
Transamidation of Amides and Amidation of Esters by Selective N–C(O)/O–C(O) Cleavage Mediated by Air- and Moisture-Stable Half-Sandwich Nickel(II)–NHC Complexes


Abstract
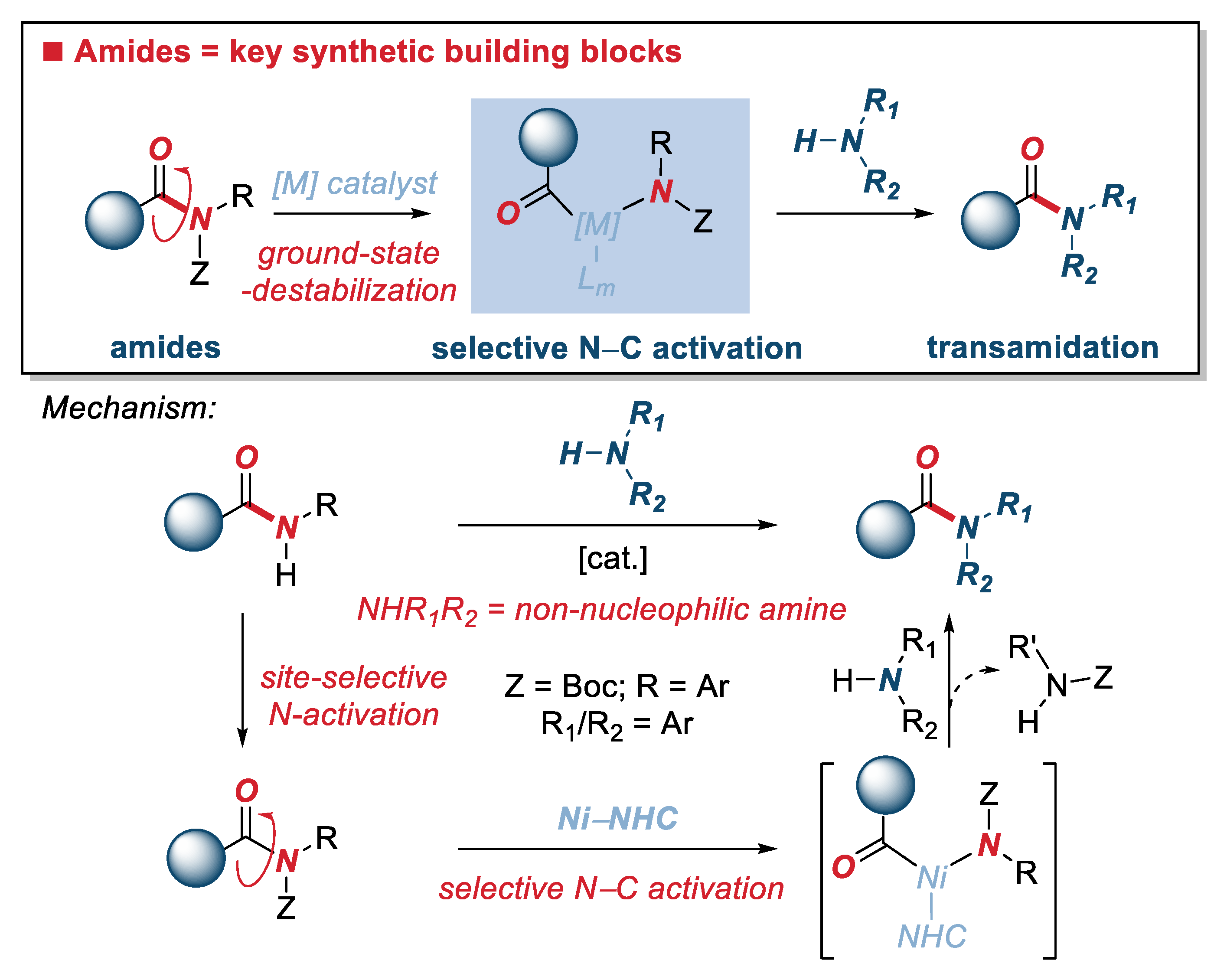
:1. Introduction
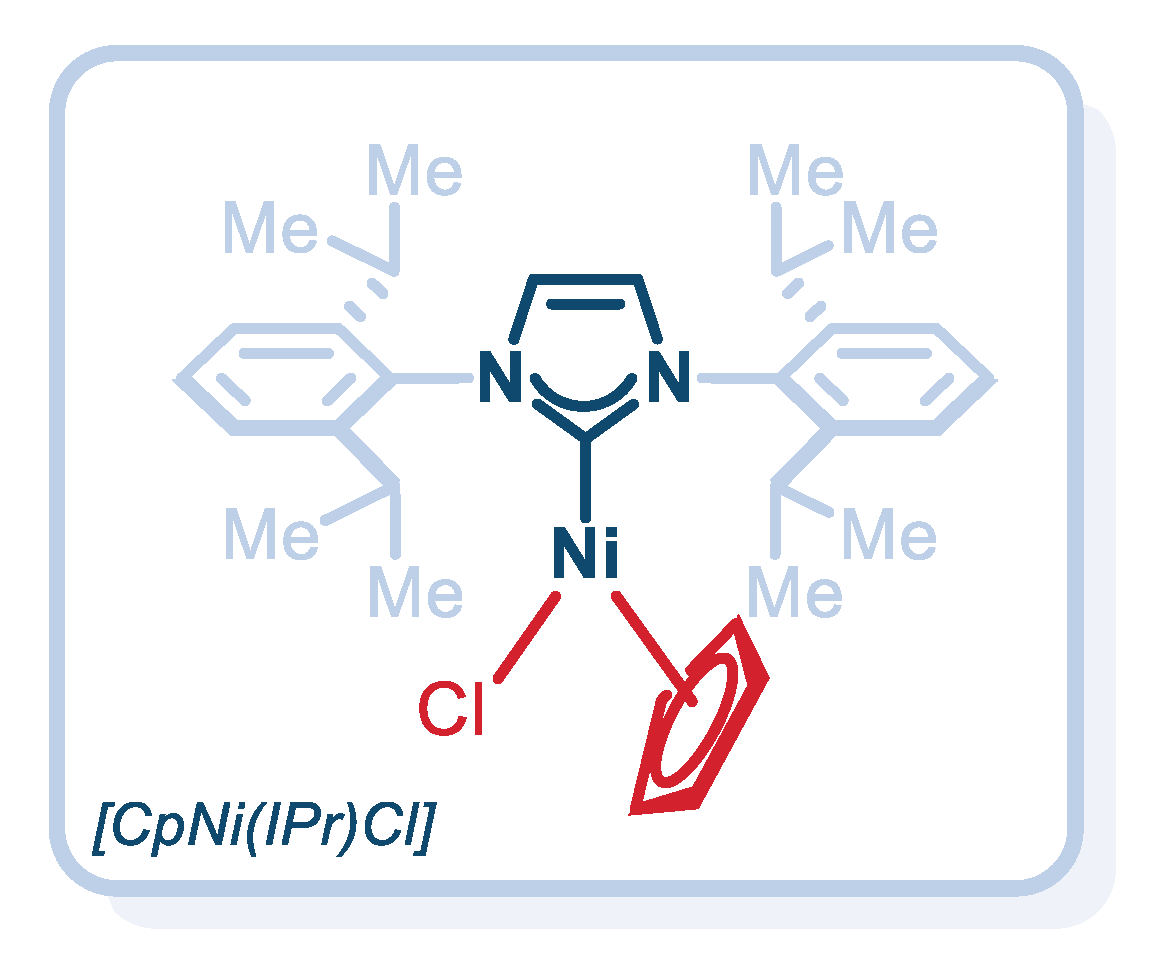
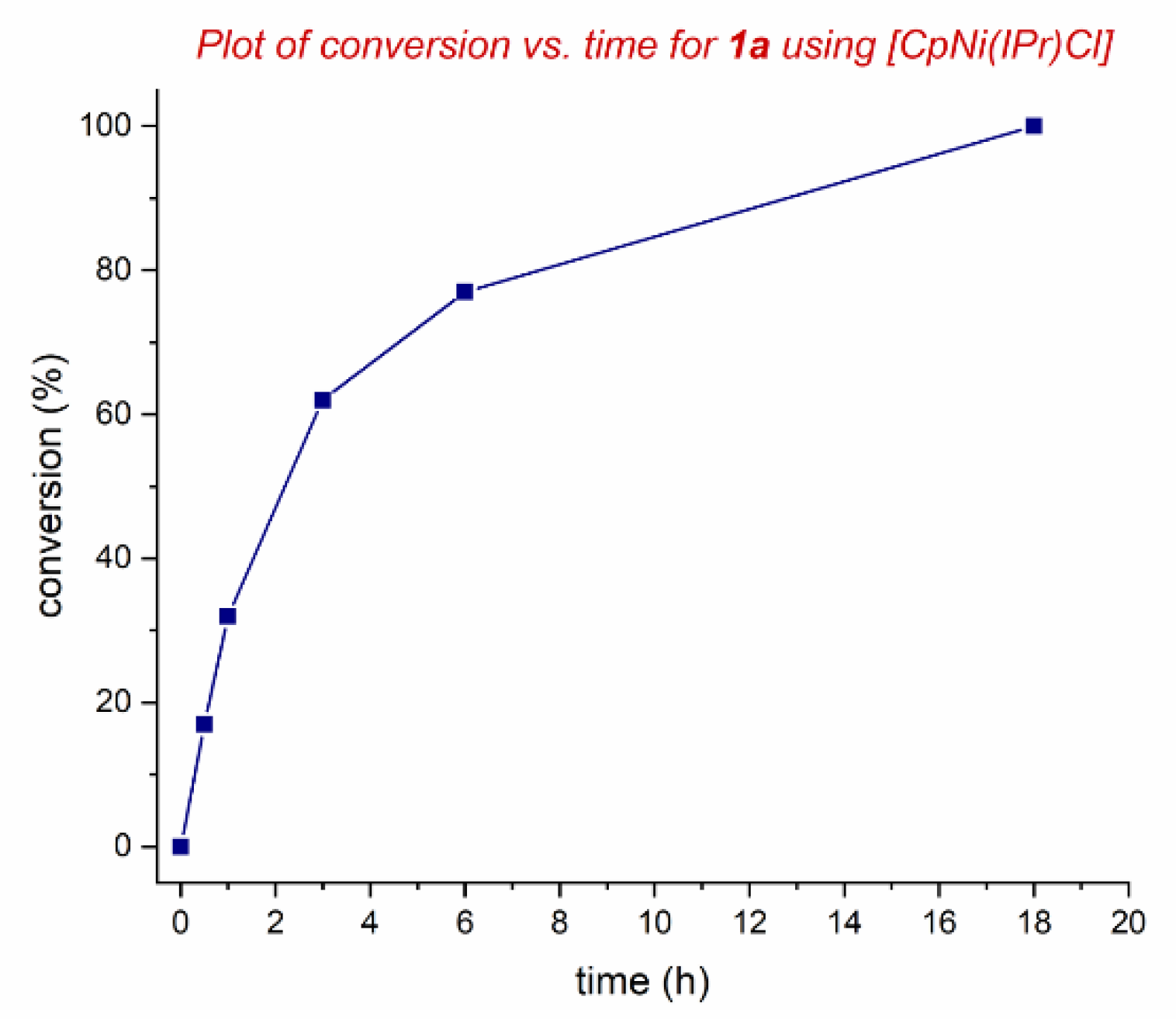
2. Results
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Hughes, B. Amino Acids, Peptides and Proteins in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef] [PubMed]
- Bryan, M.C.; Dunn, P.J.; Entwistle, D.; Gallou, F.; Koenig, S.G.; Hayler, J.D.; Hickey, M.R.; Hughes, S.; Kopach, M.E.; Moine, G.; et al. Key Green Chemistry research areas from a pharmaceutical manufacturers’ perspective revisited. Green Chem. 2018, 20, 5082–5103. [Google Scholar] [CrossRef] [Green Version]
- Loomis, W.D.; Stumpf, P.K. Transamination and Transamidation. In Nitrogen Metabolism; Allen, E.K., Allen, O.N., Böttger, I., Caspersson, T., Dillemann, G., Engel, H., Fischer, H., Guggenheim, M., Haas, P., Haurowitz, F., et al., Eds.; Springer: Berlin, Germany, 1958. [Google Scholar]
- De Figueiredo, R.M.; Suppo, J.S.; Campagne, J.M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef]
- Ojeda-Porras, A.; Gamba-Sanchez, D. Recent Developments in Amide Synthesis Using Nonactivated Starting Materials. J. Org. Chem. 2016, 81, 11548–11555. [Google Scholar] [CrossRef]
