
Influence of Gas Supply Changes on the Formation Process of Complex Mixed Gas Hydrates

Abstract
:
1. Introduction
2. Results
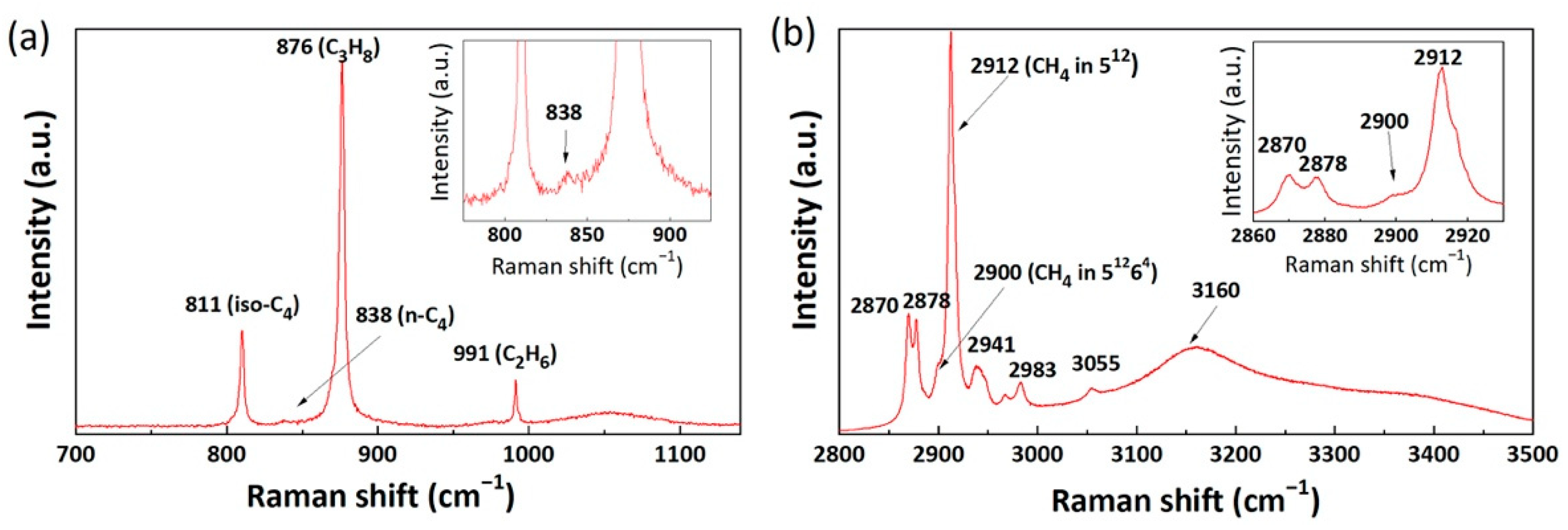
2.1. Raman Spectra of Mixed Gas Hydrates
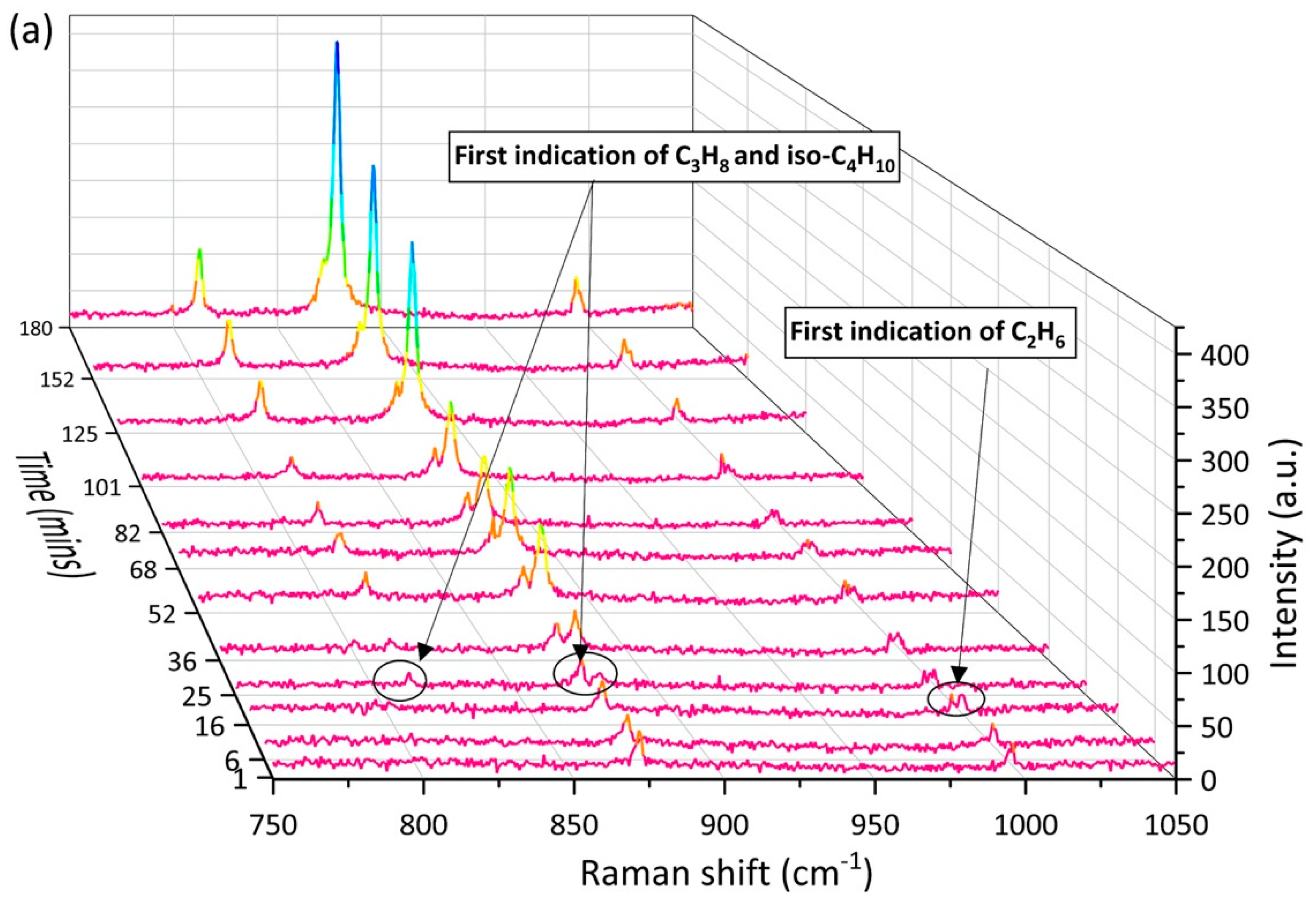
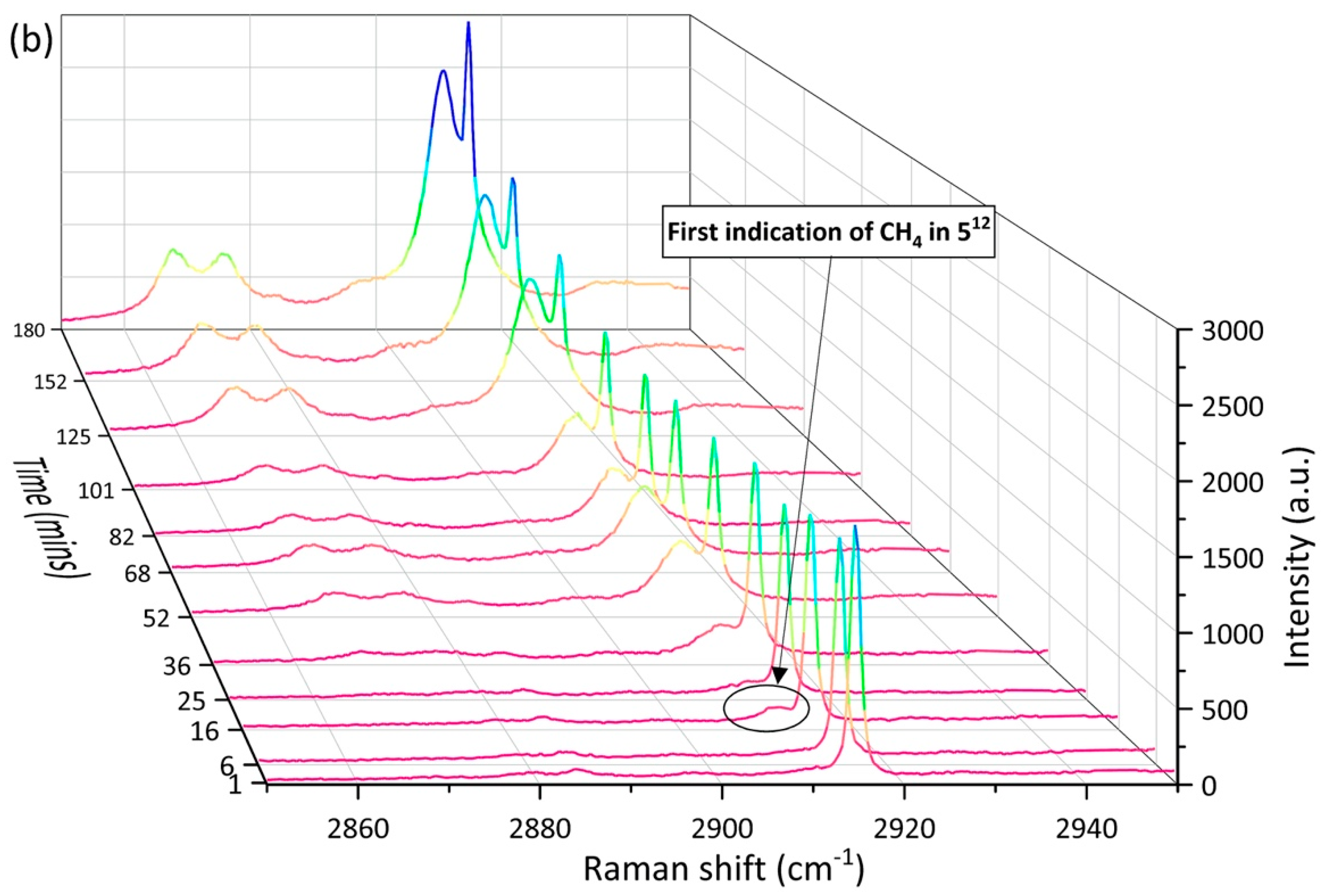
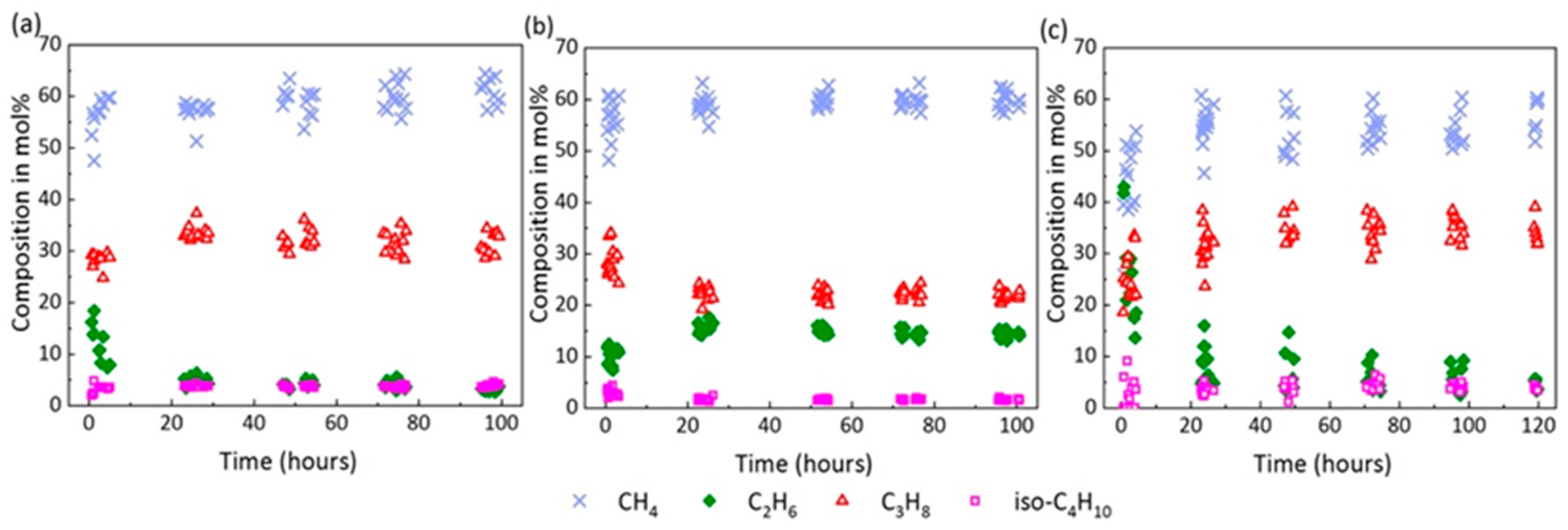
2.2. Enclathration Sequence of Gas Components into the Hydrate Structures
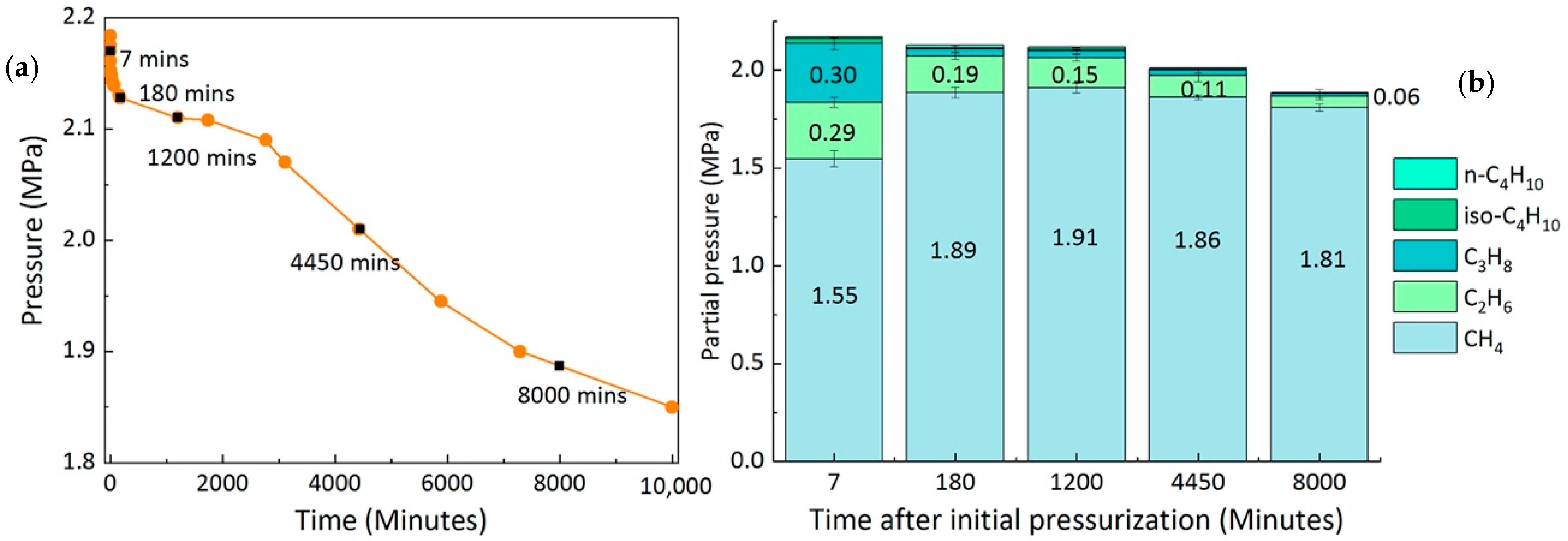
2.3. Composition of the Feed Gas Mixture
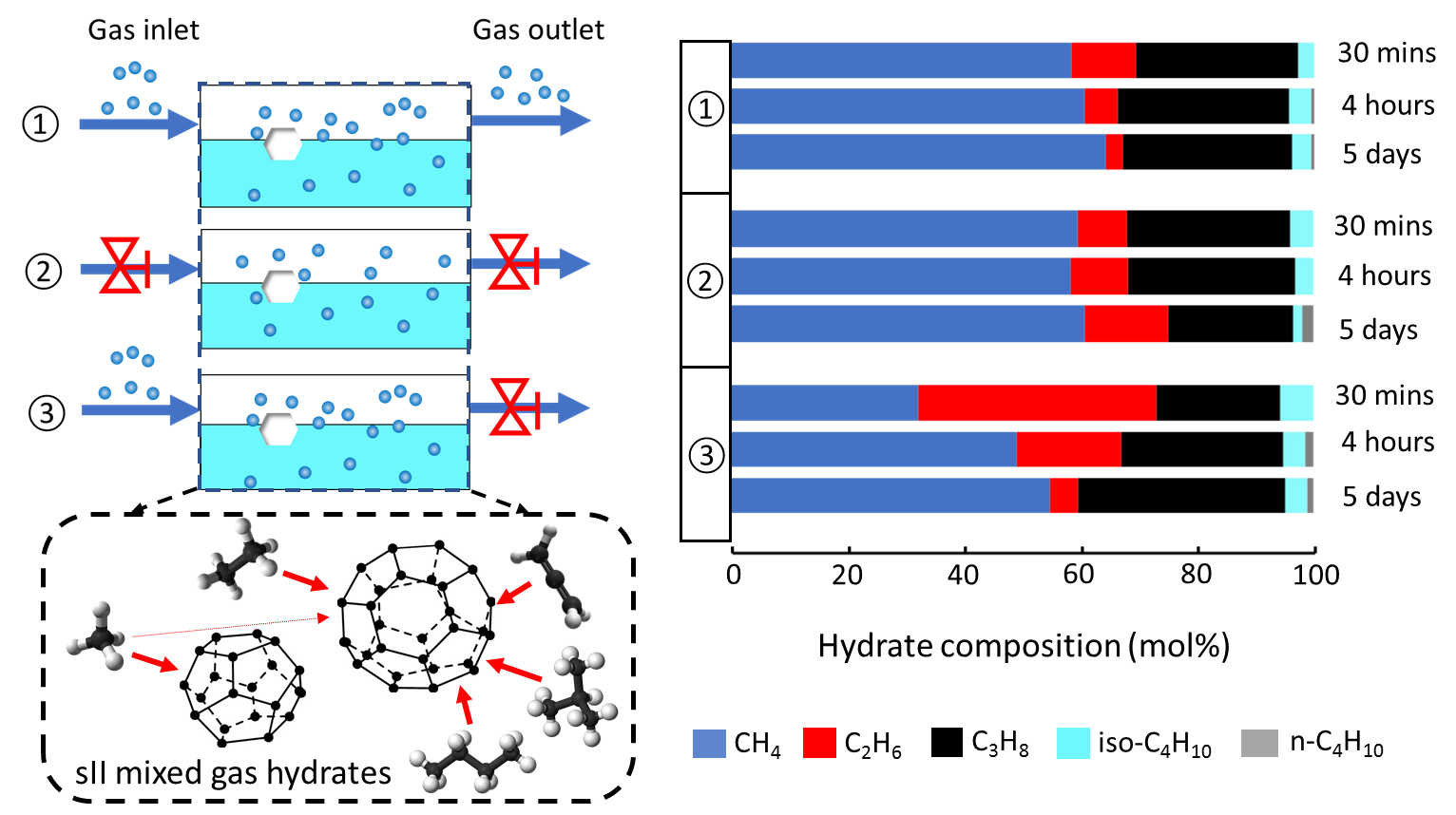
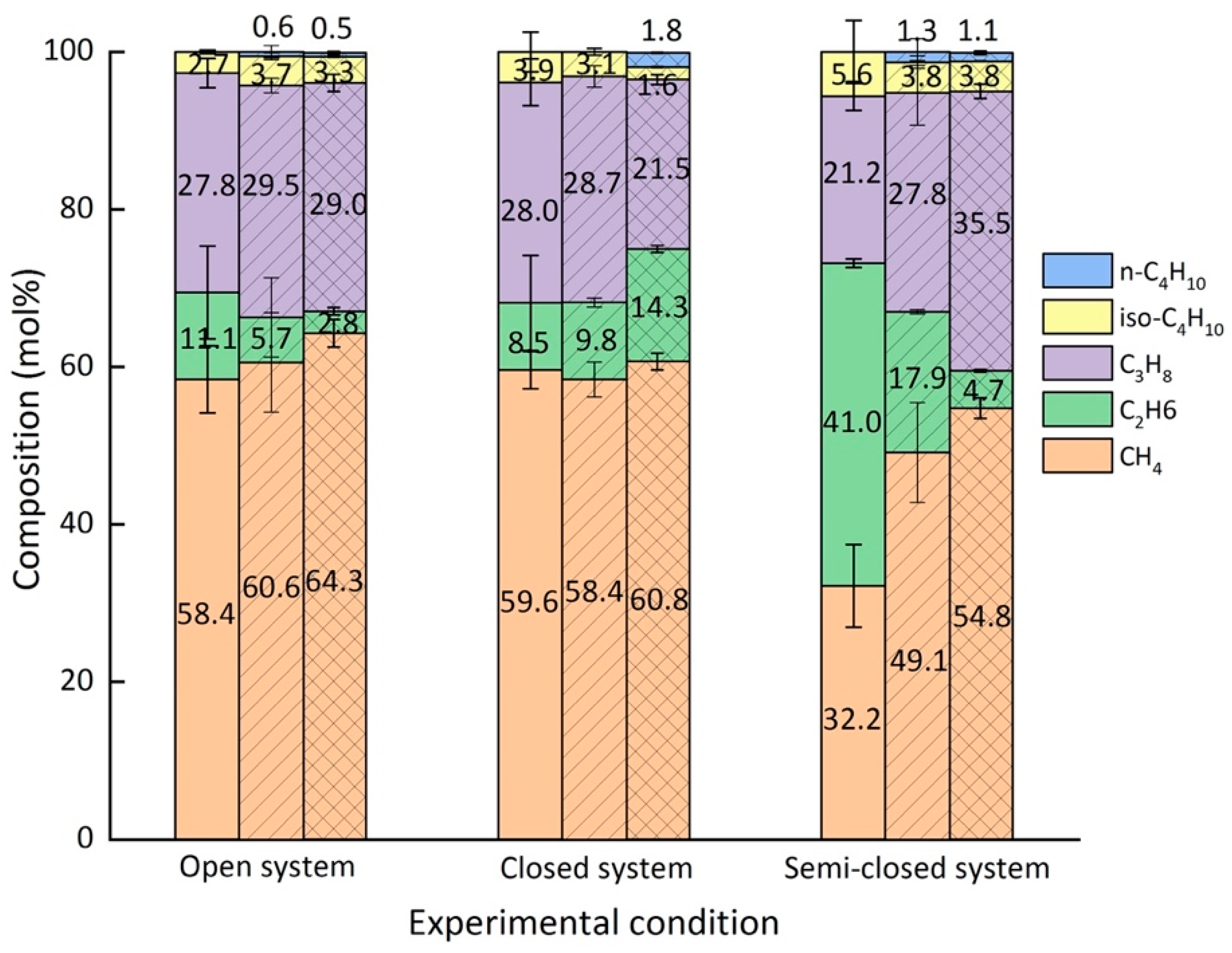
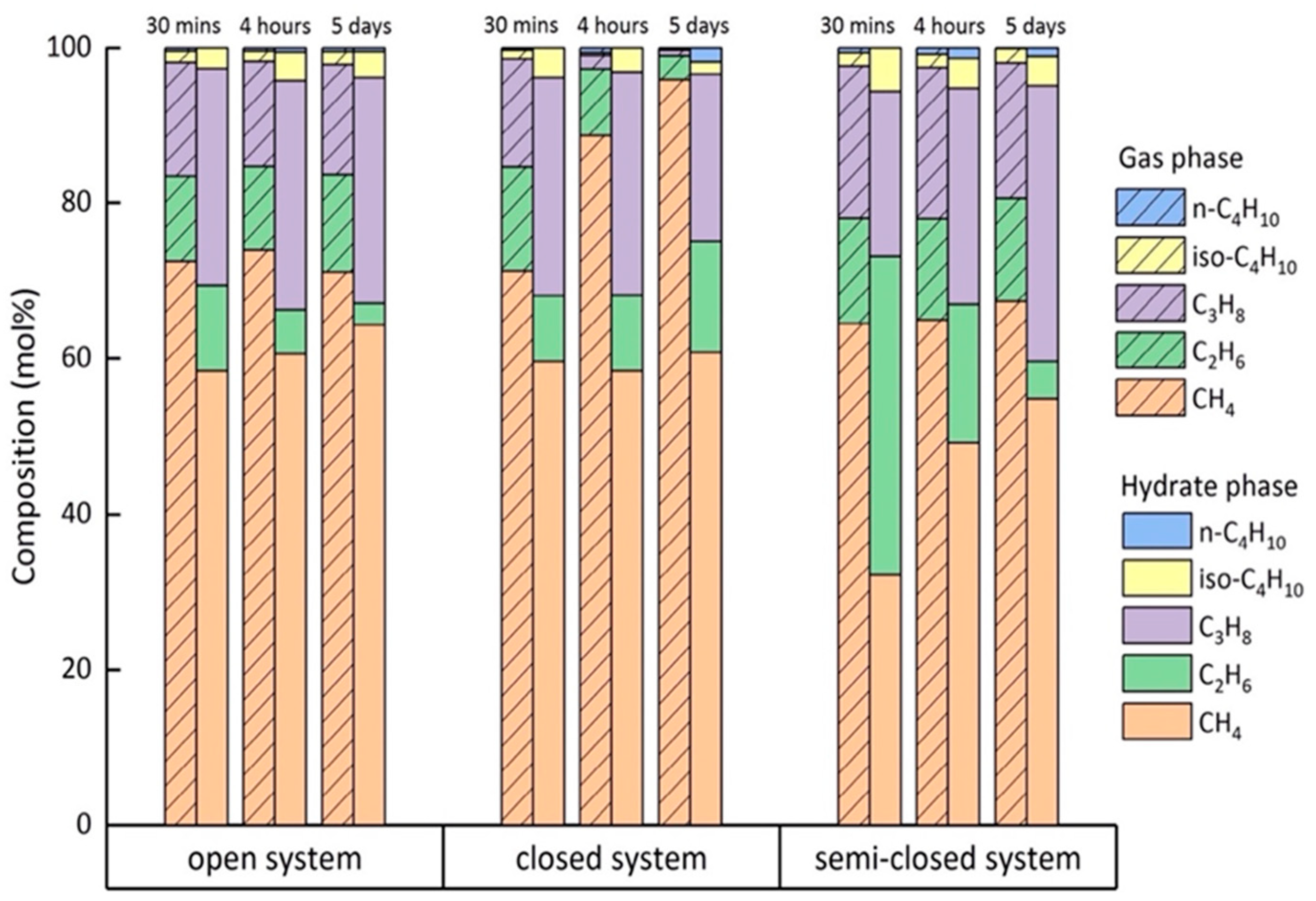
2.4. Composition of the Mixed Gas Hydrates
3. Discussion
4. Materials and Methods
4.1. Experimental Apparatus
4.2. Experimental Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- von Stackelberg, M.; Gotzen, O.; Pietuchovsky, J.; Wirtscher, O.; Fruhbuss, H.; Meinhold, W. Struktur und Formel der Gashydrate. Fortschr. Miner. 1947, 26, 122–124. [Google Scholar]
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press Taylor and Francis Group: Boca Raton, FL, USA, 2008. [Google Scholar]
- Kvenvolden, K.A. Gas hydrates—Geological perspective and gobal change. Rev. Geophys. 1993, 31, 173–187. [Google Scholar] [CrossRef]
- Kida, M.; Khlystov, O.; Zemskaya, T.; Takahashi, N.; Minami, H.; Sakagami, H.; Krylov, A.; Hachikubo, A.; Yamashita, S.; Shoji, H.; et al. Coexistence of structure I and II gas hydrates in Lake Baikal suggesting gas sources from microbial and thermogenic origin. Geophys. Res. Lett. 2006, 33, L24603. [Google Scholar] [CrossRef]
