Search for ABCB1 Modulators Among 2-Amine-5-Arylideneimidazolones as a New Perspective to Overcome Cancer Multidrug Resistance

 ,
,  , , and
, , and
Abstract
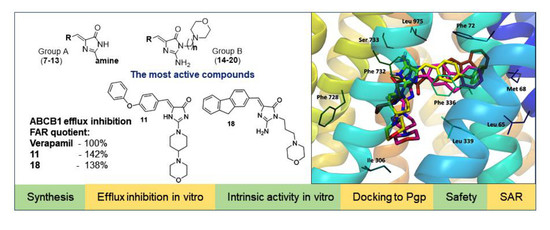
:
1. Introduction
2. Results
2.1. Synthesis
2.2. Biological Screening
2.2.1. The Rhodamine 123 Accumulation Assay
2.2.2. Cancer Cytotoxicity Assays
2.2.3. Influence on Cancer Cell Proliferation
2.3. Insight into Molecular Mechanisms of the Efflux Pump Modulation
2.3.1. Intrinsic Activity toward ABCB1 In Vitro
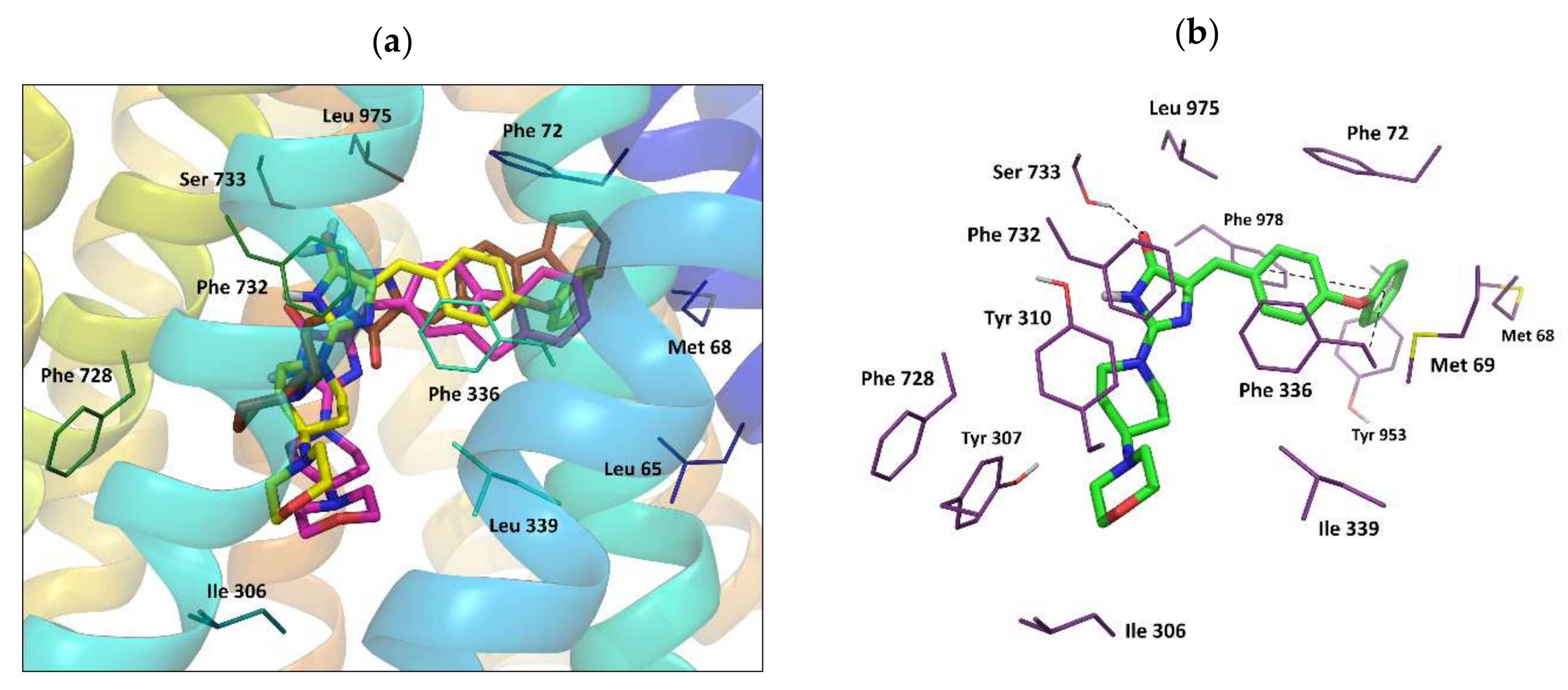
2.3.2. Molecular Modeling
Homology Model of Human Pgp
Docking Results Analysis
2.4. Safety In Vitro
3. Discussion
4. Materials and Methods
4.1. Chemistry
General Procedure for Synthesis of Final Products (7–15,17–18,20)
(Z)-5-(Biphen-4-ylmethylene)-2-morpholino-3H-imidazol-4(5H)-one hydrochloride (7)
(Z)-5-(Biphen-4-ylmethylene)-2-(4-morpholinopiperidin-1-yl)-imidazol-4(5H)-one hydrochloride (8)
(Z)-5-(Biphen-4-ylmethylene)-2-(4-(2-morpholinoethyl)piperazin-1-yl)-imidazol-4(5H)-one hydrochloride (9)
(Z)-5-(3-Phenoxybenzylidene)-2-(4-morpholinopiperidin-1-yl)-imidazol-4(5H)-one hydrochloride (10)
(Z)-5-(4-Phenoxybenzylidene)-2-(4-morpholinopiperidin-1-yl)-imidazol-4(5H)-one hydrochloride (11)
(Z)-5-((9H-Fluoren-2-yl)methylene)-2-(4-morpholinopiperidin-1-yl)-imidazol-4(5H)-one hydrochloride (12)
(Z)-5-(Anthracen-10-ylmethylene)-2-(4-morpholinopiperidin-1-yl)-imidazol-4(5H)-one hydrochloride (13)
(Z)-5-(Biphen-4-yl)-2-amino-3-(2-morpholinoethyl)-imidazol-4(5H)-one hydrochloride (14)
(Z)-5-(Biphen-4-ylmethylene)-2-amino-3-(3-morpholinopropyl)-imidazol-4(5H)-one hydrochloride (15)
(Z)-5-(4-Phenoxybenzylidene)-2-amino-3-(3-morpholinopropyl)-imidazol-4(5H)-one hydrochloride (17)
(Z)-5-((9H-Fluoren-2-yl)methylene)-2-amino-3-(3-morpholinopropyl)-imidazol-4(5H)-one hydrochloride (18)
(Z)-5-(Phenanthren-9-ylmethylene)-2-amino-3-(3-morpholinopropyl)-imidazol-4(5H)-one hydrochloride (20)
4.2. Biological Assays in Mouse T-Lymphoma Cells
4.2.1. Cell Lines
4.2.2. Evaluation of Rhodamine 123 Retention by Flow Cytometry
4.2.3. Cytotoxicity and Antiproliferative Effect
4.3. Intrinsic Activity Toward Pgp In Vitro
4.4. Molecular Modeling
4.4.1. Homology Modeling
