
Microfluidic Chip-Based Induced Phase Separation Extraction as a Fast and Efficient Miniaturized Sample Preparation Method

Abstract
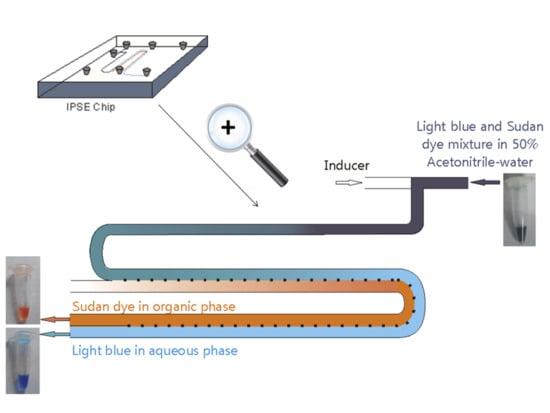
:
1. Introduction
2. Results
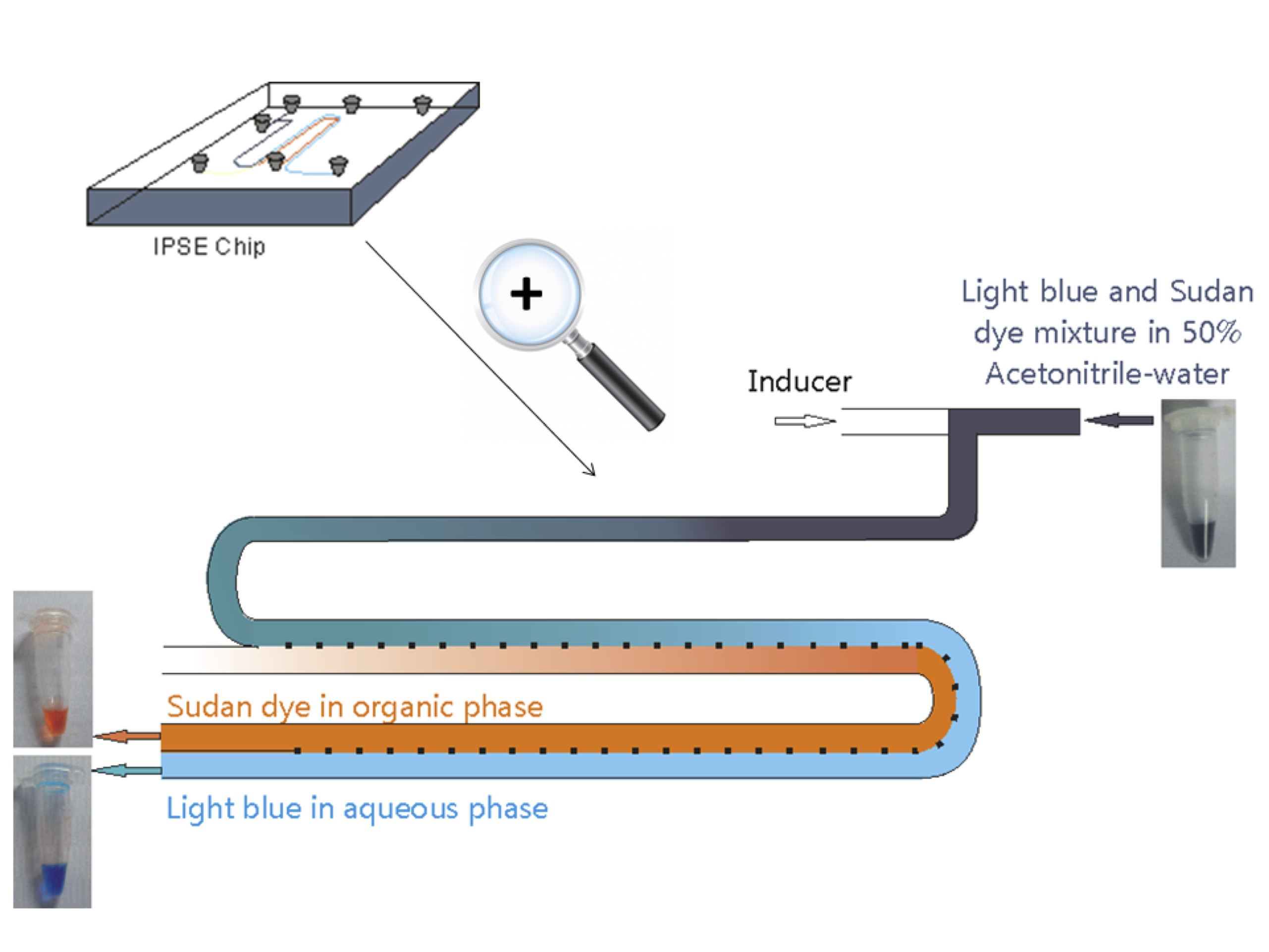
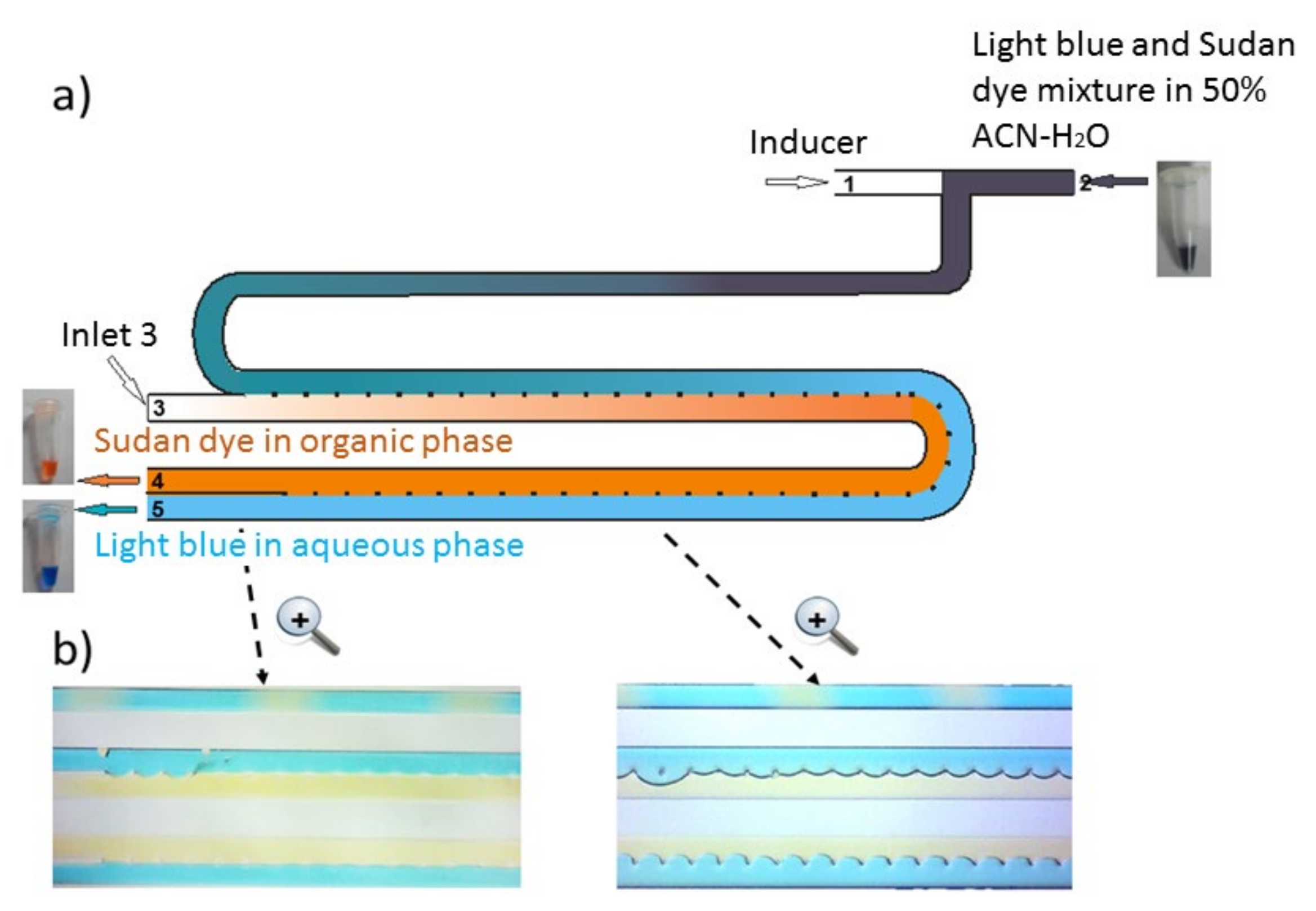
2.1. Chip Design and Proof of Principle
2.2. Inducer Optimization
2.2.1. Single Solvent Inducer
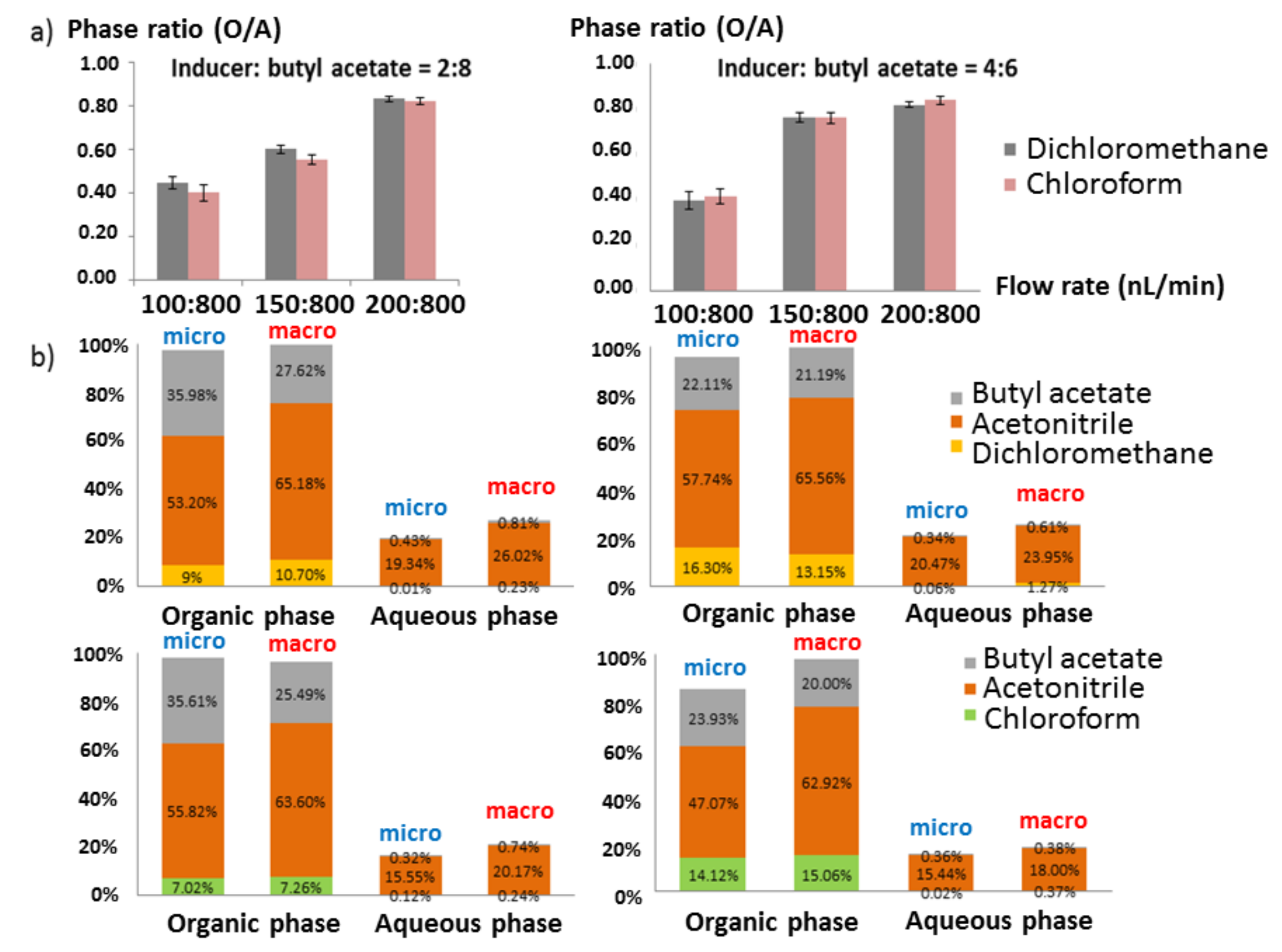
2.2.2. Mixed Solvent Inducer
2.3. Extraction Efficiency
2.3.1. Model Compounds
2.3.2. pH Effect on Acidic and Basic Compounds
2.3.3. Efficiency Comparison of Microfluidic IPSE and Microfluidic LLE
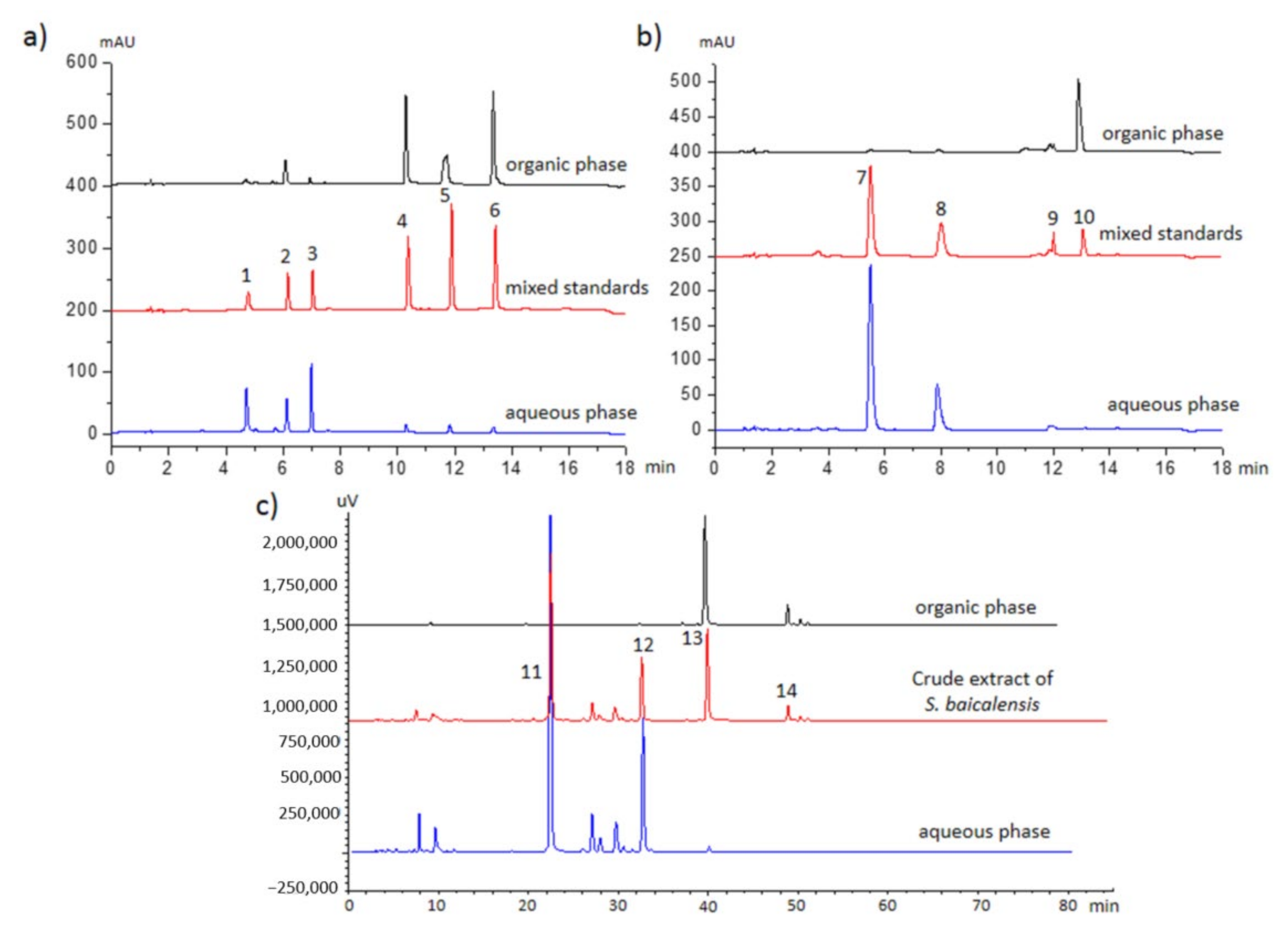
2.4. Real-Life Sample Application
3. Discussion
4. Materials and Methods
4.1. Materials and Instruments
4.2. IPSE Chip and Chip Modification
4.3. Parameter Studies
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Schug, K.A.; York, J.L. Overview of methods and considerations for handling complex samples. LCGC N. Am. 2020, 38, 241–249. [Google Scholar]
- Klein-Júnior, L.C.; Vander Heyden, Y.; Henriques, A.T. Enlarging the bottleneck in the analysis of alkaloids: A review on sample preparation in herbal matrices. Trends Anal. Chem. (TrAC) 2016, 80, 66–82. [Google Scholar] [CrossRef]
