
Functionalized Hydroperoxide Formation from the Reaction of Methacrolein-Oxide, an Isoprene-Derived Criegee Intermediate, with Formic Acid: Experiment and Theory

 ,
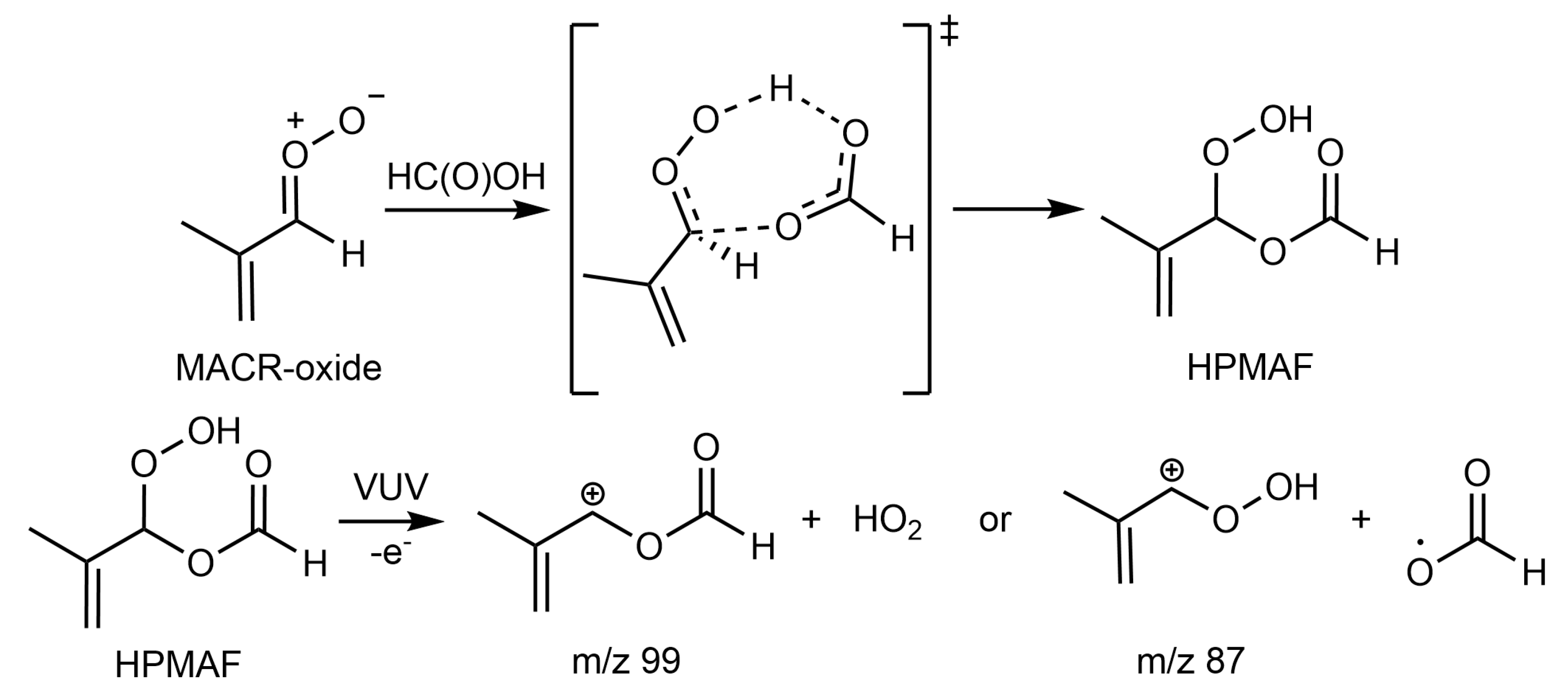
,  , ,
, ,
Abstract
:
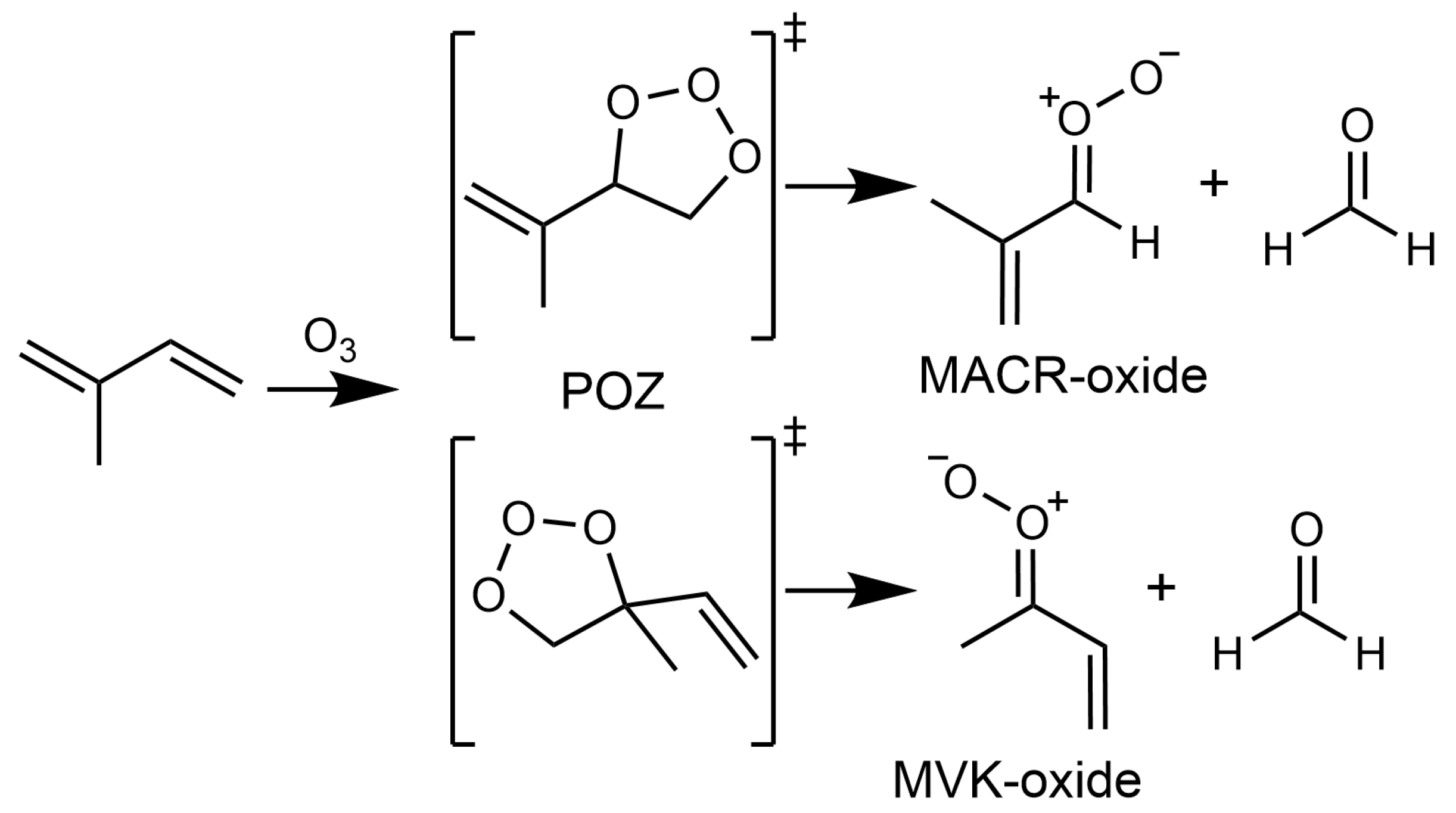
1. Introduction
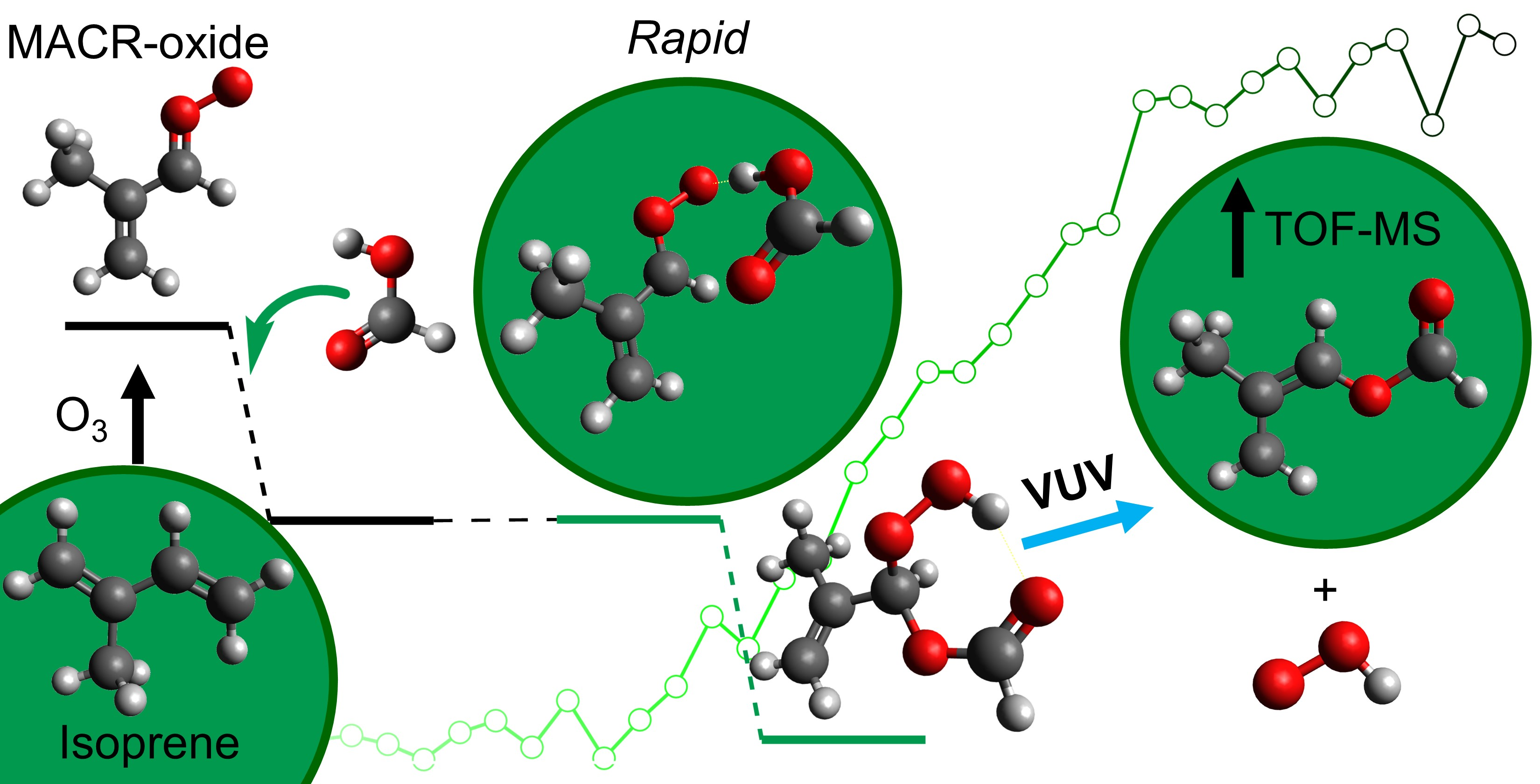
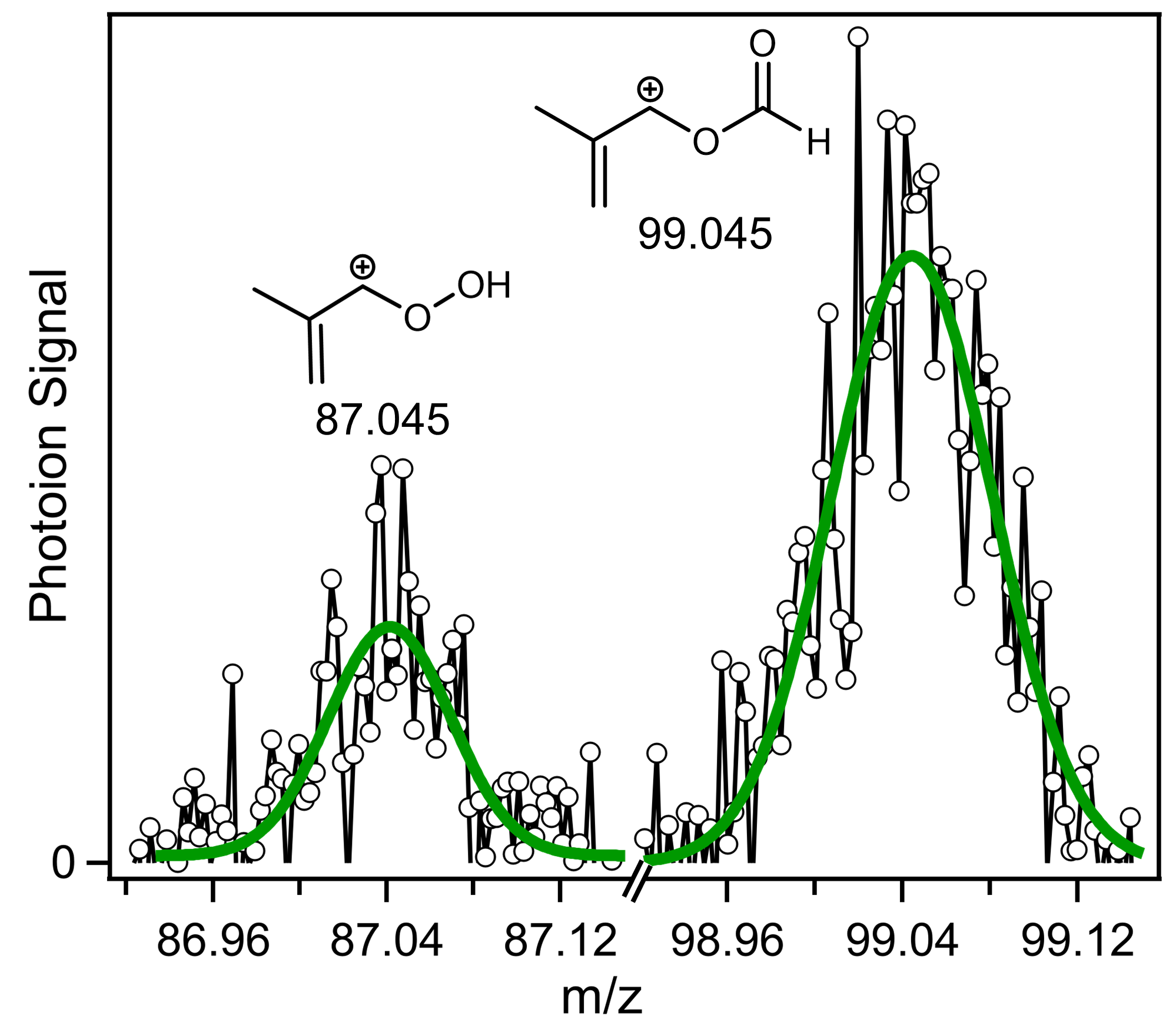
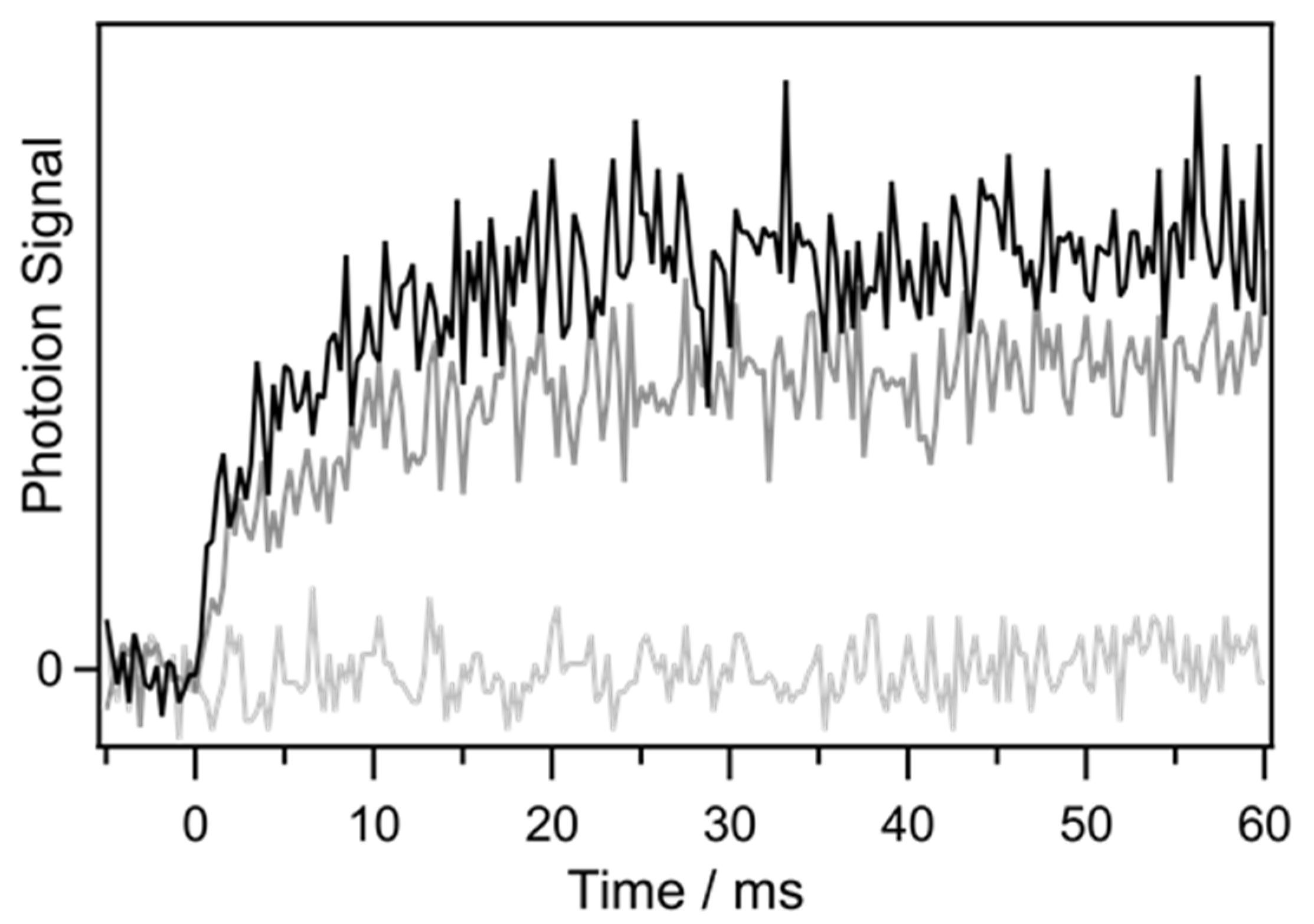
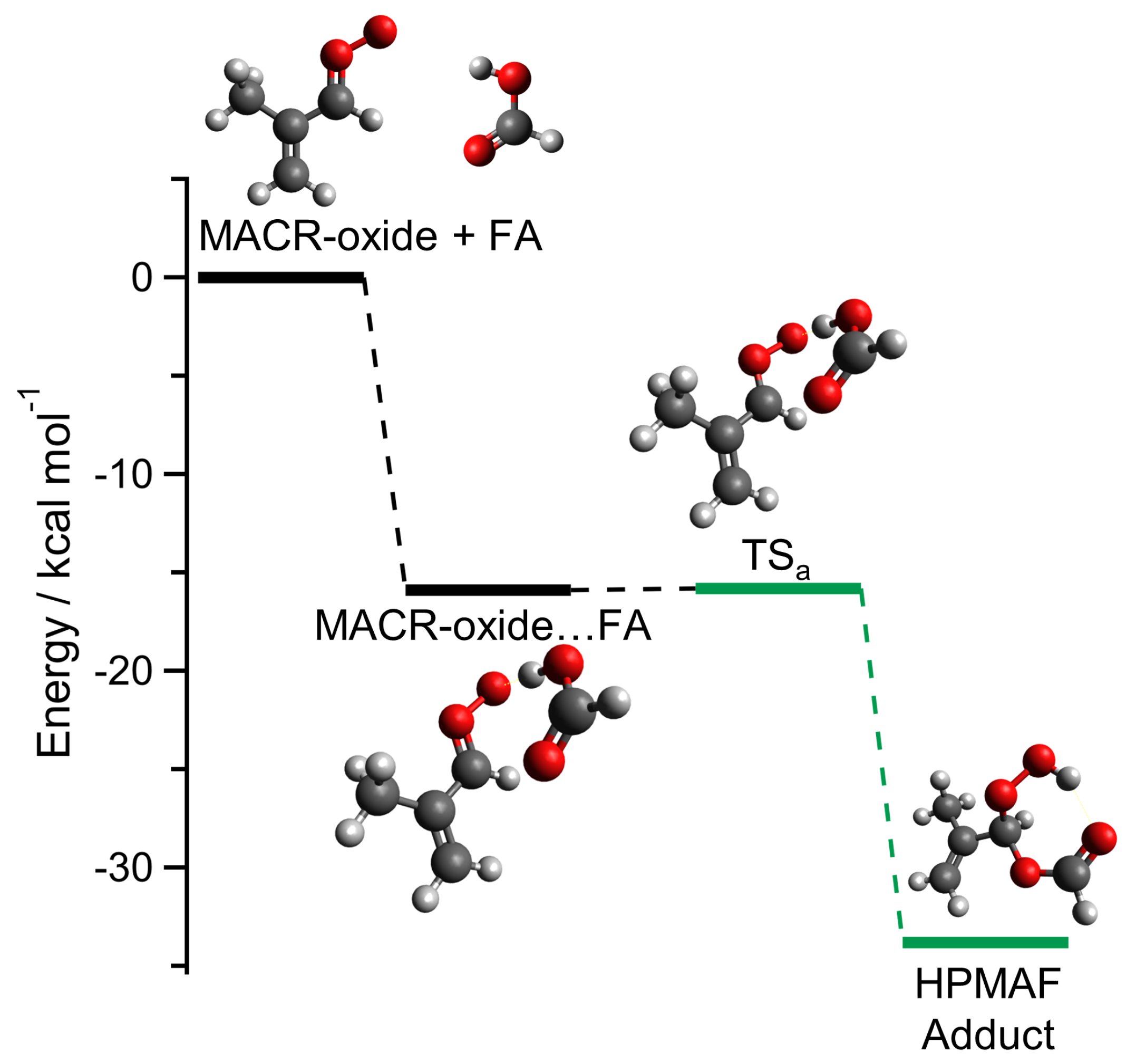
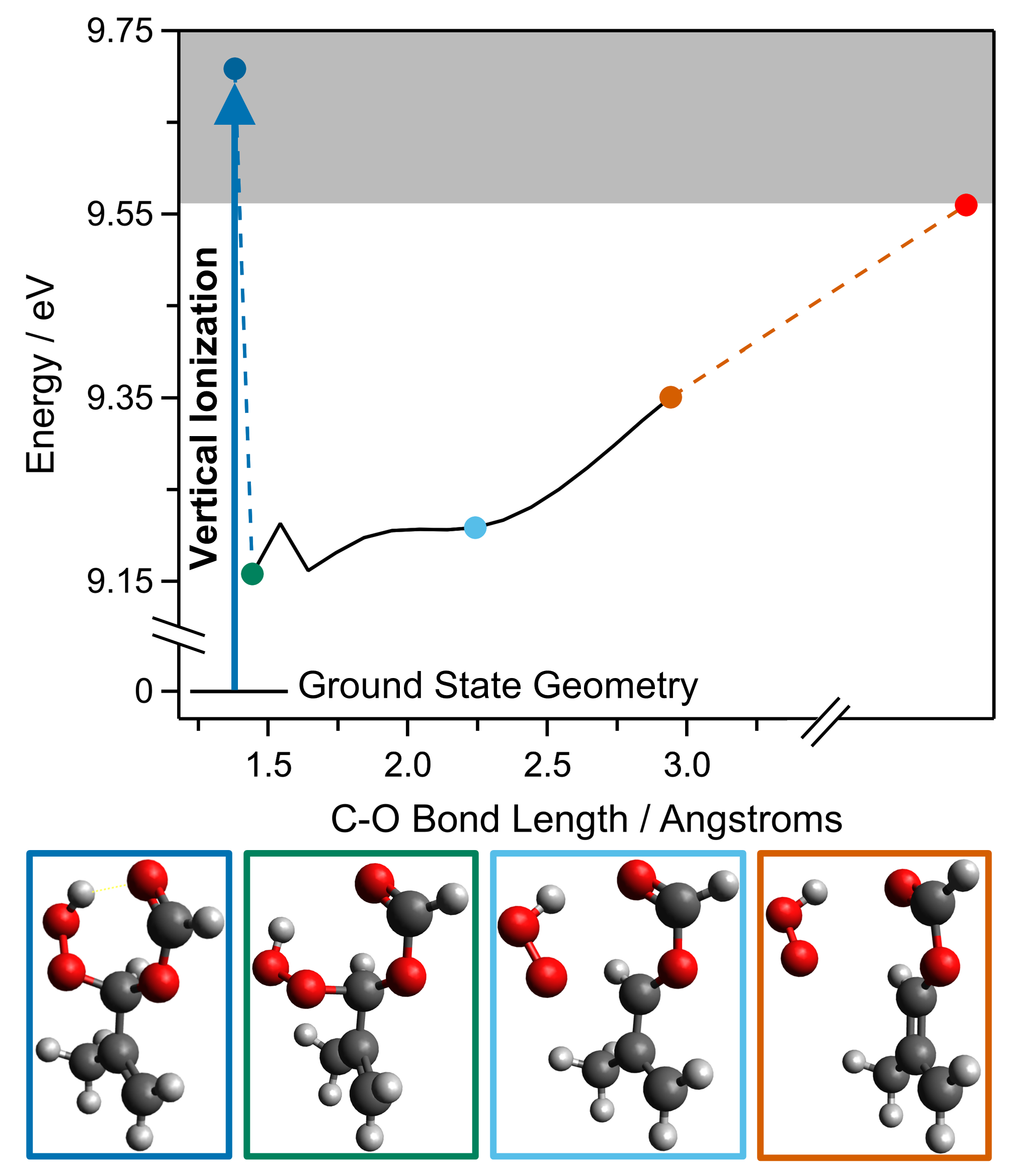
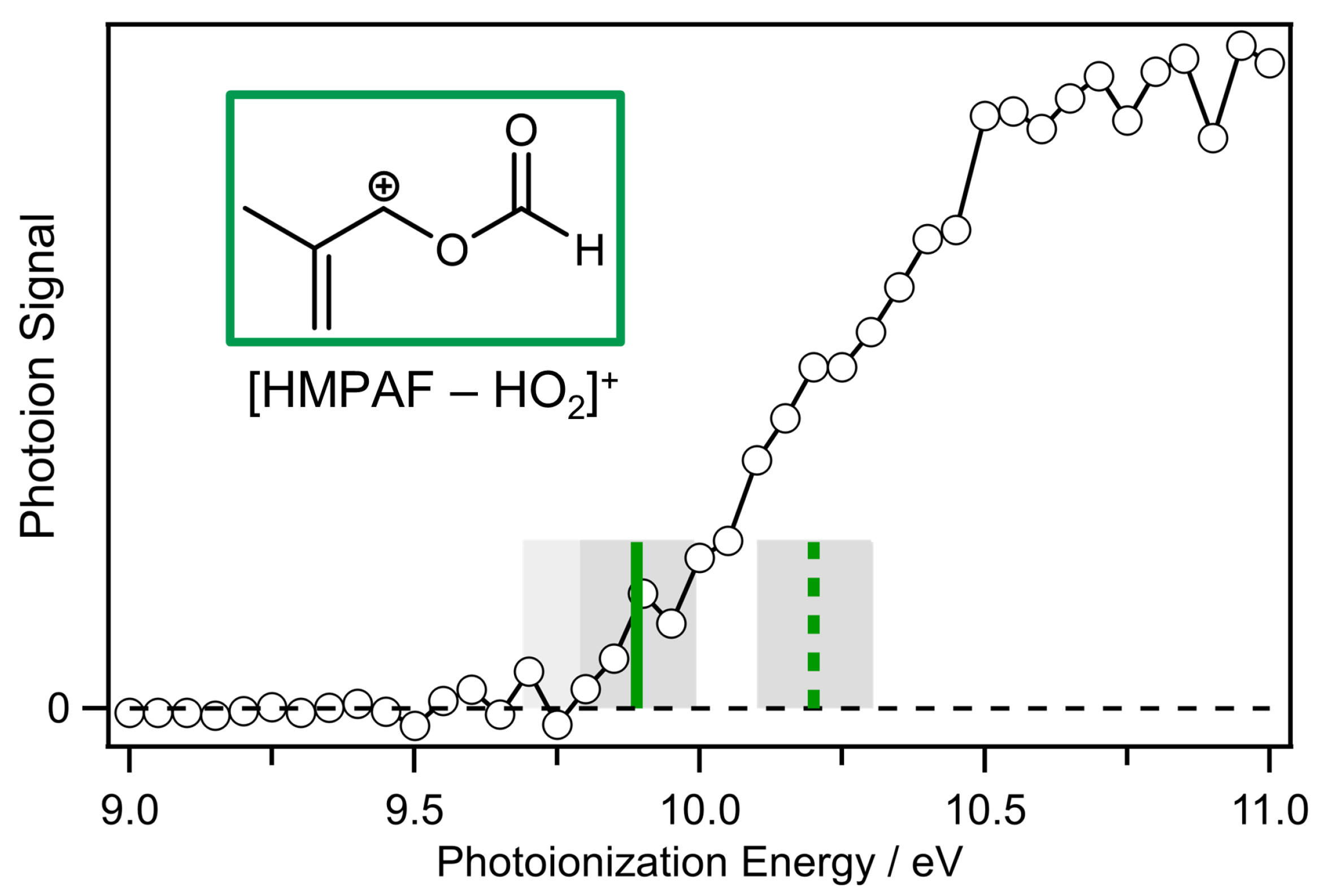
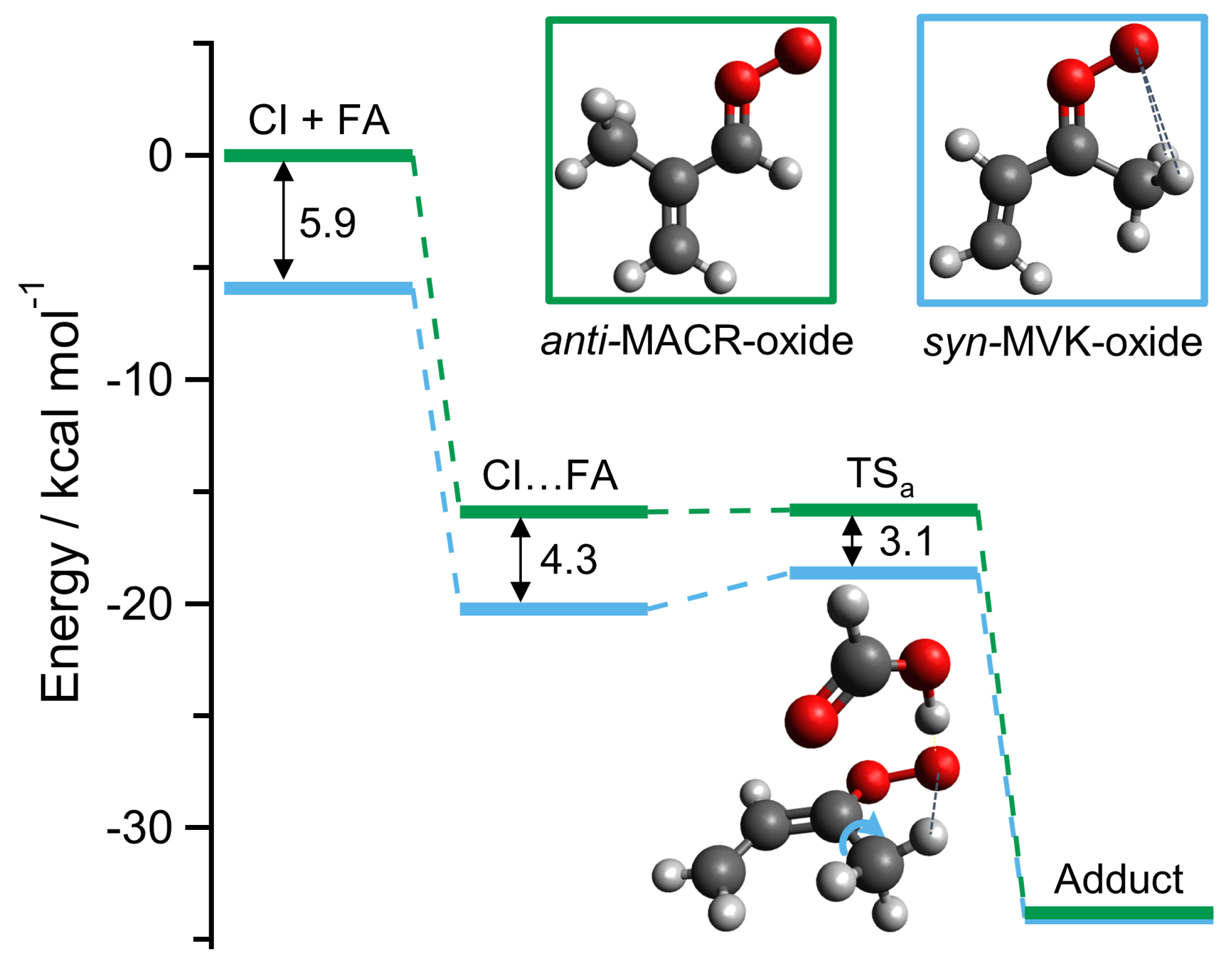
2. Results and Discussion
3. Materials and Methods
3.1. Experimental Methods
3.2. Theoretical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sindelarova, K.; Granier, C.; Bouarar, I.; Guenther, A.; Tilmes, S.; Stavrakou, T.; Müller, J.F.; Kuhn, U.; Stefani, P.; Knorr, W. Global data set of biogenic voc emissions calculated by the megan model over the last 30 years. Atmos. Chem. Phys. 2014, 14, 9317–9341. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.B.; Tyndall, G.S.; Crounse, J.D.; Teng, A.P.; Bates, K.H.; Schwantes, R.H.; Coggon, M.M.; Zhang, L.; Feiner, P.; Milller, D.O.; et al. Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene. Phys. Chem. Chem. Phys. 2016, 18, 10241–10254. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.; Marston, G. The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere. Chem. Soc. Rev. 2008, 37, 699–716. [Google Scholar] [CrossRef] [PubMed]
- Aschmann, S.M.; Atkinson, R. Formation yields of methyl vinyl ketone and methacrolein from the gas-phase reaction of o3 with isoprene. Environ. Sci. Technol. 1994, 28, 1539–1542. [Google Scholar] [CrossRef] [PubMed]
- Newland, M.J.; Nelson, B.S.; Muñoz, A.; Ródenas, M.; Vera, T.; Tárrega, J.; Rickard, A.R. Trends in stabilisation of Criegee intermediates from alkene ozonolysis. Phys. Chem. Chem. Phys. 2020, 22, 13698–13706. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, K.M.; Carslaw, N. Night-time radical chemistry during the TORCH campaign. Atmos. Environ. 2009, 43, 3220–3226. [Google Scholar] [CrossRef]