- Lu, H.; Seo, Y.; Lee, J.; Moudrakovski, I.; Ripmeester, J.A.; Chapman, N.R.; Coffin, R.B.; Gardner, G.; Pohlman, J. Complex gas hydrate from the Cascadia margin. Nature 2007, 445, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Klapp, S.A.; Murshed, M.M.; Pape, T.; Klein, H.; Bohrmann, G.; Brewer, P.G.; Kuhs, W.F. Mixed gas hydrate structures at the Chapopote Knoll, southern Gulf of Mexico. Earth Planet. Sci. Lett. 2010, 299, 207–217. [Google Scholar] [CrossRef]
- Wei, J.; Fang, Y.; Lu, H.; Lu, H.; Lu, J.; Liang, J.; Yang, S. Distribution and characteristics of natural gas hydrates in the Shenhu Sea. Mar. Pet. Geol. 2018, 98, 622–628. [Google Scholar] [CrossRef]
- Hachikubo, A.; Khlystov, O.; Manakov, A.; Kida, M.; Krylov, A.; Sakagami, H.; Minami, H.; Takahashi, N.; Shoji, H.; Kalmychkov, G.; et al. Model of formation of double structure gas hydrates in Lake Baikal based on isotopic data. Geophys. Res. Lett. 2009, 36, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhu, Y.; Lu, Z.; Huang, X.; Pang, S.; Zhang, S.; Jiang, S.; Li, Q.; Yang, K.; Li, B. Geochemistry and genesis of authigenic pyrite from gas hydrate accumulation system in the Qilian Mountain permafrost, Qinghai, northwest China. Sci. China-Earth Sci. 2014, 57, 2217–2231. [Google Scholar] [CrossRef]
- Lu, Z.; Zhu, Y.; Zhang, Y.; Wen, H.; Li, Y.; Liu, C. Gas hydrate occurrences in the Qilian Mountain permafrost, Qinghai Province, China. Cold Reg. Sci. Technol. 2011, 66, 93–104. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Y.; Wen, H.; Lu, Z.; Jia, Z.; Li, Y.; Li, Q.; Liu, C.; Wang, P.; Guo, X. Gas Hydrates in the Qilian Mountain Permafrost, Qinghai, Northwest China. Acta Geol. Sin. Ed. 2010, 84, 1–10. [Google Scholar] [CrossRef]