- Acosta-Guzmán, P.; Mateus-Gómez, A.; Gamba-Sánchez, D. Direct Transamidation Reactions: Mechanism and Recent Advances. Molecules 2018, 23, 2382. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Szostak, M. Non-Classical Amide Bond Formation: Transamidation and Amidation of Activated Amides and Esters by Selective N–C/O–C Cleavage. Synthesis 2020, 52, 2579–2599. [Google Scholar] [CrossRef]
- Li, G.; Ma, S.; Szostak, M. Amide Bond Activation: The Power of Resonance. Trends Chem. 2020, 2, 914–928. [Google Scholar] [CrossRef]
- Takise, R.; Muto, K.; Yamaguchi, J. Cross-coupling of aromatic esters and amides. Chem. Soc. Rev. 2017, 46, 5864–5888. [Google Scholar] [CrossRef]
- Dander, J.E.; Garg, N.K. Breaking Amides using Nickel Catalysis. ACS Catal. 2017, 7, 1413–1423. [Google Scholar] [CrossRef]
- Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Reversible Twisting of Primary Amides via Ground State N–C(O) Destabilization: Highly Twisted Rotationally Inverted Acyclic Amides. J. Am. Chem. Soc. 2018, 140, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Ielo, L.; Pace, V.; Holzer, W.; Rahman, M.M.; Meng, G.; Szostak, R.; Szostak, M. Electrophilicity Scale of Activated Amides: 17O NMR and 15N NMR Chemical Shifts of Acyclic Twisted Amides in N−C(O) Cross-Coupling. Chem. Eur. J. 2020, 26, 16246–16250. