
Prioritisation of Compounds for 3CLpro Inhibitor Development on SARS-CoV-2 Variants

 , , , and
, , , and
Abstract
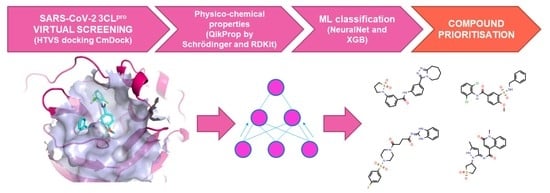
:
1. Introduction
2. Results and Discussion
2.1. Database Preparation
2.2. Binding Site Identification
2.3. HTVS
2.4. Chemical Space Prediction and Classification Supported by Machine Learning
3. Materials and Methods
3.1. Target Preparation
3.2. Virtual Screening Experiment Design
3.3. Machine Learning
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| 3CLpro | 3C-like protease |
| MD | Molecular Dynamics |
| VS | Virtual Screening |
| HTVS | High-Throughput Virtual Screening |
| LIE | Linear Interaction Energy |
| Xgb | Gradient boosting framework library |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Cao, B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Peng, Z. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Tan, W. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Widge, A.T.; Rouphael, N.G.; Jackson, L.A.; Anderson, E.J.; Roberts, P.C.; Makhene, M.; Beigel, J.H. Durability of responses after SARS-CoV-2 mRNA-1273 vaccination. N. Engl. J. Med. 2021, 384, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; Krammer, F. SARS-CoV-2 vaccines: Status report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Strych, U.; Hotez, P.J.; Bottazzi, M.E. The SARS-CoV-2 vaccine pipeline: An overview. Curr. Trop. Med. Rep. 2020, 7, 61–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Li, H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- McKee, D.L.; Sternberg, A.; Stange, U.; Laufer, S.; Naujokat, C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020, 157, 104859. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, Y.; Zhang, M.; Wang, H.; Zhao, Q.; Liu, J. Updated approaches against SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00483-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; Krogan, N.J. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Chanda, S.K. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 10, 165878. [Google Scholar] [CrossRef]
- Volz, E.; Swapnil, M.; Meera, C.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Transmission of SARS-CoV-2 Lineage, B. 1.1. 7 in England: Insights from linking epidemiological and genetic data. MedRxiv 2021, 2020-12. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. MedRxiv 2020. [Google Scholar] [CrossRef]
- Gupta, R.K. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat. Rev. Immunol. 2021, 1–2. [Google Scholar] [CrossRef]
- Hongjing, G.; Chen, Q.; Yang, G.; He, L.; Fan, H.; Deng, Y.-Q.; Wang, Y.; Teng, Y.; Zhao, Z.; Cui, Y.; et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 2020, 369, 1603–1607. [Google Scholar]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Lambson, B.E.; Vermeulen, M.; van den Berg, K.; Rossouw, T.; Boswell, M.; et al. SARS-CoV-2 501Y. V2 escapes neutralization by South African COVID-19 donor plasma. BioRxiv 2021, 27, 622–625. [Google Scholar]
- Nuno, R.F.; Morales Claro, I.; Candido, D.; Franco, L.A.M.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.M.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: Preliminary findings. Virological 2021. [Google Scholar]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of SARS-CoV-2 PLpro. Biorxiv 2020. [Google Scholar] [CrossRef]
- Sumit, K.; Sharma, P.P.; Shankar, U.; Kumar, D.; Joshi, S.K.; Pena, L.; Durvasula, R.; Kumar, A.; Kempaiah, P.; Rathi, P.; et al. Discovery of new hydroxyethylamine analogs against 3CLpro protein target of SARS-CoV-2: Molecular docking, molecular dynamics simulation, and structure–activity relationship studies. J. Chem. Inf. Model. 2020, 60, 5754–5770. [Google Scholar]
- ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Koulgi, S.; Jani, V.; Uppuladinne, M.; Sonavane, U.; Nath, A.K.; Darbari, H.; Joshi, R. Drug repurposing studies targeting SARS-CoV-2: An ensemble docking approach on drug target 3C-like protease (3CLpro). J. Biomol. Struct. Dyn. 2020, 1–21. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Inhibition of the main protease 3cl-pro of the coronavirus disease 19 via structure-based ligand design and molecular modeling. arXiv 2020, arXiv:2002.09937. [Google Scholar]
- Malcolm, B.A. The picornaviral 3C proteinases: Cysteine nucleophiles in serine proteinase folds. Prot. Sci. 1995, 4, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for Controlling the Dimer-Monomer Switch and Coupling Dimerization to Catalysis of the Severe Acute Respiratory Syndrome Coronavirus 3C-Like Protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS central science 2021, 7, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Froggatt, H.M.; Heaton, B.E.; Heaton, N.S. Development of a fluorescence-based, high-throughput SARS-CoV-2 3CLpro reporter assay. J. Virol. 2020, 94, e01265-20. [Google Scholar] [CrossRef] [PubMed]
- Mariusz, J.; Dauter, Z.; Shabalin, I.G.; Gilski, M.; Brzezinski, D.; Kowiel, M.; Rupp, B.; Wlodawer, A. Crystallographic models of SARS-CoV-2 3CLpro: In-depth assessment of structure quality and validation. IUCrJ 2021, 8, 238–256. [Google Scholar]
- Macchiagodena, M.; Pagliai, M.; Karrenbrock, M.; Guarnieri, G.; Iannone, F.; Procacci, P. Virtual Double-System Single-Box: A Nonequilibrium Alchemical Technique for Absolute Binding Free Energy Calculations: Application to Ligands of the SARS-CoV-2 Main Protease. J. Chem. Theory Comput. 2020, 16, 7160–7172. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Disc. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Baillie, T.A. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and the Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jukič, M.; Janežič, D.; Bren, U. Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules 2020, 25, 5808. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saubern, S.; Guha, R.; Baell, J.B. KNIME workflow to assess PAINS filters in SMARTS format. Comparison of RDKit and Indigo cheminformatics libraries. Mol. Inform. 2011, 30, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual screening—An overview. Drug Disc. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis: Miniperspective. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Gardner, S.P.; Sutcliffe, M.J. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally-related subfamilies. Protein Eng. 1996, 9, 1063–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Overington, J.P. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I.; Bengio, Y.; Courville, A.; Bengio, Y. Deep Learning; MIT Press: Cambridge, MA, USA, 2016; Volume 1, No. 2. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Duchesnay, E. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chen, T.; Guestrin, C. Xgboost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Chollet, F. Keras. 2015. Available online: https://keras.io (accessed on 8 May 2021).