- Lu, Z.; Zhu, Y.; Liu, H.; Zhang, Y.; Jin, C.; Huang, X.; Wang, P. Gas source for gas hydrate and its significance in the Qilian Mountain permafrost, Qinghai. Mar. Pet. Geol. 2013, 43, 341–348. [Google Scholar] [CrossRef]
- Huang, X.; Zhu, Y.; Wang, P.; Guo, X. Hydrocarbon gas composition and origin of core gas from the gas hydrate reservoir in Qilian Mountain permafrost. Geol. Bull. China 2011, 30, 1851–1856. [Google Scholar]
- Cheng, B.; Xu, J.; Lu, Z.; Li, Y.; Wang, W.; Yang, S.; Liu, H.; Wang, T.; Liao, Z. Hydrocarbon source for oil and gas indication associated with gas hydrate and its significance in the Qilian Mountain permafrost, Qinghai, Northwest China. Mar. Pet. Geol. 2018, 89, 202–215. [Google Scholar] [CrossRef]
- Liu, S.; Tan, F.; Huo, T.; Tang, S.; Zhao, W.; Chao, H. Origin of the hydrate bound gases in the Juhugeng Sag, Muli Basin, Tibetan Plateau. Int. J. Coal Sci. Technol. 2020, 7, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Zhai, G.; Zhu, Y.; Zhang, Y.; Li, Y.; Wang, W.; Wang, T.; Liu, H.; Tang, S.; Tan, P. Fault control of gas hydrate accumulation in Qilian Mountain Permafrost. Int. J. Offshore Polar Eng. 2016, 26, 199–205. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Wei, Y.; Yang, C.; Nie, J.; Cao, D. Gas migration for terrestrial gas hydrates in the Juhugeng mining area of Muli basin, Qilian Mountains, Northwest China. Energy Explor. Exploit. 2020, 38, 989–1013. [Google Scholar] [CrossRef]