- Abedi, G.; Talebpour, Z.; Jamechenarboo, F. The survey of analytical methods for sample preparation and analysis of fragrances in cosmetics and personal care products. Trends Anal. Chem. (TrAC) 2018, 102, 41–59. [Google Scholar] [CrossRef]
- Jin, W.; Zhou, T.; Li, G. Recent advances of modern sample preparation techniques for traditional Chinese medicines. J. Chromatogr. A 2019, 1606-460377, 1–16. [Google Scholar] [CrossRef]
- Majors, R.E. Overview of sample preparation. LCGC 2015, 33, 46–51. [Google Scholar]
- Chen, X.; Cui, D.-F. Microfluidic devices for sample pretreatment and applications. Microsyst. Technol. 2009, 15, 667–676. [Google Scholar] [CrossRef]
- Cui, F.; Rhee, M.; Singh, A.; Tripathi, A. Microfluidic sample preparation for medical diagnostics. Annu. Rev. Biomed. Eng. 2015, 17, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Hosic, S.; Murthy, S.K.; Koppes, A.N. Microfluidic sample preparation for single cell analysis. Anal. Chem. 2016, 88, 354–380. [Google Scholar] [CrossRef]
- Sonker, M.; Sahore, V.; Woolley, A.T. Recent advances in microfluidic sample preparation and separation techniques for molecular biomarker analysis: A critical review. Anal. Chim. Acta 2017, 986, 1–11. [Google Scholar] [CrossRef]
- Wang, K.; Luo, G. Microflow extraction: A review of recent development. Chem. Eng. Sci. 2017, 169, 18–33. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Fu, L.-M. Recent advances and applications of micromixers. Sens. Actuators B 2018, 259, 677–702. [Google Scholar] [CrossRef]
