluorescence probes. RSC Adv. 2020, 10, 10826–10847. [Google Scholar] [CrossRef]
- Courtemanche, M.-A.; Légaré, M.-A.; Maron, L.; Fontaine, F.-G. Reducing CO2 to Methanol Using Frustrated Lewis Pairs: On the Mechanism of Phosphine–Borane-Mediated Hydroboration of CO2. J. Am. Chem. Soc. 2014, 136, 10708–10717. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.-H.; Holder, A.M.; Hynes, J.T.; Musgrave, C.B. Roles of the Lewis Acid and Base in the Chemical Reduction of CO2 Catalyzed by Frustrated Lewis Pairs. Inorg. Chem. 2013, 52, 10062–10066. [Google Scholar] [CrossRef]
- Ménard, G.; Stephan, D.W. Room Temperature Reduction of CO2 to Methanol by Al-Based Frustrated Lewis Pairs and Ammonia Borane. J. Am. Chem. Soc. 2010, 132, 1796–1797. [Google Scholar] [CrossRef]
- Sgro, M.J.; Dömer, J.; Stephan, D.W. Stoichiometric CO2 reductions using a bis-borane-based frustrated Lewis pair. Chem. Commun. 2012, 48, 7253–7255. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Johnson, J.K. Design of Lewis Pair-Functionalized Metal Organic Frameworks for CO2 Hydrogenation. ACS Catal. 2015, 5, 2921–2928. [Google Scholar] [CrossRef]
- Velde, G.T.; Baerends, E.J. Precise density-functional method for periodic structures. Phys. Rev. B 1991, 44, 7888–7903. [Google Scholar] [CrossRef] [Green Version]
- BAND2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 30 March 2021).
- Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 30 March 2021).
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- van Gisbergen, S.; Snijders, J.; Baerends, E. Implementation of time-dependent density functional response equations. Comput. Phys. Commun. 1999, 118, 119–138. [Google Scholar] [CrossRef]
- Rosa, A.; Baerends, E.J.; van Gisbergen, S.J.A.; van Lenthe, E.; Groeneveld, J.A.; Snijders, J.G. Electronic Spectra of M(CO)6(M = Cr, Mo, W) Revisited by a Relativistic TDDFT Approach. J. Am. Chem. Soc. 1999, 121, 10356–10365. [Google Scholar] [CrossRef]
- Yang, L.-M.; Vajeeston, P.; Ravindran, P.; Fjellvag, H.; Tilset, M. Theoretical Investigations on the Chemical Bonding, Electronic Structure, And Optical Properties of the Metal−Organic Framework MOF-5. Inorg. Chem. 2010, 49, 10283–10290. [Google Scholar] [CrossRef]
- Gao, Z.; Ding, Y. DFT study of CO2 and H2O co-adsorption on carbon models of coal surface. J. Mol. Model. 2017, 23, 962. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Bader, R.F.W. Effect of Twisting a Polypeptide on Its Geometry and Electron Distribution. J. Phys. Chem. 1994, 98, 4473–4481. [Google Scholar] [CrossRef]
- Cheeseman, J.; Carroll, M.; Bader, R. The mechanics of hydrogen bond formation in conjugated systems. Chem. Phys. Lett. 1988, 143, 450–458. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. The X–C⋯Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Petrova, T.; Odbadrakh, K.; Nicholson, N.M.; Fuentes-Cabrera, M.; Lewis, J.P.; Hill, F.C.; Leszczynski, J. Molecular simulations of adsorption of RDX and TATP on IRMOF-1(Be). J. Mol. Model. 2012, 18, 3363–3378. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.; de Lima, G.F.; de Abreu, H.A. Structural and electronic properties of M-MOF-74 (M = Mg, Co or Mn). Chem. Phys. Lett. 2018, 691, 283–290. [Google Scholar] [CrossRef]
