
Carbon Dioxide Reduction with Hydrogen on Fe, Co Supported Alumina and Carbon Catalysts under Supercritical Conditions

 and
and
Abstract
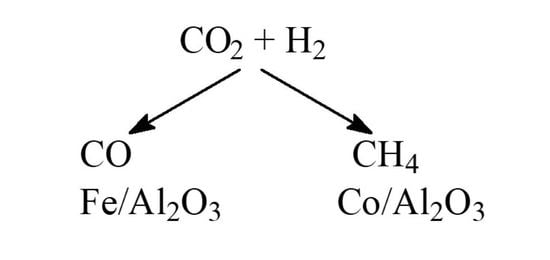
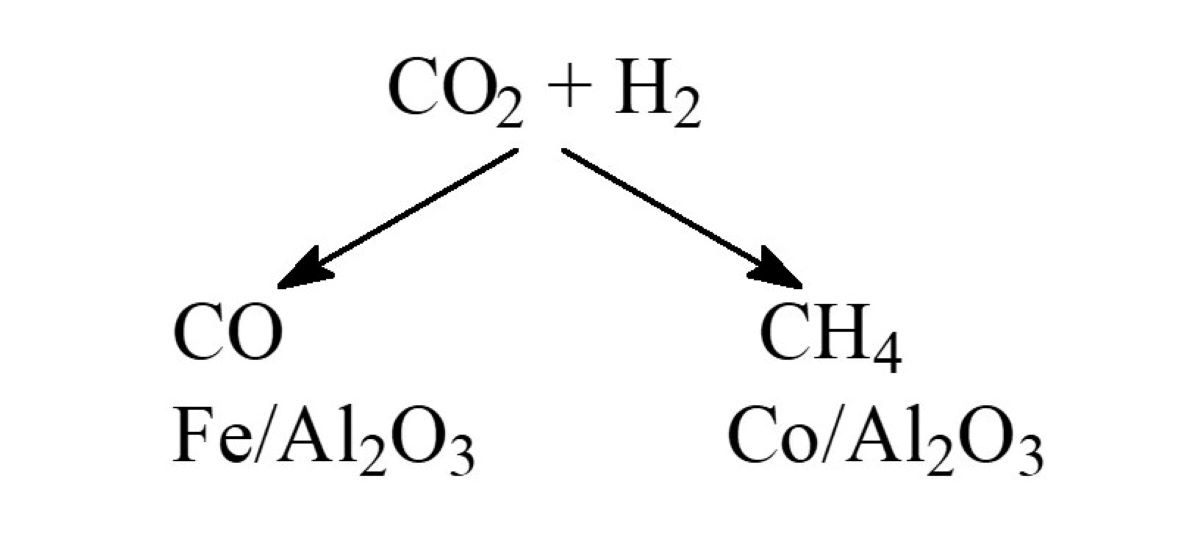
:
1. Introduction
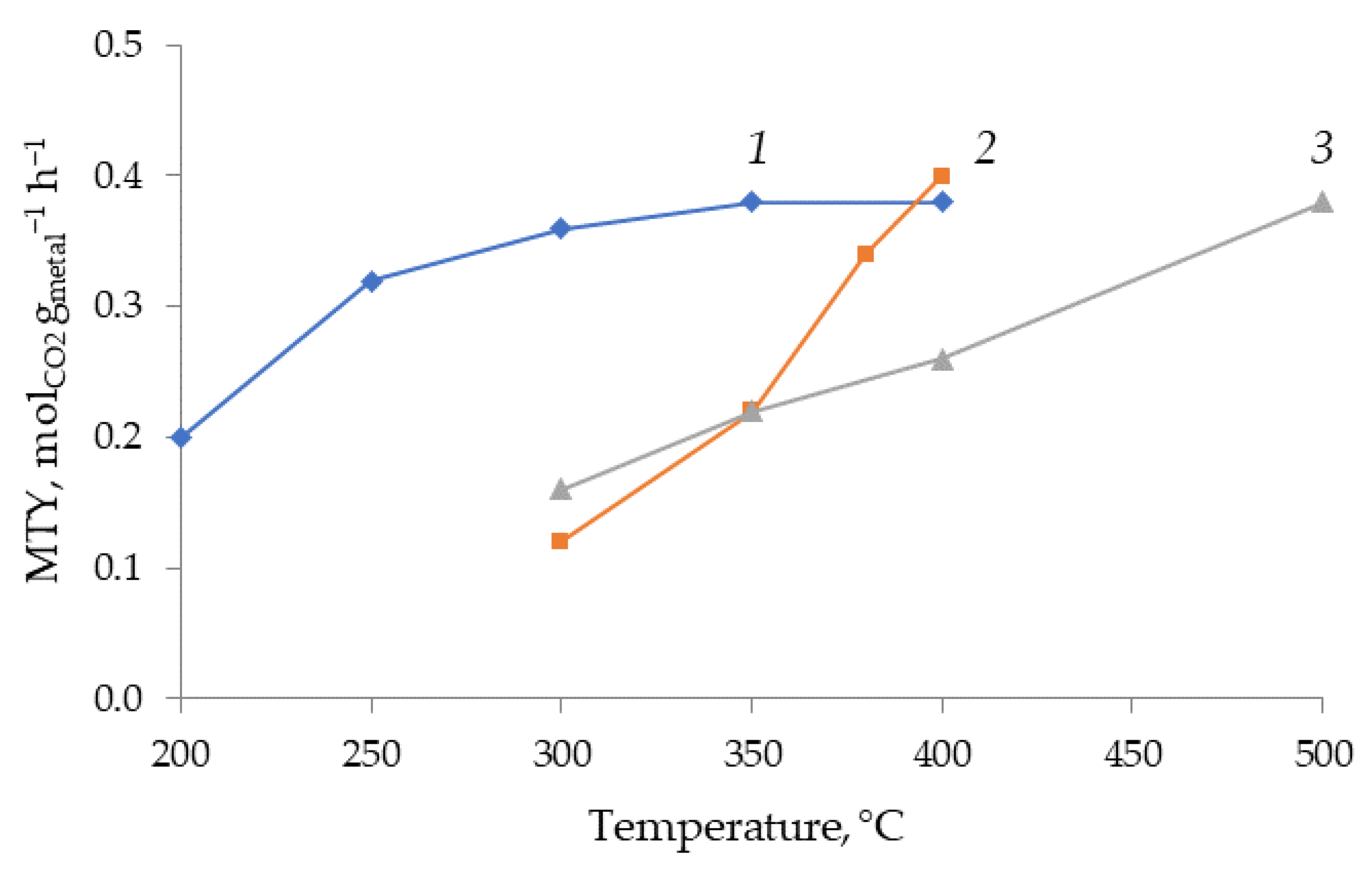
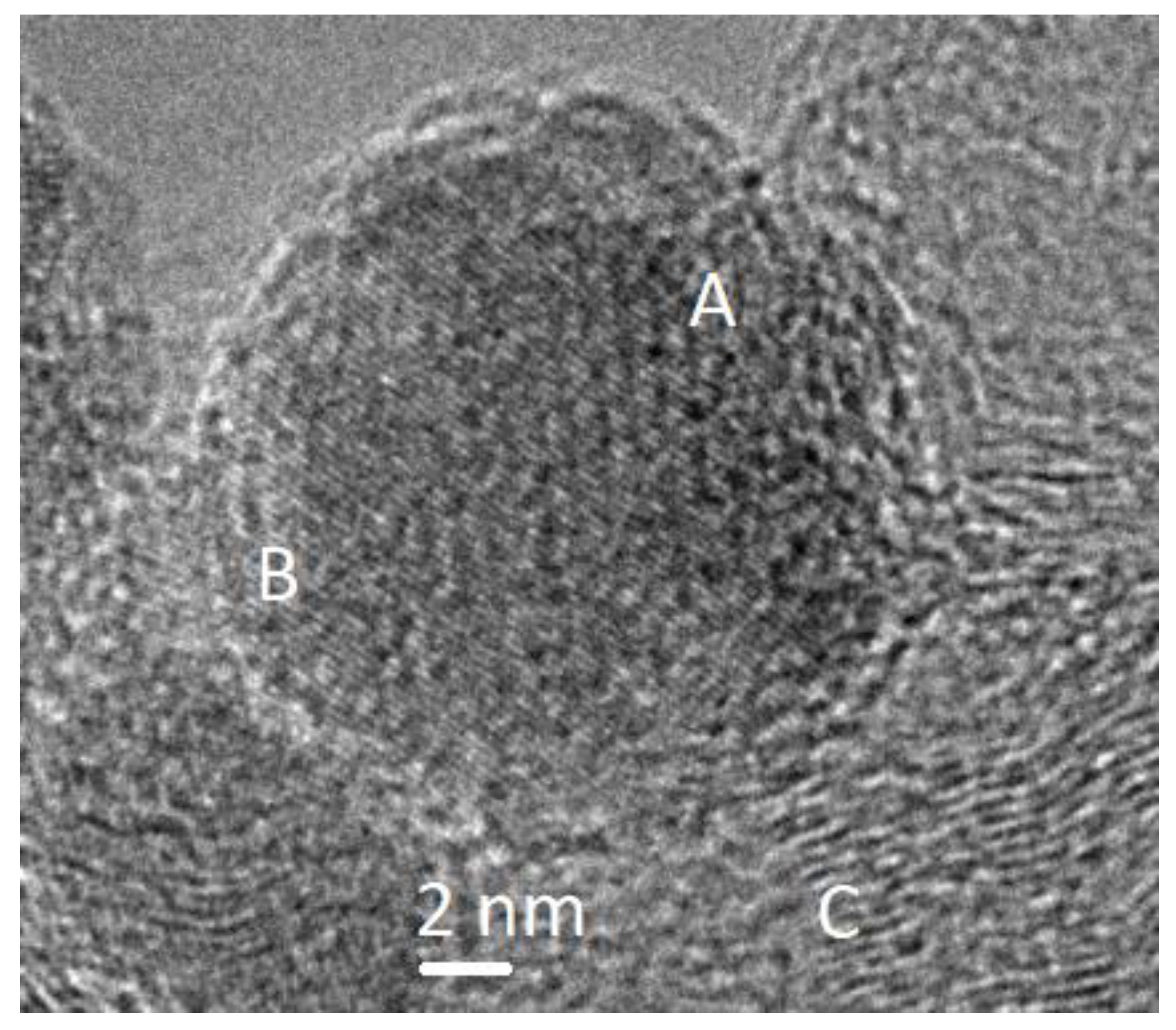
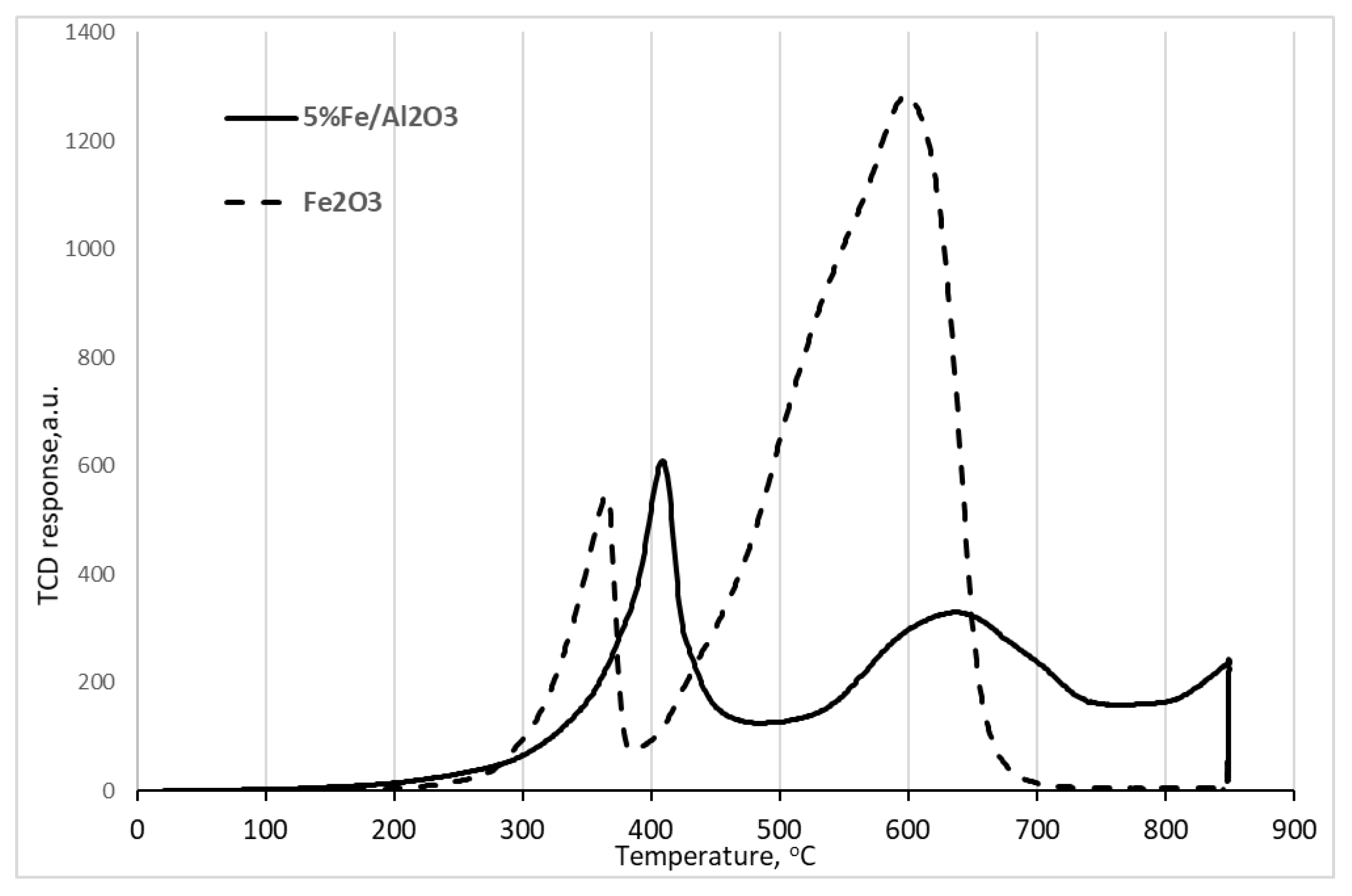
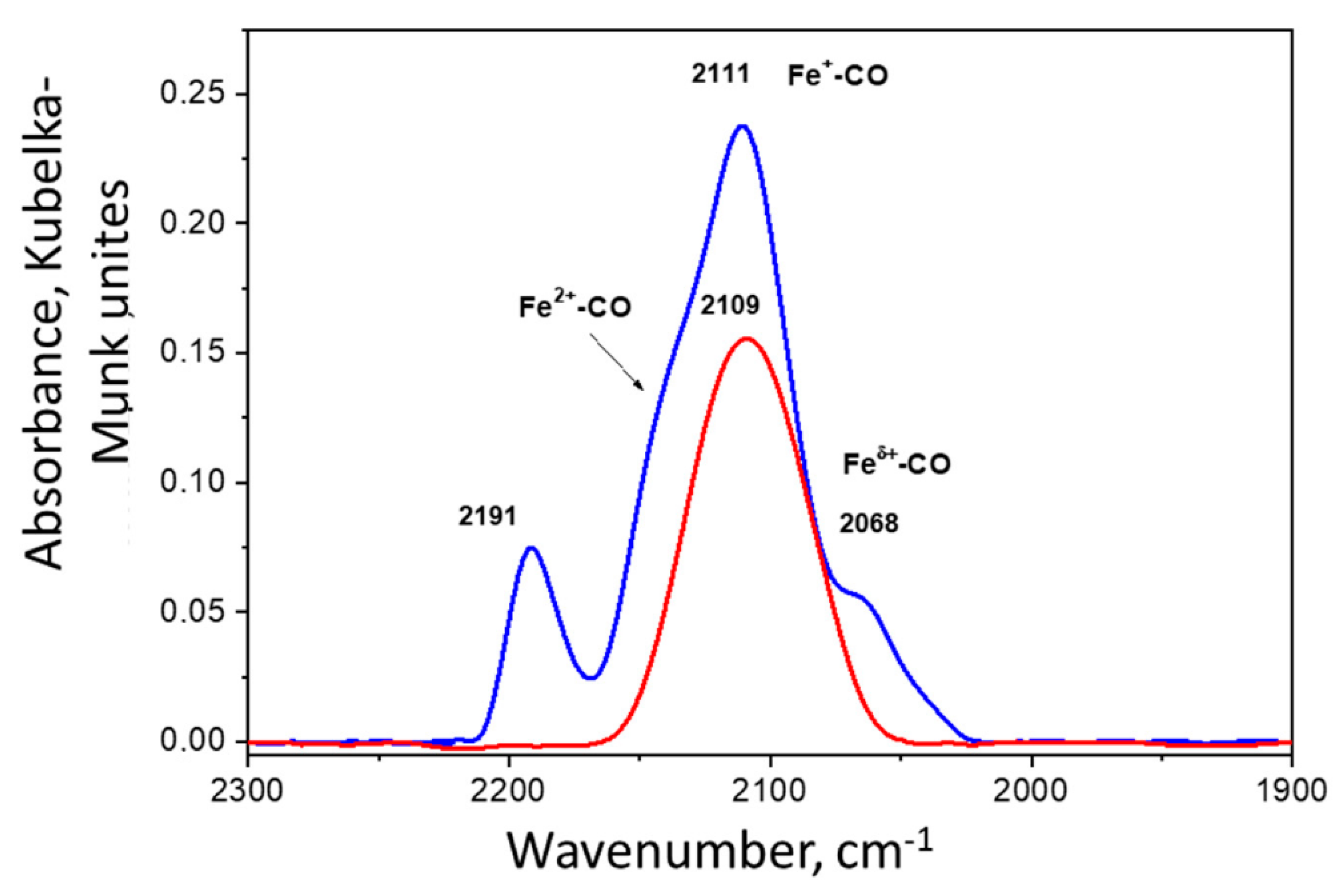
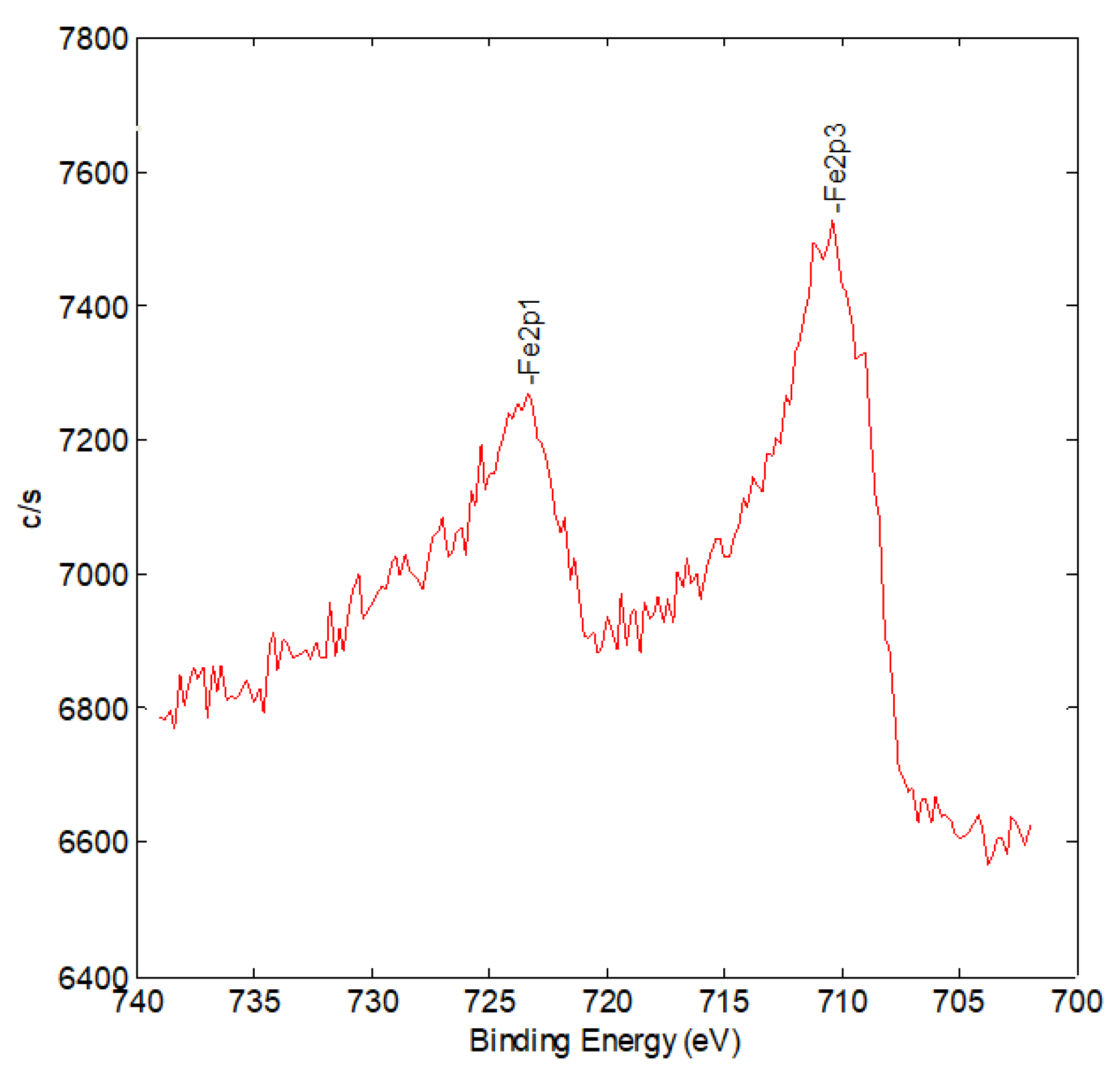
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Mikkelsen, M.; Jorgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.N.; Zhang, X.L.; Zhao, N.; Mao, F.K.; Wei, W.; Sun, Y.H. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Baiker, A. Utilization of carbon dioxide in heterogeneous catalytic synthesis. Appl. Organomet. Chem. 2000, 14, 751–762. [Google Scholar] [CrossRef]