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.L.; Yamano, M.M.; Zhou, Y.; Anthony, S.M.; Garg, N.K. A two-step approach to achieve secondary amide transamidation enabled by nickel catalysis. Nat. Commun. 2016, 7, 11554. [Google Scholar] [CrossRef] [PubMed]
- Dander, J.E.; Baker, E.L.; Garg, N.K. Nickel-catalyzed transamidation of aliphatic amide derivatives. Chem. Sci. 2017, 8, 6433–6438. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Lei, P.; Szostak, M. A General Method for Two-Step Transamidation of Secondary Amides Using Commercially Available, Air- and Moisture-Stable Palladium/NHC (N-Heterocyclic Carbene) Complexes. Org. Lett. 2017, 19, 2158–2161. [Google Scholar] [CrossRef]
- Shi, S.; Szostak, M. Pd–PEPPSI: A general Pd–NHC precatalyst for Buchwald–Hartwig cross-coupling of esters and amides (transamidation) under the same reaction conditions. Chem. Commun. 2017, 53, 10584–10587. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Li, G.; Nolan, S.P.; Szostak, M. [Pd(NHC)(acac)Cl]: Well-Defined, Air-Stable, and Readily Available Precatalysts for Suzuki and Buchwald-Hartwig Cross-coupling (Transamidation) of Amides and Esters by N-C/O-C Activation. Org. Lett. 2019, 21, 3304–3309. [Google Scholar] [CrossRef]