- Liu, C.; Meng, Q.; He, X.; Li, C.; Ye, Y.; Lu, Z.; Zhu, Y.; Li, Y.; Liang, J. Comparison of the characteristics for natural gas hydrate recovered from marine and terrestrial areas in China. J. Geochem. Explor. 2015, 152, 67–74. [Google Scholar] [CrossRef]
- Xia, N.; Liu, C.; Ye, Y.; Meng, Q.; Lin, X.; He, X. Study on determination method of natural gas hydrates by Micro-Laser Raman Spectroscopy. Rock Miner. Anal. 2011, 30, 416–422. [Google Scholar]
- Rydzy, M.B.; Schicks, J.M.; Naumann, R.; Erzinger, J. Dissociation enthalpies of synthesized multicomponent gas hydrates with respect to the guest composition and cage occupancy. J. Phys. Chem. B 2007, 111, 9539–9545. [Google Scholar] [CrossRef]
- Schicks, J.M.; Luzi-Helbing, M. Cage occupancy and structural changes during hydrate formation from initial stages to resulting hydrate phase. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 115, 528–536. [Google Scholar] [CrossRef]
- Schicks, J.M.; Luzi-Helbing, M. Kinetic and thermodynamic aspects of clathrate hydrate nucleation and growth. J. Chem. Eng. Data 2015, 60, 269–277. [Google Scholar] [CrossRef]
- Uchida, T.; Moriwaki, M.; Takeya, S.; Ikeda, I.Y.; Ohmura, R.; Nagao, J.; Minagawa, H.; Ebinuma, T.; Narita, H.; Gohara, K.; et al. Two-step formation of methane-propane mixed gas hydrates in a batch-type reactor. AIChE J. 2004, 50, 518–523. [Google Scholar] [CrossRef]