- Omae, I. Aspects of carbon dioxide utilization. Catal. Today 2006, 115, 33–52. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical re-cycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Dai, W.L.; Luo, S.L.; Yin, S.F.; Au, C.T. The direct transformation of carbon dioxide to organic carbonates over heterogeneous catalysts. Appl. Catal. A 2009, 366, 2–12. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Gong, J. Methanation of carbon dioxide: An overview. Front. Chem. Sci. Eng. 2011, 5, 2–10. [Google Scholar]
- Hoekman, S.K.; Broch, A.; Robbins, C.; Purcell, R. CO2 Recycling by Reaction with Renewably-Generated Hydrogen. Int. J. Greenh. Gas Control 2010, 4, 44–55. [Google Scholar] [CrossRef]
- Schoder, M.; Armbruster, U.; Martin, A. Heterogen katalysierte Hydrierung von Kohlendioxid zu Methan unter erhöhten Drücken. Chem. Ing. Tech. 2013, 85, 344–352. [Google Scholar] [CrossRef]
- Schoder, M.; Armbruster, U.; Martin, A. Hydrogenation of Carbon Dioxide towards Synthetic Natural Gas—A Route to Effective Future Energy Storage. DGMK Tagungsber. Conf. Proc. 2012, 3, 39–46. [Google Scholar]