- Tetala, K.K.R.; Swarts, J.W.; Chen, B.; Janssen, A.E.M.; van Beek, T.A. A three-phase microfluidic chip for rapid sample clean-up of alkaloids from plant extracts. Lab Chip 2009, 9, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Karami, M.; Yamini, Y.; Asl, Y.A. On-chip ion pair-based dispersive liquid-liquid extraction for quantitative determination of histamine H2 receptor antagonist drugs in human urine. Talanta 2020, 206-120235, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, C. Liquid-liquid-liquid three-phase microsystem: Hybrid slug flow-laminar flow. Lab Chip 2020, 20, 1891–1897. [Google Scholar] [CrossRef]
- Cai, Q.; Meng, J.; Ge, Y.; Gao, Y.; Zeng, Y.; Li, H.; Sun, Y. Fishing antitumor ingredients by G-quadruplex affinity from herbal extract on a three-phase-laminar-flow microfluidic chip. Talanta 2020, 220, 1–7. [Google Scholar] [CrossRef]
- Cai, Q.; Peng, H.; Meng, J.; Yan, Y.; Zeng, Y.; Li, P.C.H.; Sun, Y. Simultaneous determination of free and total paclitaxel in blood in a three-phase laminar flow microchip. J. Chromatogr. A 2020, 1627, 461391. [Google Scholar] [CrossRef]