- Emmerson, K.M.; Carslaw, N.; Carslaw, D.C.; Lee, J.D.; McFiggans, G.; Bloss, W.J.; Gravestock, T.; Heard, D.E.; Hopkins, J.; Ingham, T.; et al. Free radical modelling studies during the UK TORCH Campaign in Summer 2003. Atmos. Chem. Phys. 2007, 7, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.H.; Percival, C.J.; Caravan, R.L.; Taatjes, C.A.; Shallcross, D.E. Criegee intermediates and their impacts on the troposphere. Environ. Sci. Process. Impacts 2018, 20, 437–453. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, T.A.; Lester, M.I. Unimolecular decay dynamics of Criegee intermediates: Energy-resolved rates, thermal rates, and their atmospheric impact. Int. Rev. Phys. Chem. 2020, 39, 1–33. [Google Scholar] [CrossRef]
- Shallcross, D.E.; Khan, M.A.H.; Taatjes, C.A.; Percival, C.J. New insights into the role of stabilized criegee intermediates in tropospheric chemistry from direct laboratory studies. In Advances in Atmospheric Chemistry; World Scientify: Singapore, 2019; pp. 319–375. [Google Scholar]
- Osborn, D.L.; Taatjes, C.A. The physical chemistry of Criegee intermediates in the gas phase. Int. Rev. Phys. Chem. 2015, 34, 309–360. [Google Scholar] [CrossRef]
- Caravan, R.L.; Vansco, M.F.; Au, K.; Khan, M.A.H.; Li, Y.-L.; Winiberg, F.A.F.; Zuraski, K.; Lin, Y.-H.; Chao, W.; Trongsiriwat, N.; et al. Direct kinetic measurements and theoretical predictions of an isoprene-derived Criegee intermediate. Proc. Natl. Acad. Sci. USA 2020, 117, 9733–9740. [Google Scholar] [CrossRef] [PubMed]
- Taatjes, C.A.; Welz, O.; Eskola, A.J.; Savee, J.D.; Scheer, A.M.; Shallcross, D.E.; Rotavera, B.; Lee, E.P.F.; Dyke, J.M.; Mok, D.K.W.; et al. Direct measurements of conformer-dependent reactivity of the Criegee intermediate CH3CHOO. Science 2013, 340, 177–180. [Google Scholar] [CrossRef] [Green Version]
- Welz, O.; Savee, J.D.; Osborn, D.L.; Vasu, S.S.; Percival, C.J.; Shallcross, D.E.; Taatjes, C.A. Direct kinetic measurements of criegee intermediate (CH2OO) formed by reaction of CH2I with O2. Science 2012, 335, 204–207. [Google Scholar] [CrossRef]