- Lange, F.; Armbruster, U.; Martin, A. Heterogeneously-Catalyzed Hydrogenation of Carbon Dioxide to Methane using RuNi Bimetallic Catalysts. Energy Technol. 2015, 3, 55–62. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Kiwi-Minsker, L.; Roger, A.-C. Effect of Ce/Zr composition and noble metal promotion on nickel based CexZr1−xO2 catalysts for carbon dioxide methanation. Appl. Catal. A 2011, 392, 36–44. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. CO2 reduction on supported Ru/Al2O3 catalysts: Cluster size dependence of product selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, D.Z. Study of bimetallic Cu–Ni/γ-Al2O3 catalysts for carbon dioxide hydrogenation. Int. J. Hydrogen Energy 1999, 24, 351–354. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Windle, C.; Perutz, R.N. Advances in molecular photocatalytic and electrocatalytic CO2 reduction. Coord. Chem. Rev. 2012, 256, 2562–2570. [Google Scholar] [CrossRef]
- Genovese, C.; Amplelli, C.; Perathoner, S.; Centi, G. Electrocatalytic conversion of CO2 to liquid fuels using nanocarbon-based electrodes. J. Energy Chem. 2013, 22, 202–213. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. The changing paradigm in CO2 utilization. J. CO2 Util. 2013, 3–4, 65–73. [Google Scholar] [CrossRef]
- Hu, B.; Guild, C.; Suib, S.L. Thermal, electrochemical, and photochemical conversion of CO2 to fuels and value-added products. J. CO2 Util. 2013, 1, 18–27. [Google Scholar] [CrossRef]
- Ganesh, I. Conversion of carbon dioxide into methanol—A potential liquid fuel: Fundamental challenges and opportunities (a review). Renew. Sustain. Energy Rev. 2014, 31, 221–257. [Google Scholar] [CrossRef]
- Martin, O.; Martin, A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferre, D.; Perez-Ramirez, J. Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation. Angew. Chem. 2016, 55, 6261–6265. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Y.; Li, F.; Lu, X.; Dai, W.; Mori, R. Fixation and conversion of CO2 using ionic liquids. Catal. Today 2006, 115, 61–69. [Google Scholar] [CrossRef]