- Li, G.; Zhou, T.; Poater, A.; Cavallo, L.; Nolan, S.P.; Szostak, M. Buchwald-Hartwig cross-coupling of amides (transamidation) by selective N–C(O) cleavage mediated by air- and moisture-stable [Pd(NHC)(allyl)Cl] precatalysts: Catalyst evaluation and mechanism. Catal. Sci. Technol. 2020, 10, 710–716. [Google Scholar] [CrossRef]
- Ben Halima, T.; Vandavasi, J.K.; Shkoor, M.; Newman, S.G. A Cross-Coupling Approach to Amide Bond Formation from Esters. ACS Catal. 2017, 7, 2176–2180. [Google Scholar] [CrossRef]
- Halima, B.T.; Masson-Makdissi, J.; Newman, S.G. Nickel-Catalyzed Amide Bond Formation from Methyl Esters. Angew. Chem. Int. Ed. 2018, 57, 12925–12929. [Google Scholar] [CrossRef]
- Zheng, Y.L.; Newman, S.G. Methyl Esters as Cross-Coupling Electrophiles: Direct Synthesis of Amide Bonds. ACS Catal. 2019, 9, 4426–4433. [Google Scholar] [CrossRef]
- Li, G.; Ji, C.L.; Hong, X.; Szostak, M. Highly Chemoselective, Transition-Metal-Free Transamidation of Unactivated Amides and Direct Amidation of Alkyl Esters by N–C/O–C Cleavage. J. Am. Chem. Soc. 2019, 141, 11161–11172. [Google Scholar] [CrossRef] [PubMed]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananikov, V.P. Nickel: The “Spirited Horse” of Transition Metal Catalysis. ACS Catal. 2015, 5, 1964–1971. [Google Scholar] [CrossRef]
- Diccianni, J.B.; Diao, T. Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends Chem. 2019, 1, 830–844. [Google Scholar] [CrossRef]