- Welz, O.; Eskola, A.J.; Sheps, L.; Rotavera, B.; Savee, J.D.; Scheer, A.M.; Osborn, D.L.; Lowe, D.; Murray Booth, A.; Xiao, P.; et al. Rate Coefficients of C1 and C2 Criegee intermediate reactions with formic and acetic acid near the collision limit: Direct kinetics measurements and atmospheric implications. Angew. Chem. Int. Ed. 2014, 53, 4547–4550. [Google Scholar] [CrossRef] [PubMed]
- Chhantyal-Pun, R.; Rotavera, B.; McGillen, M.R.; Khan, M.A.H.; Eskola, A.J.; Caravan, R.L.; Blacker, L.; Tew, D.P.; Osborn, D.L.; Percival, C.J.; et al. Criegee Intermediate Reactions with Carboxylic Acids: A Potential Source of Secondary Organic Aerosol in the Atmosphere. ACS Earth Space Chem. 2018, 2, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Appel, B.R.; Tokiwa, Y.; Hsu, J.; Kothny, E.L.; Hahn, E. Visibility as related to atmospheric aerosol constituents. Atmos. Environ. 1985, 19, 1525–1534. [Google Scholar] [CrossRef]
- Seaton, A.; Godden, D.; Macnee, W.; Donaldson, K. Particulate air pollution and acute health effects. Lancet 1995, 345, 176–178. [Google Scholar] [CrossRef]
- Bates, J.T.; Weber, R.J.; Abrams, J.; Verma, V.; Fang, T.; Klein, M.; Strickland, M.J.; Sarnat, S.E.; Chang, H.H.; Mulholland, J.A.; et al. Reactive oxygen species generation linked to sources of atmospheric particulate matter and cardiorespiratory effects. Environ. Sci. Technol. 2015, 49, 13605–13612. [Google Scholar] [CrossRef] [PubMed]
- Stocker, T.F.; Qin, D.; Plattner, G.-K.; Tignor, M.; Allen, S.K.; Boschung, J.; Nauels, A.; Xia, Y.; Bex, V.; Midgley, P.M. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge: New York, NY, USA, 2013. [Google Scholar]
- Pilinis, C.; Pandis, S.N.; Seinfeld, J.H. Sensitivity of direct climate forcing by atmospheric aerosols to aerosol size and composition. J. Geophys. Res. 1995, 100, 18739. [Google Scholar] [CrossRef]
- Cox, R.A.; Penkett, S.A. Aerosol formation from sulphur dioxide in the presence of ozone and olefinic hydrocarbons. J. Chem. Soc. Faraday Trans. 1 1972, 68, 1735–1753. [Google Scholar] [CrossRef]
- Cox, R.A.; Penkett, S.A. Oxidation of atmospheric SO2 by products of the ozone–olefin reaction. Nature 1971, 230, 321–322. [Google Scholar] [CrossRef]