- Foan, L.; El Sabahy, J.; Ricoul, F.; Bourlon, B.; Vignoud, S. Development of a new phase for lab-on-a-chip extraction of polycyclic aromatic hydrocarbons from water. Sens. Actuators B 2018, 255, 1039–1047. [Google Scholar] [CrossRef]
- Alidoust, M.; Yamini, Y.; Baharfar, M.; Seidi, S.; Rasouli, F. Microfluidic-enabled versatile hyphenation of electromembrane extraction and thin film solid phase microextraction. Talanta 2020, 224, 121864. [Google Scholar] [CrossRef]
- De Jong, J.; Lammertink, R.; Wessling, M. Membranes and microfluidics: A review. Lab Chip 2006, 6, 1125–1139. [Google Scholar] [CrossRef]
- Ramos Payán, M.D.; Jensen, H.; Petersen, N.J.; Hansen, S.H.; Pedersen-Bjergaard, S. Liquid-phase microextraction in a microfluidic-chip—High enrichment and sample clean-up from small sample volumes based on three-phase extraction. Anal. Chim. Acta 2012, 735, 46–53. [Google Scholar] [CrossRef]
- Yan, Z.; Du, C.; Chen, Y.; Luo, G. A novel hollow-fiber membrane embedded co-axial microdevice for simultaneous extraction and stripping. Solv. Extr. Ion Exch. 2020, 38, 103–119. [Google Scholar] [CrossRef]
- Shen, Y.; van Beek, T.A.; Zuilhof, H.; Chen, B. Hyphenation of optimized microfluidic sample preparation with nano liquid chromatography for faster and greener alkaloid analysis. Anal. Chim. Acta 2013, 797, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yang, H.; Zhang, M.; Li, S.; Chen, B.; Yao, S. Novel isolation of phytochemical