- Danopoulos, A.A.; Simler, T.; Braunstein, P. N-Heterocyclic Carbene Complexes of Copper, Nickel, and Cobalt. Chem. Rev. 2019, 119, 3730–3961. [Google Scholar] [CrossRef]
- Henrion, M.; Ritleng, V.; Chetcuti, M.J. Nickel N-Heterocyclic Carbene-Catalyzed C–C Bond Formation: Reactions and Mechanistic Aspects. ACS Catal. 2015, 5, 1283–1302. [Google Scholar] [CrossRef]
- Ritleng, V.; Henrion, M.; Chetcuti, M.J. Nickel N-Heterocyclic Carbene-Catalyzed C–Heteroatom Bond Formation, Reduction, and Oxidation: Reactions and Mechanistic Aspects. ACS Catal. 2016, 6, 890–906. [Google Scholar] [CrossRef]
- Banach, Ł.; Guńka, P.A.; Zachara, J.; Buchowicz, W. Half-sandwich Ni(II) complexes [Ni(Cp)(X)(NHC)]: From an underestimated discovery to a new chapter in organonickel chemistry. Coord. Chem. Rev. 2019, 389, 19–58. [Google Scholar] [CrossRef]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-Heterocyclic Carbene Complexes in C–H Activation Reactions. Chem. Rev. 2020, 120, 1981–2048. [Google Scholar] [CrossRef]
- Ritleng, V.; Oertel, A.M.; Chetcuti, M.J. Half-sandwich NHC-nickel(ii) complexes as pre-catalysts for the fast Suzuki coupling of aryl halides: A comparative study. Dalton Trans. 2010, 39, 8153–8160. [Google Scholar] [CrossRef] [PubMed]
- Oertel, A.M.; Ritleng, V.; Burr, L.; Chetcuti, M.J. Synthesis and Structural Characterization of Half-Sandwich Nickel Complexes Bearing Two Different N-Heterocyclic Carbene Ligands. Organometallics 2011, 30, 6685–6691. [Google Scholar] [CrossRef]