- Boy, M.; Mogensen, D.; Smolander, S.; Zhou, L.; Nieminen, T.; Paasonen, P.; Plass-Dülmer, C.; Sipilä, M.J.; Petaja, T.; Mauldin, R., III; et al. Oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals as a crucial source for atmospheric sulfuric acid concentrations. Atmos. Chem. Phys. 2013, 13, 1680–7316. [Google Scholar] [CrossRef] [Green Version]
- Mauldin III, R.L.; Berndt, T.; Sipilä, M.; Paasonen, P.; Petäjä, T.; Kim, S.; Kurtén, T.; Stratmann, F.; Kerminen, V.M.; Kulmala, M. A new atmospherically relevant oxidant of 3sulphur dioxide. Nature 2012, 488, 193. [Google Scholar] [CrossRef] [PubMed]
- Vansco, M.F.; Marchetti, B.; Lester, M.I. Electronic spectroscopy of methyl vinyl ketone oxide: A four-carbon unsaturated Criegee intermediate from isoprene ozonolysis. J. Chem. Phys. 2018, 149, 244309. [Google Scholar] [CrossRef] [PubMed]
- Vansco, M.F.; Marchetti, B.; Trongsiriwat, N.; Wang, G.; Bhagde, T.; Walsh, P.J.; Klippenstein, S.J.; Lester, M.I. Synthesis, electronic spectroscopy and photochemistry of methacrolein oxide: A four carbon unsaturated Criegee intermediate from isoprene ozonolysis. J. Am. Chem. Soc. 2019, 141, 15058–15069. [Google Scholar] [CrossRef]
- Barber, V.P.; Pandit, S.; Green, A.M.; Trongsiriwat, N.; Walsh, P.J.; Klippenstein, S.J.; Lester, M.I. Four-carbon criegee intermediate from isoprene ozonolysis: Methyl vinyl ketone oxide synthesis, infrared spectrum, and oh production. J. Am. Chem. Soc. 2018, 140, 10866–10880. [Google Scholar] [CrossRef]
- Hansen, A.S.; Liu, Z.; Chen, S.; Schumer, M.G.; Walsh, P.J.; Lester, M.I. Unraveling conformer-specific sources of hydroxyl radical production from an isoprene-derived criegee intermediate by deuteration. J. Phys. Chem. A 2020, 124, 4929–4938. [Google Scholar] [CrossRef]
- Chung, C.-A.; Lee, Y.-P. Infrared characterization of formation and resonance stabilization of the Criegee intermediate methyl vinyl ketone oxide. Comm. Chem. 2021, 4, 8. [Google Scholar] [CrossRef]