- Tian, Q.; Li, R.; Sun, H.; Xue, Z.; Mu, T. Theoretical and experimental study on the interaction between 1-butyl-3-methylimidazolium acetate and CO2. J. Mol. Liq. 2015, 208, 259–268. [Google Scholar] [CrossRef]
- Carroll, M.T.; Bader, R.F. An analysis of the hydrogen bond in BASE-HF complexes using the theory of atoms in molecules. Mol. Phys. 1988, 65, 695–722. [Google Scholar] [CrossRef]
- Carroll, M.T.; Chang, C.; Bader, R.F. Prediction of the structures of hydrogen-bonded complexes using the laplacian of the charge density. Mol. Phys. 1988, 63, 387–405. [Google Scholar] [CrossRef]
- Shahi, A.; Arunan, E. Hydrogen bonding, halogen bonding and lithium bonding: An atoms in molecules and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular bonding. Phys. Chem. Chem. Phys. 2014, 16, 22935–22952. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essen, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Keith, T.A. TK Gristmill Software AIMAll. 2019. Available online: http://aim.tkgristmill.com/index.html (accessed on 30 March 2021).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Accounts Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Outeiral, C.; Vincent, M.A.; Pendás, Á.M.; Popelier, P.L.A. Revitalizing the concept of bond order through delocalization measures in real space. Chem. Sci. 2018, 9, 5517–5529. [Google Scholar] [CrossRef] [Green Version]
- Hugas, D.; Guillaumes, L.; Duran, M.; Simon, S. Delocalization indices for non-covalent interaction: Hydrogen and DiHydrogen bond. Comput. Theor. Chem. 2012, 998, 113–119. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Characterization of a Dihydrogen Bond on the Basis of the Electron Density. J. Phys. Chem. A 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Bakhmutov, V.I. Dihydrogen Bond: Principles, Experiments and Applications; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2008. [Google Scholar]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Baker, E.; Hubbard, R. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 1984, 44, 97–179. [Google Scholar] [CrossRef]
- Grabowski, S.J. A new measure of hydrogen bonding strength – ab initio and atoms in molecules studies. Chem. Phys. Lett. 2001, 338, 361–366. [Google Scholar] [CrossRef]
- Muñoz, J.; Fradera, X.; Orozco, M.; Luque, J. Topological Analysis of Hydrogen Bonded Complexes; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2002. [Google Scholar]
- Kuc, A.; Enyashin, A.; Seifert, G. Metal−Organic Frameworks: Structural, Energetic, Electronic, and Mechanical Properties. J. Phys. Chem. B 2007, 111, 8179–8186. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.Q.; Mai, T.; Pham-Tran, N.-N.; Kawazoe, Y.; Mizuseki, H.; Nguyen-Manh, D. Engineering of Band Gap in Metal–Organic Frameworks by Functionalizing Organic Linker: A Systematic Density Functional Theory Investigation. J. Phys. Chem. C 2014, 118, 4567–4577. [Google Scholar] [CrossRef]