- Sun, J.M.; Fujita, S.; Arai, M. Development in the green synthesis of cyclic carbonate from carbon dioxide using ionic liquids. J. Organomet. Chem. 2005, 690, 3490–3497. [Google Scholar] [CrossRef]
- Jessop, P.G. Homogeneous catalysis using supercritical fluids: Recent trends and systems studied. J. Supercrit. Fluids 2006, 38, 211–231. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.; Zhao, Y.; Ma, X. Hydrogenation of scCO2 to Formic Acid Catalyzed by Heterogeneous Ruthenium(III)/Al2O3 Catalysts. Chem. Lett. 2016, 45, 555–557. [Google Scholar] [CrossRef]
- Bogdan, V.I.; Pokusaeva, Y.A.; Koklin, A.E.; Savoliv, S.V.; Chernyak, S.A.; Lunin, V.V.; Kustov, L.M. Carbon Dioxide Reduction with Hydrogen on Carbon-Nanotube-Supported Catalysts under Supercritical Conditions. Energy Technol. 2019, 7, 1900174. [Google Scholar] [CrossRef]
- Chernyak, S.A.; Ivanov, A.S.; Stolbov, D.N.; Maksimov, S.V.; Maslakov, K.I.; Chernavskii, P.A.; Pokusaeva, Y.A.; Koklin, A.E.; Bogdan, V.I.; Savilov, S.V. Sintered Fe/CNT framework catalysts for CO2 hydrogenation into hydrocarbons. Carbon 2020, 168, 475–484. [Google Scholar] [CrossRef]
- Pokusaeva, Y.A.; Koklin, A.E.; Lunin, V.V.; Bogdan, V.I. CO2 hydrogenation on Fe-based catalysts doped with potassium in gas phase and under supercritical conditions. Mendeleev Commun. 2019, 29, 382–384. [Google Scholar] [CrossRef]
- Bogdan, V.I.; Koklin, A.E.; Nikolaev, S.A.; Kustov, L.M. Carbon Dioxide Hydrogenation on Au Nanoparticles Supported on TiO2, ZrO2 and Sulfated ZrO2 Under Supercritical Conditions. Top. Catal. 2016, 59, 1104–1109. [Google Scholar] [CrossRef]
- Bogdan, V.I.; Kustov, L.M. Reduction of carbon dioxide with hydrogen on a CuO–ZnO mixed catalyst under supercritical conditions. Mendeleev Commun. 2015, 25, 446–448. [Google Scholar] [CrossRef]