- Vansco, M.F.; Caravan, R.L.; Zuraski, K.; Winiberg, F.A.F.; Au, K.; Trongsiriwat, N.; Walsh, P.J.; Osborn, D.L.; Percival, C.J.; Khan, M.A.H.; et al. Experimental evidence of dioxole unimolecular decay pathway for isoprene-derived criegee intermediates. J. Phys. Chem. A 2020, 124, 3542–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-H.; Yang, C.-H.; Takahashi, K.; Lin, J.J.-M. Kinetics of unimolecular decay of methyl vinyl ketone oxide, an isoprene-derived criegee intermediate, under atmospherically relevant conditions. J. Phys. Chem. A 2020, 124, 9375–9381. [Google Scholar] [CrossRef]
- Vansco, M.F.; Caravan, R.L.; Pandit, S.; Zuraski, K.; Winiberg, F.A.F.; Au, K.; Bhagde, T.; Trongsiriwat, N.; Walsh, P.J.; Osborn, D.L.; et al. Formic acid catalyzed isomerization and adduct formation of an isoprene-derived Criegee intermediate: Experiment and theory. Phys. Chem. Chem. Phys. 2020, 22, 26796. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-H.; Yin, C.; Takahashi, K.; Lin, J.J.-M. Surprisingly long lifetime of methacrolein oxide, an isoprene derived Criegee intermediate, under humid conditions. Comm. Chem. 2021, 4, 12. [Google Scholar] [CrossRef]
- Kuo, M.-T.; Weber, I.; Fittschen, C.; Vereecken, L.; Lin, J.J.-M. Kinetics of dimethyl sulfide (DMS) reactions with isoprene-derived Criegee intermediates studied with direct UV absorption. Atmos. Chem. Phys. 2020, 20, 12983–12993. [Google Scholar] [CrossRef]
- Caravan, R.L.; Vansco, M.F.; Lester, M.I. Open questions on the reactivity of Criegee intermediates. Comm. Chem. 2021, 4, 44. [Google Scholar] [CrossRef]
- Kuwata, K.T.; Valin, L.C. Quantum chemical and RRKM/Master equation studies of isoprene ozonolysis: Methacrolein and methacrolein oxide. Chem. Phys. Lett. 2008, 451, 186–191. [Google Scholar] [CrossRef]
- Vereecken, L.; Novelli, A.; Taraborrelli, D. Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates. Phys. Chem. Chem. Phys. 2017, 19, 31599–31612. [Google Scholar] [CrossRef] [Green Version]
- Kuwata, K.T.; Valin, L.C.; Converse, A.D. Quantum chemical and master equation studies of the methyl vinyl carbonyl oxides formed in isoprene ozonolysis. J. Phys. Chem. A 2005, 109, 10725. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Takahashi, K. Effect of unsaturated substituents in the reaction of Criegee intermediates with water vapor. Phys. Chem. Chem. Phys. 2018, 20, 20217–20227. [Google Scholar] [CrossRef]