- Uchida, T.; Takeya, S.; Kamata, Y.; Ikeda, I.Y.; Nagao, J.; Ebinuma, T.; Narita, H.; Zatsepina, O.; Buffett, B.A. Spectroscopic observations and thermodynamic calculations on clathrate hydrates of mixed gas containing methane and ethane: Determination of structure, composition and cage occupancy. J. Phys. Chem. B 2002, 106, 12426–12431. [Google Scholar] [CrossRef]
- Subramanian, S.; Kini, R.A.; Dec, S.F.; Sloan, E.D. Structural transition studies in methane + ethane hydrates using Raman and NMR. Ann. N. Y. Acad. Sci. 2000, 912, 873–886. [Google Scholar] [CrossRef]
- Subramanian, S.; Sloan, E.D. Trends in vibrational frequencies of guests trapped in clathrate hydrate cages. J. Phys. Chem. B 2002, 106, 4348–4355. [Google Scholar] [CrossRef]
- Uchida, T.; Takeya, S.; Kamata, Y.; Ohmura, R.; Narita, H. Spectroscopic measurements on binary, ternary, and quaternary mixed-gas molecules in clathrate structures. Ind. Eng. Chem. Res. 2007, 46, 5080–5087. [Google Scholar] [CrossRef]
- Chazallon, B.; Focsa, C.; Charlou, J.L.; Bourry, C.; Donval, J.P. A comparative Raman spectroscopic study of natural gas hydrates collected at different geological sites. Chem. Geol. 2007, 244, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Sum, A.K.; Burruss, R.C.; Sloan, E.D. Measurement of clathrate hydrates via Raman spectroscopy. J. Phys. Chem. B 1997, 101, 7371–7377. [Google Scholar] [CrossRef]
- Walrafen, G.E.; Yang, W.H.; Chu, Y.C. Raman evidence for the clathratelike structure of highly supercooled water. ACS Symp. Ser. 1997, 676, 287–308. [Google Scholar]