- Espinosa, E.; Lecomte, C.; Molins, E. Experimental electron density overlapping in hydrogen bonds: Topology vs. energetics. Chem. Phys. Lett. 1999, 300, 745–748. [Google Scholar] [CrossRef]
- Espinosa, E.; Souhassou, M.; Lachekar, H.; LeComte, C. Topological analysis of the electron density in hydrogen bonds. Acta Crystallogr. Sect. B Struct. Sci. 1999, 55, 563–572. [Google Scholar] [CrossRef]
- Hafizovic, J.; Bjørgen, M.; Olsbye, U.; Dietzel, P.; Bordiga, S.; Prestipino, C.; Lamberti, C.; Lillerud, K.P. The Inconsistency in Adsorption Properties and Powder XRD Data of MOF-5 Is Rationalized by Framework Interpenetration and the Presence of Organic and Inorganic Species in the Nanocavities. J. Am. Chem. Soc. 2007, 129, 3612–3620. [Google Scholar] [CrossRef]
- Hendon, C.H.; Tiana, D.; Fontecave, M.; Sanchez, C.; D’Arras, L.; Sassoye, C.; Rozes, L.; Mellot-Draznieks, C.; Walsh, A. Engineering the Optical Response of the Titanium-MIL-125 Metal–Organic Framework through Ligand Functionalization. J. Am. Chem. Soc. 2013, 135, 10942–10945. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Guest | Bond a X---Y | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) |
|---|---|---|---|---|---|---|---|---|
| IRMOF-C-BF2-CO2 | CO2 | O---O | 3.07 | 0.00771 | 0.02996 | 0.03868 | 91.27 | −0.00446 |
| O---O | 3.26 | 0.00381 | 0.01612 | 0.02143 | 92.83 | −0.02477 | ||
| O---H | 2.97 | 0.00379 | 0.01349 | 0.01334 | 92.83 | −0.02477 | ||
| C---O | 3.16 | 0.00528 | 0.02320 | 0.00999 | 37.41 | 0.03874 | ||
| IRMOF-C-BF2-H2O | H2O | O---H | 2.32 | 0.01168 | 0.04255 | 0.04561 | 71.77 b | −0.00224 b |
| IRMOF-C-BF2-H2 | H2 | H---C | 3.03 | 0.00456 | 0.01150 | 0.01614 | 12.24 | −0.00126 |
| IRMOF-C-BF2-CO2 H2O | H2O | O---O | 3.01 | 0.00823 | 0.03263 | 0.04285 | 69.81 | 0.02101 |
| O---O | 3.22 | 0.00593 | 0.02241 | 0.03059 | 69.81 | 0.02101 | ||
| CO2 | O---O | 3.30 | 0.00348 | 0.01514 | 0.01894 | 91.68 | −0.03609 | |
| O---O | 3.09 | 0.00599 | 0.02348 | 0.02581 | 89.88 | 0.01492 | ||
| O---O | 3.55 | 0.00250 | 0.01147 | 0.00836 | 89.88 | 0.01492 | ||
| O---H | 2.99 | 0.00373 | 0.01351 | 0.01197 | 91.68 | −0.03609 | ||
| C---O | 2.95 | 0.00798 | 0.03175 | 0.01660 | 43.12 | 0.03087 | ||
| C---O | 3.02 | 0.00725 | 0.03000 | 0.01374 | 43.12 | 0.03087 | ||
| IRMOF-C-BF2-H2 CO2 | H2 | H---H | 2.70 | 0.00302 | 0.01053 | 0.00854 | 14.47 | −0.01376 |
| CO2 | C---C | 3.26 | 0.00559 | 0.01996 | 0.00869 | 29.53 | 0.04988 | |
| C---O | 3.49 | 0.00264 | 0.01230 | 0.01181 | 29.53 | 0.04988 | ||
| IRMOF-C-BF2-H2 CO2 H2O | H2 | H---H | 3.14 | 0.00105 | 0.00417 | 0.00314 | 14.95 | −0.01398 |
| H2O | H---O | 2.03 | 0.01879 | 0.07680 | 0.05326 | 16.71 b | −0.03286 b | |
| CO2 | C---C | 3.27 | 0.00539 | 0.01927 | 0.00828 | 36.07 | 0.04204 |
| Name | Guest | Angle | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) | ΔE(Y) kcal/mol | ΔN(Y) |
|---|---|---|---|---|---|---|---|---|---|---|
| IRMOF-C-BF2-CO2 H2O | HH2O-OCO2 | 169.24 | 2.34 | 0.00848 | 0.03246 | 0.02483 | 16.09 | −0.02908 | 89.88 | 0.01492 |
| IRMOF-C-BF2-H2 CO2 | HH2-OCO2 | 164.35 | 2.90 | 0.00324 | 0.01161 | 0.01484 | 14.47 | −0.01376 | 92.89 | −0.01609 |