4.4.2. Induced Fit Docking of Verapamil and Synthesized Compounds
4.5. Safety In Vitro
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ren, F.; Shen, J.; Shi, H.; Hornicek, F.J.; Kan, Q.; Duan, Z. Novel mechanisms and approaches to overcome multidrug resistance in the treatment of ovarian cancer. Biochim. Biophys. Acta 2016, 1866, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Aynacioglu, A.; Bilir, A.; Kadomatsu, K. Dual inhibition of P-glycoprotein and midkine may increase therapeutic effects of anticancer drugs. Med. Hypotheses 2017, 107, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, S.; Meng, X.; Shang, H.; Guan, Y. Inhibition of mdr1 by G-quadruplex oligonucleotides and reversal of paclitaxel resistance in human ovarian cancer cells. Tumour Biol. 2015, 36, 6433–6443. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef]
- Hano, M.; Tomasova, L.; Seres, M.; Pavlikova, L.; Breier, A.; Sulova, Z. Interplay between P-glycoprotein expression and resistance to endoplasmic reticulum stressors. Molecules 2018, 23, 337. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 446, 749–757. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281. [Google Scholar] [CrossRef]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; De Lourdes Bastos, M.; Remiao, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharm. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef]
- Padowski, J.M.; Pollack, G.M. Pharmacokinetic and pharmacodynamics implications of P-glycoprotein modulation. Methods Mol. Biol. 2010, 596, 359–384. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or evade multidrug resistance P-glycoprotein in cancer treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Yang, Z.; Tang, S.; Han, Y.; Ma, J. Effects of P-glycoprotein and its inhibitors on apoptosis in K562 cells. Molecules 2014, 19, 13061–13075. [Google Scholar] [CrossRef]
- Li, X.; Li, J.P.; Yuan, H.Y.; Gao, X.; Qu, X.J.; Xu, W.F.; Tang, W. Recent advances in P-glycoprotein-mediated multidrug resistance reversal mechanisms. Methods Find. Exp. Clin. Pharm. 2007, 29, 607–617. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Ozols, R.F.; Cunnion, R.E.; Klecker, R.W., Jr.; Hamilton, T.C.; Ostchega, Y.; Parrillo, J.E.; Young, R.C. Verapamil and adriamycin in the treatment of drug-resistant ovarian cancer patients. J. Clin. Oncol. 1987, 5, 641–647. [Google Scholar] [CrossRef]