compositions by phase transition extraction with acetonitrile. J. Sep. Sci. 2011, 34, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, S.; Chen, J.; Liu, G.; Chen, B.; Yao, S. Optimization of sample pretreatment methods for simultaneous determination of dolasetron and hydrodolasetron in human plasma by HPLC-ESI-MS. J. Chromatogr. Sci. 2012, 50, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Tao, Y.; Luo, D.; Feng, J.; Wang, L.; Yu, M.; Li, Y.; Covaci, A.; Mei, S. Simultaneous biomonitoring of 15 organophosphate flame retardants metabolites in urine samples by solvent induced phase transition extraction coupled with ultra-performance liquid chromatography-tandem mass spectrometry. Chemosphere 2019, 233, 724–732. [Google Scholar] [CrossRef]
- Wen, R.; Yu, M.; Jiang, L.; Feng, L.; Deng, W.; Chen, B. A proton nuclear magnetic resonance (1H NMR) investigation of NaCl-induced phase separation of acetonitrile-water mixtures. Am. J. Anal. Chem. 2017, 8, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.-T.; Cai, B.-D.; Yu, L.; Ding, J.; Feng, Y.-Q. Sensitive determination of brassinosteroids by solid phase boronate affinity labeling coupled with liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1546, 10–17. [Google Scholar] [CrossRef]