| IRMOF-C-BF2-H2 CO2 H2O | HH2-OCO2 | 174.57 | 2.82 | 0.00372 | 0.01317 | 0.01749 | 14.95 | −0.01399 | 90.62 | 0.00061 |
| Structure Name | Energy/eV | f·10−3 |
|---|---|---|
| IRMOF-1 | 4.2 | 0.57 |
| IRMOF-C-BF2 | 5.4 | 8.4 |
| C-BF2 | 4.0 | 210 |
| IRMOF-C’-BF2 | 5.4 | 6.3 |
| C’-BF2 | 3.0 | 305 |
| IRMOF-C-CH2BF2 | 2.5 | 5.02 |
| C-CH2BF2 | 4.0 | 331 |
| Structure Name | Guest Molecule(s) | Relative Energy/eV |
|---|---|---|
| IRMOF-C-BF2 | - | 0.000 |
| IRMOF-C-BF2-CO2 | CO2 | −22.912 |
| IRMOF-C-BF2-H2 | H2 | −6.803 |
| IRMOF-C-BF2-H2O | H2O | −14.096 |
| IRMOF-C-BF2-CO2-H2 | CO2 + H2 | −29.661 |
| IRMOF-C-BF2-CO2-H2O | CO2 + H2O | −37.117 |
| Guest Molecules | IRMOF-C-BF2 | IRMOF-1 | ||
|---|---|---|---|---|
| Energy/eV | f·103 | Energy/eV | f·10−3 | |
| - | 2.7 | 8.3 | 4.1 | 0.64 |
| CO2 | 2.7 | 5.6 | 4.1 | 0.42 |
| H2 | 2.7 | 8.3 | 4.1 | 0.69 |
| H2O | 2.7 | 8.3 | 4.2 | 0.56 |
| CO2 H2 | 2.7 | 8.4 | 4.2 | 0.57 |
| CO2 H2O | 2.5 | 4.3 | 4.2 | 0.43 |
| Name | Guest | Bond a X---Y | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) |
|---|---|---|---|---|---|---|---|---|
| IRMOF-C-BF2---CO2 H2 | H2 | H--C | 2.92 | 0.00624 | 0.01902 | 0.01642 | 7.96 | 0.01584 |
| CO2 | C---O | 3.16 | 0.00526 | 0.02315 | 0.00998 | 37.96 | 0.03723 | |
| O---H | 2.97 | 0.00378 | 0.01325 | 0.01322 | 1.34 b | −0.00624 | ||
| O---O | 3.26 | 0.00379 | 0.01606 | 0.02123 | 92.68 | −0.02581 | ||
| O---O | 3.08 | 0.00768 | 0.02994 | 0.03838 | 91.29 | −0.00058 |
| Name | Guest | Angle | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) | ΔE(Y) kcal/mol | ΔN(Y) |
|---|---|---|---|---|---|---|---|---|---|---|
| IRMOF-C-BF2---CO2 H2 | HH2O-OCO2 | 157.24 | 3.16 | 0.00163 | 0.00708 | 0.00766 | 15.89 | −0.01790 | 91.29 | −0.00058 |
| Structure | Guest Molecules | Band Gap/eV | Valence Electrons | Valence Band Index | Conduction Band Index | Bottom of Valence Band/eV | Top of Conduction Band/eV |
|---|---|---|---|---|---|---|---|
| IRMOF-1 | - | 3.4 | - | - | - | - | - |
| IRMOF-C-BF2 | - | 0.403 | 548 | 274 | 275 | −4.789 | −4.408 |
| IRMOF-C-BF2 | CO2 | 0.378 | 570 | 285 | 286 | −4.789 | −4.408 |
| IRMOF-C-BF2 | CO2, H2 | 0.714 | 572 | 286 | - | −5.089 | −4.354 |
| IRMOF-C’-BF2 | - | 2.413 | 548 | 274 | - | −6.476 | −4.082 |
| Structure | Guest | Bond a X---Y | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) |
|---|---|---|---|---|---|---|---|---|
| IRMOF-C-BF2 unit cell CO2 | CO2 | O---C | 3.62 | 0.00282 | 0.01015 | 0.01167 | 98.13 | −0.02345 |
| O---O | 3.67 | 0.00118 | 0.00666 | 0.00844 | 101.69 | −0.04197 | ||
| O---O | 4.78 | 0.00014 | 0.00068 | 0.00149 | 98.13 | −0.02345 | ||
| IRMOF-C-BF2 unit cell CO2 H2 | CO2 | O---C | 4.91 | 0.0001 | 0.00043 | 0.00033 | 76.99 | −0.02534 |
| O---H | 2.93 | 0.00298 | 0.01232 | 0.00889 | 76.99 | −0.02534 | ||
| H2 | H---O | 2.93 | 0.00283 | 0.01044 | 0.01512 | 18.43 b | −0.02876 b | |
| H---O | 3.43 | 0.00137 | 0.00639 | 0.00569 | 6.65 b | 0.02471 b | ||
| H---C | 3.42 | 0.00177 | 0.00538 | 0.00534 | 6.65 | 0.02471 |
| Name | Guest | Angle | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) | ΔE(Y) kcal/mol | ΔN(Y) |