- Oertel, A.M.; Ritleng, V.; Chetcuti, M.J. Synthesis and Catalytic Activity in Suzuki Coupling of Nickel Complexes Bearing n-Butyl- and Triethoxysilylpropyl-Substituted NHC Ligands: Toward the Heterogenization of Molecular Catalysts. Organometallics 2012, 31, 2829–2840. [Google Scholar] [CrossRef]
- Henrion, M.; Chetcuti, M.J.; Ritleng, V. From acetone metalation to the catalytic α-arylation of acyclic ketones with NHC–nickel(ii) complexes. Chem. Commun. 2014, 50, 4624–4627. [Google Scholar] [CrossRef]
- Bheeter, L.P.; Henrion, M.; Brelot, L.; Darcel, C.; Chetcuti, M.J.; Sortais, J.B.; Ritleng, V. Hydrosilylation of Aldehydes and Ketones Catalyzed by an N-Heterocyclic Carbene-Nickel Hydride Complex under Mild Conditions. Adv. Synth. Catal. 2012, 354, 2619–2624. [Google Scholar] [CrossRef]
- Bheeter, L.P.; Henrion, M.; Chetcuti, M.J.; Darcel, C.; Ritleng, V.; Sortais, J.B. Cyclopentadienyl N-heterocyclic carbene–nickel complexes as efficient pre-catalysts for the hydrosilylation of imines. Catal. Sci. Technol. 2013, 3, 3111–3116. [Google Scholar] [CrossRef]
- Oertel, A.M.; Freudenreich, J.; Gein, J.; Ritleng, V.; Veiros, L.F.; Chetcuti, M.J. Intramolecular Nitrile C–H Bond Activation in Nickel NHC Complexes: A Route to New Nickelacycles. Organometallics 2011, 30, 3400–3411. [Google Scholar] [CrossRef]