- Barber, V.P.; Hansen, A.S.; Georgievskii, Y.; Klippenstein, S.J.; Lester, M.I. Experimental and theoretical studies of the double substituted methyl-ethyl criegee intermediate: Infrared action spectroscopy and unimolecular decay to OH products. J. Chem. Phys. 2020, 152, 094301. [Google Scholar] [CrossRef]
- Anglada, J.M.; Solé, A. Impact of the water dimer on the atmospheric reactivity of carbonyl oxides. Phys. Chem. Chem. Phys. 2016, 18, 17698–17712. [Google Scholar] [CrossRef] [Green Version]
- Watson, N.A.I.; Black, J.A.; Stonelake, T.M.; Knowles, P.J.; Beames, J.M. An Extended Computational Study of Criegee Intermediate–Alcohol Reactions. J. Phys. Chem. A 2019, 123, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Stone, D.; Blitz, M.; Daubney, L.; Howes, N.U.M.; Seakins, P. Kinetics of CH2OO reactions with SO2, NO2, NO, H2O and CH3CHO as a function of pressure. Phys. Chem. Chem. Phys. 2014, 16, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltola, J.; Seal, P.; Inkilä, A.; Eskola, A. Time-resolved, broadband UV-absorption spectrometry measurements of Criegee intermediate kinetics using a new photolytic precursor: Unimolecular decomposition of CH2OO and its reaction with formic acid. Phys. Chem. Chem. Phys. 2020, 22, 11797–11808. [Google Scholar] [CrossRef] [Green Version]
- Vereecken, L. The reaction of Criegee intermediates with acids and enols. Phys. Chem. Chem. Phys. 2017, 19, 28630–28640. [Google Scholar] [CrossRef] [PubMed]
- Moshammer, K.; Jasper, A.W.; Popolan-Vaida, D.M.; Lucassen, A.; Diévart, P.; Selim, H.; Eskola, A.J.; Taatjes, C.A.; Leone, S.R.; Sarathy, S.M.; et al. Detection and Identification of the Keto-Hydroperoxide (HOOCH2OCHO) and other intermediates during low-temperature oxidation of dimethyl ether. J. Phys. Chem. A 2015, 119, 7361–7374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezas, C.; Endo, Y. The reactivity of the Criegee intermediate CH3CHOO with water probed by FTMW spectroscopy. J. Chem. Phys. 2018, 148, 014308. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, K.T.; Hermes, M.R.; Carlson, M.J.; Zogg, C.K. Computational studies of the isomerization and hydration reactions of acetaldehyde oxide and methyl vinyl carbonyl oxide. J. Phys. Chem. A 2010, 114, 9192–9204. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-C.; Chao, W.; Chang, C.-H.; Takahashi, K.; Lin, J.J.-M. Temperature dependence of the reaction of anti-CH3CHOO with water vapor. Phys. Chem. Chem. Phys. 2016, 18, 28189–28197. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-C.; Chang, H.-T.; Chang, C.-H.; Chao, W.; Smith, M.C.; Chang, C.-H.; Jr-Min Lin, J.; Takahashi, K. Competition between H2O and (H2O)2 reactions with CH2OO/CH3CHOO. Phys. Chem. Chem. Phys. 2016, 18, 4557–4568. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Bao, J.L.; Truhlar, D.G. Atmospheric chemistry of Criegee intermediates: Unimolecular reactions and reactions with water. J. Am. Chem. Soc. 2016, 138, 14409–14422. [Google Scholar] [CrossRef]
- Chao, W.; Lin, Y.-H.; Yin, C.T.; Lin, W.H.; Takahashi, K.; Lin, J.J.M. Temperature and isotope effects in the reaction of CH3CHOO with methanol. Phys. Chem. Chem. Phys. 2019, 21, 13633–13640. [Google Scholar] [CrossRef]
- Stone, D.; Au, K.; Sime, S.; Medeiros, D.J.; Blitz, M.; Seakins, P.W.; Decker, Z.; Sheps, L. Unimolecular decomposition kinetics of the stabilised Criegee intermediates CH2OO and CD2OO. Phys. Chem. Chem. Phys. 2018, 20, 24940–24954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Liu, F.; Barber, V.P.; Klippenstein, S.J.; McCoy, A.B.; Lester, M.I. Communication: Real time observation of unimolecular decay of Criegee intermediates to OH radical products. J. Chem. Phys. 2016, 144, 061102. [Google Scholar] [CrossRef] [Green Version]
- Sheps, L.; Rotavera, B.; Eskola, A.J.; Osborn, D.L.; Taatjes, C.A.; Au, K.; Shallcross, D.E.; Khan, M.A.H.; Percival, C.J. The reaction of Criegee intermediate CH2OO with water dimer: Primary products and atmospheric impact. Phys. Chem. Chem. Phys. 2017, 19, 21970–21979. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Liu, Y.; Dong, W.; Yang, X. Unimolecular Reaction Rate Measurement of syn-CH3CHOO. J. Phys. Chem. Lett. 2019, 10, 4817–4821. [Google Scholar] [CrossRef] [PubMed]