- Bogdan, V.I.; Koklin, A.E.; Kozak, D.O.; Kustov, L.M. Reduction of carbon dioxide by hydrogen on metal-carbon catalysts under supercritical conditions. Russ. J. Phys. Chem. A 2016, 90, 2352–2357. [Google Scholar] [CrossRef]
- Pokusaeva, Y.A.; Koklin, A.E.; Eliseev, O.L.; Kazantsev, R.V.; Bogdan, V.I. Hydrogenation of carbon oxides over the Fe-based catalysts on the carbon support. Russ. Chem. Bull. 2020, 69, 237–240. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | T, °C | CO2 Conversion, % | Selectivity, vol. % | ||
|---|---|---|---|---|---|
| CO | CH4 | CxHy * | |||
| Co/Al2O3 | 200 | 10 | 0 | 96 | 4 |
| 250 | 16 | 0 | 96 | 4 | |
| 300 | 18 | 0 | 98 | 2 | |
| 350 | 19 | 0 | 98 | 2 | |
| 400 | 19 | 0 | 98 | 2 | |
| Fe/Al2O3 | 250 | 0 | - | - | - |
| 300 | 6 | 45 | 40 | 15 | |
| 320 | 7 | 45 | 43 | 11 | |
| 350 | 11 | 40 | 44 | 16 | |
| 380 | 17 | 60 | 29 | 11 | |
| 400 | 20 | 66 | 25 | 9 | |
| Fe/C | 300 | 8 | 62 | 21 | 17 |
| 350 | 11 | 58 | 15 | 27 | |