- Kelly, R.A.; Scott, N.M.; Díez-González, S.; Stevens, E.D.; Nolan, S.P. Simple Synthesis of CpNi(NHC)Cl Complexes (Cp=Cyclopentadienyl; NHC=N-Heterocyclic Carbene). Organometallics 2005, 24, 3442–3447. [Google Scholar] [CrossRef]
- Martin, A.R.; Makida, Y.; Meiries, S.; Slawin, A.M.Z.; Nolan, S.P. Enhanced Activity of [Ni(NHC)CpCl] Complexes in Arylamination Catalysis. Organometallics 2013, 32, 6265–6270. [Google Scholar] [CrossRef]
- Buchspies, J.; Rahman, M.; Szostak, M. Suzuki–Miyaura Cross-Coupling of Amides Using Well-Defined, Air- and Moisture-Stable Nickel/NHC (NHC=N-Heterocyclic Carbene) Complexes. Catalysts 2020, 10, 372. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
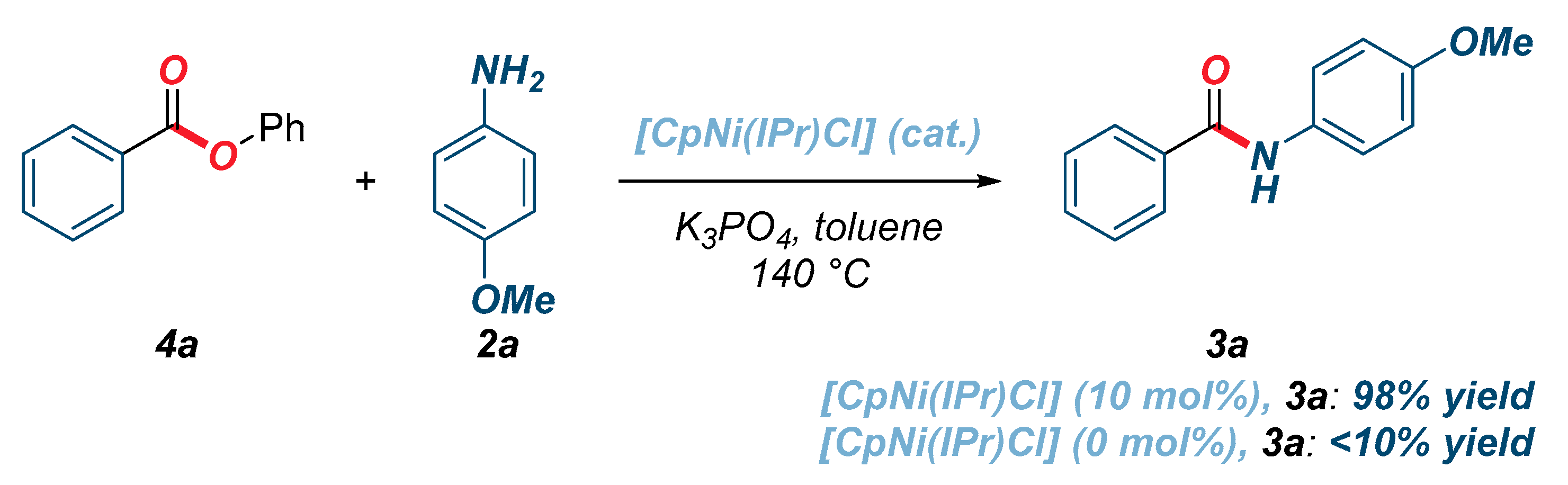{kind=link}
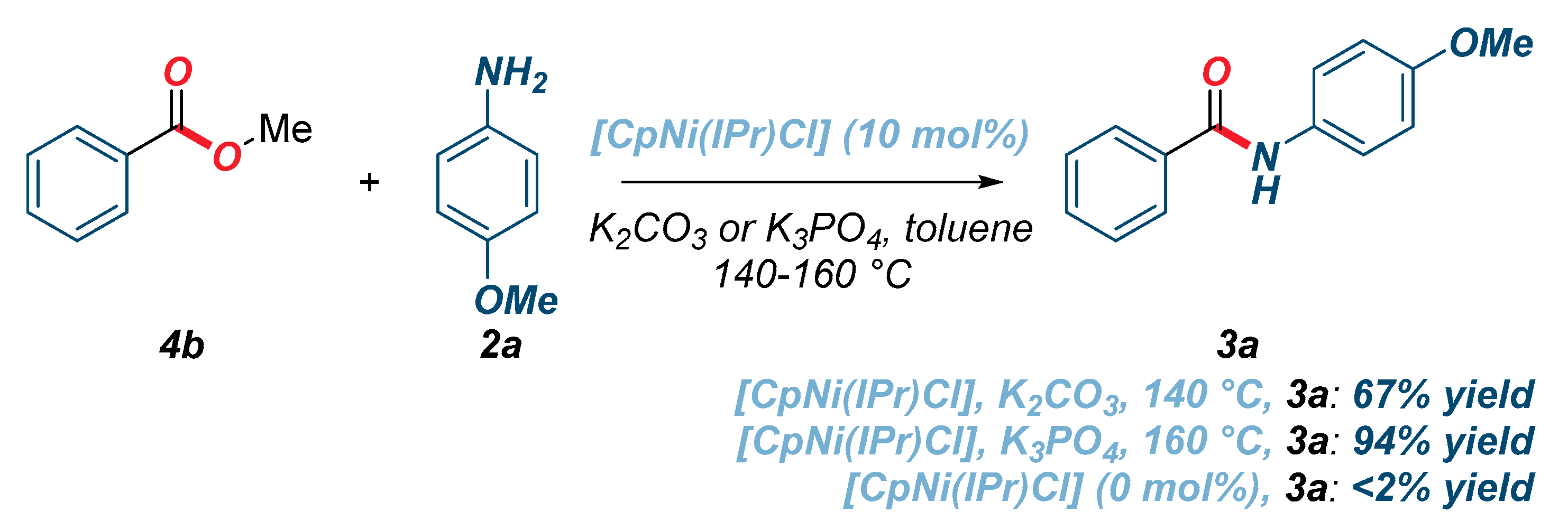{kind=link}

| Entry | Catalyst | [Ni] (mol%) | Base | Solvent | Yield (%) 2 |
|---|---|---|---|---|---|
| 1 | [CpNi(IPr)Cl] | 10 | K2CO3 | toluene | >98 |
| 2 | [CpNi(IPr)Cl] | 10 | K3PO4 | toluene | >98 |
| 3 | [CpNi(IPr)Cl] | 5 | K2CO3 | toluene | 74 |
| 4 | [CpNi(IPr)Cl] | 5 | K3PO4 | toluene | 52 |
| 5 | [CpNi(IPr)Cl] | - | K2CO3 | toluene | <10 |
| 6 | [CpNi(IPr)Cl] | - | K3PO4 | toluene | <10 |

| Entry | Amide | Ar-NH2 | 3 | Yield (%) 2 |
|---|---|---|---|---|
| 1 | C6H5 | 4-MeO-C6H4 | 3a | 98 |
| 2 | C6H5 | 4-Me-C6H4 | 3b | 97 |