- Yoshida, M.; Akane, A.; Nishikawa, M.; Watabiki, T.; Tsuchihashi, H. Extraction of thiamylal in serum using hydrophilic acetonitrile with subzero-temperature and salting-out methods. Anal. Chem. 2004, 76, 4672–4675. [Google Scholar] [CrossRef]
- Liu, G.; Zhou, N.; Zhang, M.; Li, S.; Tian, Q.; Chen, J.; Chen, B.; Wu, Y.; Yao, S. Hydrophobic solvent induced phase transition extraction to extract drugs from plasma for high performance liquid chromatography-mass spectrometric analysis. J. Chromatogr. A 2010, 1217, 243–249. [Google Scholar] [CrossRef]
- Cai, B.-D.; Ye, E.-C.; Yuan, B.-F.; Feng, Y.-Q. Sequential solvent induced phase transition extraction for profiling of endogenous phytohormones in plants by liquid chromatography-mass spectrometry. J. Chromatogr. B 2015, 1004, 23–29. [Google Scholar] [CrossRef]
- Wen, R.; Yang, Z.; Jiang, L.; Ma, M.; Chen, B.; Wen, R.; Zeng, D.; van Beek, T.A. Rapid analysis of illegal cationic dyes in foods and surface waters using high temperature Direct Analysis in Real Time high-resolution mass spectrometry. J. Agric. Food Chem. 2018, 66, 7542–7549. [Google Scholar] [CrossRef] [PubMed]
- Žnidaršič-Plazl, P.; Plazl, I. Steroid extraction in a microchannel system-mathematical modelling and experiments. Lab Chip 2007, 7, 883–889. [Google Scholar] [CrossRef] [PubMed]
- SciFinder. Available online: https://scifinder.cas.org (accessed on 26 November 2020).