| 400 | 13 | 73 | 10 | 17 | |
| 500 | 19 | 90 | 6 | 4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogdan, V.I.; Koklin, A.E.; Kustov, A.L.; Pokusaeva, Y.A.; Bogdan, T.V.; Kustov, L.M. Carbon Dioxide Reduction with Hydrogen on Fe, Co Supported Alumina and Carbon Catalysts under Supercritical Conditions. Molecules 2021, 26, 2883. https://doi.org/10.3390/molecules26102883
Bogdan VI, Koklin AE, Kustov AL, Pokusaeva YA, Bogdan TV, Kustov LM. Carbon Dioxide Reduction with Hydrogen on Fe, Co Supported Alumina and Carbon Catalysts under Supercritical Conditions. Molecules. 2021; 26(10):2883. https://doi.org/10.3390/molecules26102883
Chicago/Turabian StyleBogdan, Viktor I., Aleksey E. Koklin, Alexander L. Kustov, Yana A. Pokusaeva, Tatiana V. Bogdan, and Leonid M. Kustov. 2021. "Carbon Dioxide Reduction with Hydrogen on Fe, Co Supported Alumina and Carbon Catalysts under Supercritical Conditions" Molecules 26, no. 10: 2883. https://doi.org/10.3390/molecules26102883