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Liu, J.; Shang, W.J.; Xie, H.; Ke, C.; Hu, H.; Gao, M.; et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, A.D.; Zheng, J.; Kim, Y.; Perera, K.D.; Mackin, S.; Meyerholz, D.K.; Kashipathy, M.M.; Battaile, K.P.; Lovell, S.; Perlman, S.; et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV–infected mice. Sci. Transl. Med. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Morley, S.D.; Afshar, M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using RiboDock. J. Comput. Aided Mol. Des. 2004, 18, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, B.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Graf, M.M.; Bren, U.; Haltrich, D.; Oostenbrink, C. Molecular dynamics simulations give insight into D-glucose dioxidation at C2 and C3 by Agaricus meleagris pyranose dehydrogenase. Comput. Aided Mol. Des. 2013, 27, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Jukic, M.; Ilc, N.; Sluga, D.; Tomšič, G.; Podlipnik, Č. CmDock. 2021. Available online: https://gitlab.com/Jukic/cmdock/ (accessed on 8 May 2021).
- Tosco, P.; Stiefl, N.; Landrum, G. Bringing the MMFF force field to the RDKit: Implementation and validation. J. Cheminformatics 2014, 6, 1–4. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant 1 | Alternative Name | Sprot/ All Mutations | Key Mutations | Comment | 3CLpro/PLpro Mutations 2 |
|---|---|---|---|---|---|
| B.1.1.7 | UK Variant | 8/23 | E69/70 del 144Y del N501Y (RBD interface) A570D P681H | higher transmissibility | none/A1708D |
| B.1.351 | South African Variant | 9/21 | K417N (RBD) E484K (RBD) N501Y (RBD) orf1b del | escape host immune response | none/K1655N |
| P.1 | Brasil Variant | 10/17 | K417N/T (RBD) E484K (RBD) N501Y (RBD) orf1b del | under research | none/K1795Q |
| B.1.617 | Indian Variant | 7/23 | G142D E154K L452R (RBD) E484Q (RBD) D614G P681R Q1071H | under research | none/under research |
| no | Structure | Mr (g/mol) | Cluster/QPlogS 1 | CmDock Docking Score 2 | Classification 3 |
|---|---|---|---|---|---|
| 1 |  | 451.54 | 5/−6.42 | −32.51 | general |
| 2 |  | 465.35 | 5/−6.22 | −29.02 | general |
| 3 |  | 459.49 | 5/−3.89 | −26.80 | viral_cys_prot |
| 4 |  | 400.45 | 4/−4.54 | −25.58 | viral_cys_prot |
| 5 |  | 325.37 | 5/−3.59 | −25.53 | viral_cys_prot |
| 6 |  | 396.85 | 5/−6.47 | −25.05 | general |
| 7 |  | 399.83 | 2/−3.44 | −24.76 | general |
| 8 |  | 425.50 | 4/−5.38 | −24.51 | general |
| 9 |  | 353.44 | 5/−4.40 | −24.17 | general |
| 10 |  | 494.55 | 5/−5.60 | −24.01 | general |
| 11 |  | 490.60 | 5/−2.87 | −23.98 | general |
| 12 |  | 337.37 | 5/−4.75 | −23.61 | viral_cys_prot |
| 13 |  | 425.52 | 5/−5.66 | −23.53 | general |
| 14 |  | 335.34 | 5/−3.90 | −23.26 | general |
| 15 |  | 401.44 | 4/−5.00 | −23.18 | general |
| Macro F1/mse 1 | NeuralNet 2 | XGB 2 | Linear 2 | Majority/Average 3 |
|---|---|---|---|---|
| Classification | 0.895 ± 0.05 | 0.889 ± 0.014 | 0.667 ± 0.015 | 0.283 ± 0.001 |
| Regression | 0.002 ± 0.001 | 0.003 ±0.001 | 0.012 ± 0.002 | 0.005 ± 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jukič, M.; Škrlj, B.; Tomšič, G.; Pleško, S.; Podlipnik, Č.; Bren, U. Prioritisation of Compounds for 3CLpro Inhibitor Development on SARS-CoV-2 Variants. Molecules 2021, 26, 3003. https://doi.org/10.3390/molecules26103003
Jukič M, Škrlj B, Tomšič G, Pleško S, Podlipnik Č, Bren U. Prioritisation of Compounds for 3CLpro Inhibitor Development on SARS-CoV-2 Variants. Molecules. 2021; 26(10):3003. https://doi.org/10.3390/molecules26103003
Chicago/Turabian StyleJukič, Marko, Blaž Škrlj, Gašper Tomšič, Sebastian Pleško, Črtomir Podlipnik, and Urban Bren. 2021. "Prioritisation of Compounds for 3CLpro Inhibitor Development on SARS-CoV-2 Variants" Molecules 26, no. 10: 3003. https://doi.org/10.3390/molecules26103003