- Schicks, J.M. Der Einfluss Molekularer Eigenschaften von Gasen auf Die Hydratbildungsprozesse, Die Thermodynamischen Eigenschaften und Die Reaktionen von Einfachen und Gemischten Gashydraten. Ph.D. Thesis, University of Potsdam, Potsdam, Germany, 2013. [Google Scholar]
- Radhakrishnan, R.; Trout, B.L. A new approach for studying nucleation phenomena using molecular simulations: Application to CO2 hydrate clathrates. J. Chem. Phys. 2002, 117, 1786–1796. [Google Scholar] [CrossRef] [Green Version]
- Long, J. Gas Hydrate Formation Mechanism and its Kinetic Inhibition. Ph.D. Thesis, Colorado School of Mines, Golden, CO, USA, 1994. Available online: https://tinyurl.com/yjcuh8xm (accessed on 3 March 2021).
- Kvamme, B. A new theorie for kinetics of hydrate formation. In Proceedings of the 2nd International Conference on Natural Gas Hydrates, Toulouse, France, 2–6 June 1996; pp. 139–146. [Google Scholar]
- Sloan, E.D.; Fleyfel, F. A molecular mechanism for gas hydrate nucleation from ice. AIChE J. 1991, 37, 1281–1292. [Google Scholar] [CrossRef]
- Geiger, A.; Mausbach, P.; Schnitker, J. Computer simulation study of the hydrogen bond network in metastable water. In Water and Aqueous Solutions; Neilson, G.W., Enderby, J.E., Eds.; Hilger, A: Bristol, UK, 1986; pp. 15–30. Available online: https://www.academia.edu/34422752/Computer_Simulation_Study_of_the_Hydrogen_Bond_Network_in_Metastable_Water (accessed on 6 January 2021).
- Ludwig, R. The importance of tetrahedrally coordinated molecules for the explanation of liquid water properties. ChemPhysChem 2007, 8, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Kashchiev, D.; Firoozabadi, A. Driving force for crystallization of gas hydrates. J. Cryst. Growth 2002, 241, 220–230. [Google Scholar] [CrossRef]