- Osborn, D.L.; Zou, P.; Johnsen, H.; Hayden, C.C.; Taatjes, C.A.; Knyazev, V.D.; North, S.W.; Peterka, D.S.; Ahmed, M.; Leone, S.R. The multiplexed chemical kinetic photoionization mass spectrometer: A new approach to isomer-resolved chemical kinetics. Rev. Sci. Instrum. 2008, 79, 104103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01, Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. MOLPRO, Version 2015 ed. a Package of ab Initio Programs, 2019. Available online: https://www.molpro.net/ (accessed on 20 May 2021).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ωB97XD 6–31 + G* | B2PLYP-D3 cc-pVTZ | CCSD(T)-F12 TZF | |
|---|---|---|---|
| VIE (HPMAF) | 9.71 | 9.94 | 10.18 |
| AIE (HPMAF) | 9.16 | 9.32 | 9.73 |
| CH3C(=CH2)C+HOOH (HO2-loss) | 9.58 | 9.56 | 9.89 |
| CH3C(=CH2)C+HOC(O)H (HCO2-loss) | 10.17 | 10.05 | 10.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vansco, M.F.; Zuraski, K.; Winiberg, F.A.F.; Au, K.; Trongsiriwat, N.; Walsh, P.J.; Osborn, D.L.; Percival, C.J.; Klippenstein, S.J.; Taatjes, C.A.; et al. Functionalized Hydroperoxide Formation from the Reaction of Methacrolein-Oxide, an Isoprene-Derived Criegee Intermediate, with Formic Acid: Experiment and Theory. Molecules 2021, 26, 3058. https://doi.org/10.3390/molecules26103058
Vansco MF, Zuraski K, Winiberg FAF, Au K, Trongsiriwat N, Walsh PJ, Osborn DL, Percival CJ, Klippenstein SJ, Taatjes CA, et al. Functionalized Hydroperoxide Formation from the Reaction of Methacrolein-Oxide, an Isoprene-Derived Criegee Intermediate, with Formic Acid: Experiment and Theory. Molecules. 2021; 26(10):3058. https://doi.org/10.3390/molecules26103058
Chicago/Turabian StyleVansco, Michael F., Kristen Zuraski, Frank A. F. Winiberg, Kendrew Au, Nisalak Trongsiriwat, Patrick J. Walsh, David L. Osborn, Carl J. Percival, Stephen J. Klippenstein, Craig A. Taatjes, and et al. 2021. "Functionalized Hydroperoxide Formation from the Reaction of Methacrolein-Oxide, an Isoprene-Derived Criegee Intermediate, with Formic Acid: Experiment and Theory" Molecules 26, no. 10: 3058. https://doi.org/10.3390/molecules26103058