- Mu, X.; Liang, Q.; Hu, P.; Ren, K.; Wang, Y.; Luo, G. Selectively modified microfluidic chip for solvent extraction of Radix Salvia Miltiorrhiza using three-phase laminar flow to provide double liquid–liquid interface area. Microfluid. Nanofluid. 2010, 9, 365–373. [Google Scholar] [CrossRef]
- Hu, Y.; Peng, H.; Yan, Y.; Guan, S.; Wang, S.; Li, P.C.H.; Sun, Y. Integration of laminar flow extraction and capillary electrophoretic separation in one microfluidic chip for detection of plant alkaloids in blood samples. Anal. Chim. Acta 2017, 985, 121–128. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
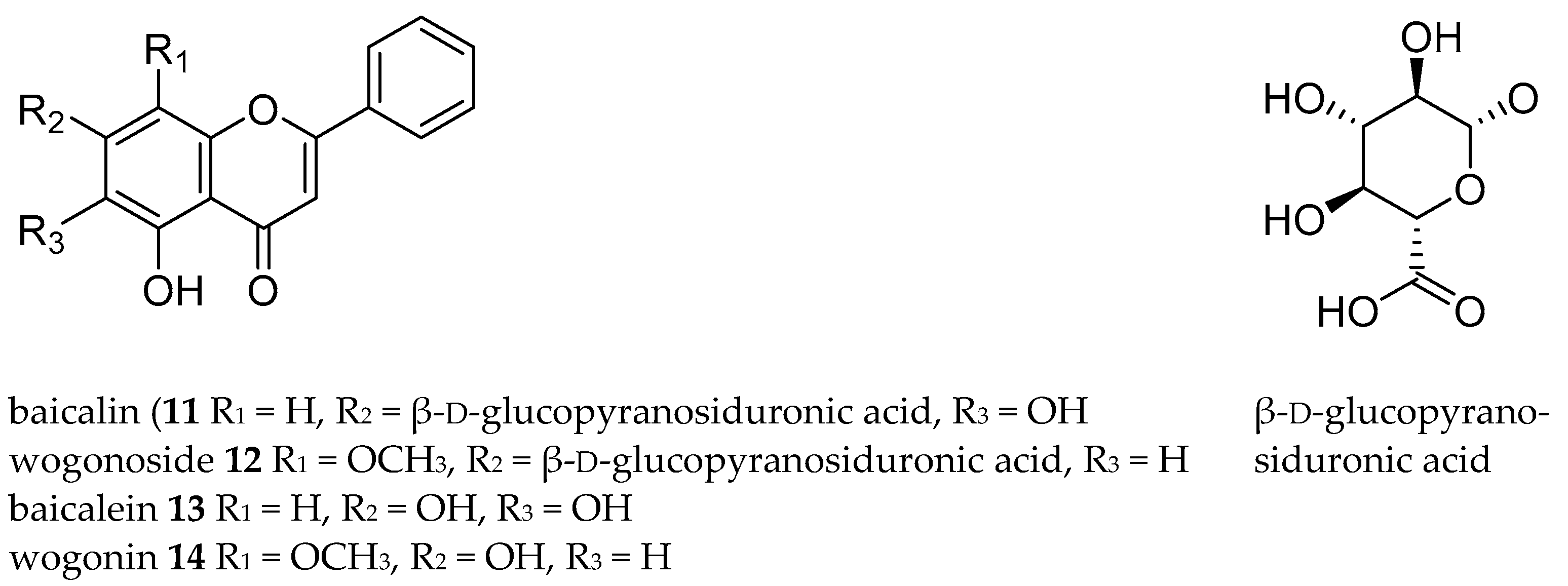{kind=link}
| Compounds | Extraction Efficiency of IPSE Chip in % (SD, n = 3) | Extraction Efficiency of IPSE in Eppendorf Tube in % (SD, n = 3) | ||
|---|---|---|---|---|
| Organic Phase | Aqueous Phase | Organic Phase | Aqueous Phase | |
| Chlorogenic acid | 10.4 (0.89) | 89.6 (1.3) | 19.1 (0.62) | 80.9 (0.64) |
| Epigallocatechin gallate | 43.2 (0.69) | 56.8 (1.0) | 45.5 (2.5) | 54.5 (2.7) |
| Rutin | 6.10 (0.21) | 93.9 (1.5) | 6.60 (0.039) | 93.4 (0.46) |
| Quercetin | 94.0 (2.2) | 6.00 (0.37) | 94.4 (0.094) | 5.60 (0.16) |
| Santonin | 90.7 (1.9) | 9.30 (0.37) | 93.9 (0.46) | 6.10 (0.11) |
| Alizarin | 96.2 (1.9) | 3.80 (0.29) | 96.8 (0.38) | 3.20 (0.10) |
| Compounds | Extraction Efficiency at pH 3.07 in % (SD, n = 3) | Extraction Efficiency at pH 9.95 in % (SD, n = 3) | ||||||
|---|---|---|---|---|---|---|---|---|