- Walsh, M.R.; Koh, C.A.; Sloan, E.D.; Sum, A.K.; Wu, D.T. Microsecond simulations of spontaneous methane hydrate nucleation and growth. Science (80-) 2009, 326, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L.C.; Hujo, W.; Molinero, V. Nucleation pathways of clathrate hydrates: Effect of guest size and solubility. J. Phys. Chem. B 2010, 114, 13796–13807. [Google Scholar] [CrossRef]
- Jacobson, L.C.; Hujo, W.; Molinero, V. Amorphous precursors in the nucleation of clathrate hydrates. J. Am. Chem. Soc. 2010, 132, 11806–11811. [Google Scholar] [CrossRef]
- Lederhos, J.P.; Christiansen, R.L.; Sloan, E.D. A first order method of hydrate equilibrium estimation and its use with new structures. Fluid Phase Equilib. 1993, 83, 445–454. [Google Scholar] [CrossRef]
- Szasz, G.J.; Sheppard, N.; Rank, D.H. Spectroscopic studies of rotational isomerism. I. Liquid n-butane and the assignment of the normal modes of vibration. J. Chem. Phys. 1948, 16, 704–711. [Google Scholar] [CrossRef]
- Luzi, M.; Schicks, J.M.; Naumann, R.; Erzinger, J.; Udachin, K.; Moudrakowski, I.; Ripmeester, J.A.; Ludwig, R. Investigations on the Influence of Guest Molecule characteristics and the presence of multicomponent gas mixtures on gas hydrate properties. In Proceedings of the 6th International Conference on Gas Hydrates, Vancouver, BC, Canada, 6–10 July 2008; Available online: https://open.library.ubc.ca/cIRcle/collections/59278/items/1.0040949 (accessed on 13 December 2020).