| 3 | C6H5 | 2-Me-C6H4 | 3c | 77 |
| 4 | C6H5 | 3,5-Me2-C6H3 | 3d | 71 |
| 5 | C6H5 | 4-F-C6H4 | 3e | 64 |
| 6 | C6H5 | 4-CF3-C6H4 | 3f | 43 |

| Entry | Amide | Ar-NH2 | 3 | Yield (%) 2 |
|---|---|---|---|---|
| 1 | n-C9H19 | 4-MeO-C6H4 | 3g | 92 |
| 2 | Cyclohexyl | 4-MeO-C6H4 | 3h | 98 |
| 3 | 4-Me-C6H4 | 4-MeO-C6H4 | 3i | 86 |
| 4 | 4-MeO-C6H4 | 4-MeO-C6H4 | 3j | 62 |
| 5 | 4-CF3-C6H4 | 4-MeO-C6H4 | 3k | 62 |
| 6 | Ph-CH=CH | 4-MeO-C6H4 | 3l | 73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchspies, J.; Rahman, M.M.; Szostak, M. Transamidation of Amides and Amidation of Esters by Selective N–C(O)/O–C(O) Cleavage Mediated by Air- and Moisture-Stable Half-Sandwich Nickel(II)–NHC Complexes. Molecules 2021, 26, 188. https://doi.org/10.3390/molecules26010188
Buchspies J, Rahman MM, Szostak M. Transamidation of Amides and Amidation of Esters by Selective N–C(O)/O–C(O) Cleavage Mediated by Air- and Moisture-Stable Half-Sandwich Nickel(II)–NHC Complexes. Molecules. 2021; 26(1):188. https://doi.org/10.3390/molecules26010188
Chicago/Turabian StyleBuchspies, Jonathan, Md. Mahbubur Rahman, and Michal Szostak. 2021. "Transamidation of Amides and Amidation of Esters by Selective N–C(O)/O–C(O) Cleavage Mediated by Air- and Moisture-Stable Half-Sandwich Nickel(II)–NHC Complexes" Molecules 26, no. 1: 188. https://doi.org/10.3390/molecules26010188