| IPSE Chip | Eppendorf Tube | IPSE Chip | Eppendorf Tube | |||||
| Organic Phase | Aqueous Phase | Organic Phase | Aqueous Phase | Organic Phase | Aqueous Phase | Organic Phase | Aqueous Phase | |
| 4-Hydroxy-benzoic acid | 78.7 (9.5) | 21.3 (0.76) | 72.7 (0.014) | 27.3 (0.46) | 1.20 (0.085) | 98.8 (0.82) | 2.20 (0.53) | 97.8 (0.91) |
| Syringic acid | 73.5 (9.2) | 26.5 (0.64) | 67.4 (2.6) | 32.6 (0.35) | 1.40 (0.056) | 98.6 (3.1) | 3.00 (0.72) | 97.0 (2.5) |
| Emetine | 47.2 (1.8) | 52.8 (4.4) | 33.6 (0.93) | 66.4 (23) | 48.5 (10) | 51.6 (3.9) | 43.1 (4.5) | 56.9 (2.4) |
| Vincamine | 19.4 (0.66) | 80.6 (2.0) | 16.7 (0.18) | 83.4 (0.41) | 94.00 (7.8) | 5.96 (1.8) | 97.2 (1.2) | 2.81 (0.66) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Chen, B.; Zuilhof, H.; van Beek, T.A. Microfluidic Chip-Based Induced Phase Separation Extraction as a Fast and Efficient Miniaturized Sample Preparation Method. Molecules 2021, 26, 38. https://doi.org/10.3390/molecules26010038
Shen Y, Chen B, Zuilhof H, van Beek TA. Microfluidic Chip-Based Induced Phase Separation Extraction as a Fast and Efficient Miniaturized Sample Preparation Method. Molecules. 2021; 26(1):38. https://doi.org/10.3390/molecules26010038
Chicago/Turabian StyleShen, Yao, Bo Chen, Han Zuilhof, and Teris A. van Beek. 2021. "Microfluidic Chip-Based Induced Phase Separation Extraction as a Fast and Efficient Miniaturized Sample Preparation Method" Molecules 26, no. 1: 38. https://doi.org/10.3390/molecules26010038