- Chaouachi, M.; Neher, S.H.; Falenty, A.; Kuhs, W.F. Time Resolved Coarsening of Clathrate Crystals: The Case of Gas Hydrates. Cryst. Growth Des. 2017, 17, 2458–2472. [Google Scholar] [CrossRef]
- Chen, X.; Espinoza, D.N. Ostwald ripening changes the pore habit and spatial variability of clathrate hydrate. Fuel 2018, 214, 614–622. [Google Scholar] [CrossRef]
- Schicks, J.M.; Pan, M.; Giese, R.; Poser, M.; Aminatulmimi, N.I.; Luzi-Helbing, M.; Bleisteiner, B.; Lenz, C. A new high-pressure cell for systematic in situ investigations of micro-scale processes in gas hydrates using confocal micro-Raman spectroscopy. Rev. Sci. Instrum. 2020, 91, 115103. [Google Scholar] [CrossRef] [PubMed]
- Wedler, G.; Freund, H.-J. Lehrbuch der Physikalischen Chemie, 6th ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Pan, M.; Schicks, J.M. Raman Spectroscopic Data from Gas Hydrates Formed from a Complex Gas Mixture with Different Gas Supply Conditions. GFZ Data Services, 2021. Available online: https://doi.org/10.5880/GFZ.3.1.2021.003 (accessed on 24 May 2021).
- Schrötter, H.W.; Klöckner, H.W. Raman scattering cross sections in gases and liquids. In Raman Spectroscopy of Gases and Liquids. Topics in Current Physics, vol 11; Weber, A., Ed.; Springer: Berlin/Heidelberg, Germany, 1979; pp. 123–166. [Google Scholar] [CrossRef]
- Schrader, B. Infrared and Raman Spectroscopy: Methods and Applications; VCH Verlagsgesellschaft mbH: Weinheim, Germany, 1995. [Google Scholar]
- Burke, E.A.J. Raman microspectrometry of fluid inclusions. Lithos 2001, 55, 139–158. [Google Scholar] [CrossRef]
- Beeskow-Strauch, B.; Schicks, J.M.; Spangenberg, E.; Erzinger, J. The influence of SO2 and NO2 impurities on CO2 gas hydrate formation and stability. Chem. A Eur. J. 2011, 17, 4376–4384. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Kuhs, W.F. Quantitative analysis of gas hydrates using Raman spectroscopy. AIChE J. 2013, 59, 2155–2167. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Vibrational Mode | Cavity Type | νmeasured (cm−1) | νliterature (cm−1) | References |
|---|---|---|---|---|---|
| CH4 | C–H symmetric stretching | sII 512 | 2912 | 2912 | [27] |
| sII 51264 | 2901 | 2901 | |||
| C2H6 | C–C symmetric stretching | sII 51264 | 991 | 991 | [10] |
| C3H8 | C–C symmetric stretching | sII 51264 | 876 | 876 | [10] |
| iso-C4H10 | C−C symmetric stretching | sII 51264 | 811 | 811 | [27] |
| n-C4H10 | C–C symmetric stretching | sII 51264 | 838 (gauche form) | 838 | [26] |
| Experimental Condition | Time | Average Concentration of Each Component in the Hydrate Phase (mol%) | ||||
|---|---|---|---|---|---|---|
| CH4 | C2H6 | C3H8 | iso-C4H10 | n-C4H10 1 | ||
| Open | After 30 min | 58.4 | 11.1 | 27.8 | 2.7 | 0.0 |
| After 4 h | 60.6 | 5.7 | 29.5 | 3.7 | 0.6 | |
| After 5 days | 64.3 | 2.8 | 29.0 | 3.3 | 0.5 | |
| Closed | After 30 min | 59.6 | 8.5 | 28.0 | 3.9 | 0.0 |
| After 4 h | 58.4 | 9.8 | 28.7 | 3.1 | 0.0 | |
| After 5 days | 60.8 | 14.3 | 21.5 | 1.6 | 1.8 | |
| Semi-closed | After 30 min | 32.2 | 41.0 | 21.2 | 5.6 | 0.0 |
| After 4 h | 49.1 | 17.9 | 27.8 | 3.9 | 1.3 | |
| After 5 days | 54.8 | 4.7 | 35.5 | 3.8 | 1.1 | |
| Molecule | Structure | Solubility (mmol/L) 1 | Guest Diameter (Å) | Guest-to-Cavity Ratio | |
|---|---|---|---|---|---|
| 512 (sII) | 51264 (sII) | ||||
| CH4 |  | 1.6 | 4.36 | 0.87 | 0.66 |
| C2H6 |  | 2.2 | 5.50 | 1.10 | 0.83 |
| C3H8 |  | 1.8 | 6.28 | 1.25 | 0.94 |
| iso-C4H10 |  | 1.0 | 6.50 | 1.29 | 0.98 |
| n-C4H10 |  | 1.6 | 7.10 | 1.41 | 1.07 |
 | 7.86 | 1.57 | 1.18 | ||
| Gas Inlet | Gas Outlet | System | |
|---|---|---|---|
| Test Scenario 1 | Open | Open | Open system |
| Test Scenario 2 | Closed | Closed | Closed system |
| Test Scenario 3 | Open | Closed | Semi-closed system |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, M.; Schicks, J.M. Influence of Gas Supply Changes on the Formation Process of Complex Mixed Gas Hydrates. Molecules 2021, 26, 3039. https://doi.org/10.3390/molecules26103039
Pan M, Schicks JM. Influence of Gas Supply Changes on the Formation Process of Complex Mixed Gas Hydrates. Molecules. 2021; 26(10):3039. https://doi.org/10.3390/molecules26103039
Chicago/Turabian StylePan, Mengdi, and Judith M. Schicks. 2021. "Influence of Gas Supply Changes on the Formation Process of Complex Mixed Gas Hydrates" Molecules 26, no. 10: 3039. https://doi.org/10.3390/molecules26103039