|---|---|---|---|---|---|---|---|---|---|---|
| IRMOF-C-BF2 unit cell CO2 H2 | HH2-OCO2 | 89.14 | 3.40 | 0.00171 | 0.00742 | 0.00738 | 18.43 | −0.02876 | 76.99 | −0.02534 |
| Structure Name | Energy/eV | f·103 |
|---|---|---|
| IRMOF-C-BF2 | 2.7 | 8.4 |
| IRMOF-C-(2)-BF2 | 1.96 | 2.5 |
| IRMOF-C-BF2 | 3.0 | 6.3 |
| Structure | Guest | Bond a X---Y | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) |
|---|---|---|---|---|---|---|---|---|
| IRMOF-C-(2)-BF2 | H2 | H---C | 3.07 | 0.00529 | 0.01608 | 0.01332 | 13.98 | −0.01306 |
| CO2 | O---C | 3.36 | 0.00443 | 0.01573 | 0.01788 | 87.19 | −0.01574 | |
| O---C | 3.18 | 0.00481 | 0.02022 | 0.01158 | 84.24 | −0.00994 | ||
| C---O | 3.01 | 0.00703 | 0.02939 | 0.01498 | 34.01 | 0.03829 | ||
| IRMOF-C’-BF2 | H2 | H---O | 2.71 | 0.00524 | 0.01680 | 0.02629 | 19.67 b | −0.03312 b |
| CO2 | H---C | 3.07 | 0.00393 | 0.01011 | 0.01476 | 5.33 | 0.02927 | |
| O---O | 3.12 | 0.00610 | 0.02394 | 0.02616 | 86.48 | −0.01103 | ||
| O---O | 3.03 | 0.00700 | 0.02624 | 0.03210 | 87.84 | −0.01642 |
| Name | Guest | Angle | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) | ΔE(X) kcal/mol | ΔN(Y) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IRMOF-C-(2)-BF2 | HH2-OCO2 | 141.45 | 2.81 | 0.00429 | 0.01474 | 0.02012 | 13.98 | −0.01306 | 84.24 | −0.00994 | |
| IRMOF-C’-BF2 | HH2-OCO2 | 112.87 | 3.12 | 0.00270 | 0.01030 | 0.01307 | 5.33 | 0.02927 | 86.48 | −0.01103 | |
| Structure | Guest | Bond a X---Y | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) |
|---|---|---|---|---|---|---|---|---|
| IRMOF-C-CH2BF2-H2 CO2 | H2 | H---O | 3.13 | 0.00216 | 0.00855 | 0.01077 | 13.40 b | −0.00557 b |
| H2 | H---O | 2.92 | 0.00432 | 0.01430 | 0.01754 | 11.98 | −0.00067 | |
| H2 | H---O | 3.47 | 0.00128 | 0.00549 | 0.00566 | 11.98 | −0.00067 | |
| CO2 | O---O | 3.19 | 0.00516 | 0.01941 | 0.02875 | 92.17 | −0.01350 | |
| CO2 | O---C | 3.43 | 0.00477 | 0.01459 | 0.01971 | 91.03 | −0.01823 |
| Name | Guest | Angle | Distance (Å) | ρ | ∇2ρ | DI(X,Y) | ΔE(X) kcal/mol | ΔN(X) | ΔE(Y) kcal/mol | ΔN(Y) |
|---|---|---|---|---|---|---|---|---|---|---|
| IRMOF-C-CH2 BF2 H2 CO2 | HH2---OCO2 | 90.48 | 3.32 | 0.00204 | 0.00855 | 0.00873 | 13.40 | −0.00557 | 91.03 | −0.01823 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chacón, P.; Hernández-Lima, J.G.; Bazán-Jiménez, A.; García-Revilla, M.A. Modeling Adsorption and Optical Properties for the Design of CO2 Photocatalytic Metal-Organic Frameworks. Molecules 2021, 26, 3060. https://doi.org/10.3390/molecules26103060
Chacón P, Hernández-Lima JG, Bazán-Jiménez A, García-Revilla MA. Modeling Adsorption and Optical Properties for the Design of CO2 Photocatalytic Metal-Organic Frameworks. Molecules. 2021; 26(10):3060. https://doi.org/10.3390/molecules26103060
Chicago/Turabian StyleChacón, Priscila, Joseelyne G. Hernández-Lima, Adán Bazán-Jiménez, and Marco A. García-Revilla. 2021. "Modeling Adsorption and Optical Properties for the Design of CO2 Photocatalytic Metal-Organic Frameworks" Molecules 26, no. 10: 3060. https://doi.org/10.3390/molecules26103060