- Dyląg, T.; Zygmunt, M.; Maciąg, D.; Handzlik, J.; Bednarski, M.; Filipek, B.; Kieć-Kononowicz, K. Synthesis and evaluation of in vivo activity of diphenylhydantoin basic derivatives. Eur. J. Med. Chem. 2004, 39, 1013–1027. [Google Scholar] [CrossRef]
- Lu, H.B.; Kong, D.J.; Wu, B.; Wang, S.; Wang, Y. Synthesis and evaluation of anti-inflammatory and antitussive activity of hydantion derivatives. Lett. Drug Des. Discov. 2012, 9, 638–642. [Google Scholar] [CrossRef]
- Handzlik, J.; Bojarski, A.J.; Satała, G.; Kubacka, M.; Sadek, B.; Ashoor, A.; Siwek, A.; Więcek, M.; Kucwaj, K.; Filipek, B.; et al. SAR-studies on the importance of aromatic ring topologies in search for selective 5-HT7 receptor ligands among phenylpiperazine hydantoin derivatives. Eur. J. Med. Chem. 2014, 78, 324–339. [Google Scholar] [CrossRef]
- Bohnert, J.A.; Schuster, S.; Kern, W.V.; Karcz, T.; Olejarz, A.; Kaczor, A.; Handzlik, J.; Kieć-Kononowicz, K. Novel piperazine arylideneimidazolones inhibit the AcrAB-TolC pump in Escherichia coli and simultaneously act as fluorescent membrane probes in a combined real-time influx and efflux assay. Antimicrob. Agents Chemother. 2016, 60, 1974–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczor, A.; Witek, K.; Podlewska, S.; Czekajewska, J.; Lubelska, A.; Żesławska, E.; Nitek, W.; Latacz, G.; Alibert, S.; Pagès, J.M.; et al. 5-arylideneimidazolones with amine at position 3 as potential antibiotic adjuvants against multidrug resistant bacteria. Molecules 2019, 24, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spengler, G.; Evaristo, M.; Handzlik, J.; Serly, J.; Molnár, J.; Viveiros, M.; Kiéc-Kononowicz, K.; Amaral, L. Biological activity of hydantoin derivatives on P-glycoprotein (ABCB1) of mouse lymphoma cells. Anticancer Res. 2010, 30, 4867–4871. [Google Scholar]
- Handzlik, J.; Spengler, G.; Mastek, B.; Dela, A.; Molnar, J.; Amaral, L.; Kieć-Kononowicz, K. 5-arylidene (thio) hydantoin derivatives as modulators of cancer efflux pump. Acta Pol. Pharm. Drug Res. 2012, 69, 149–153. [Google Scholar]
- Żesławska, E.; Kincses, A.; Spengler, G.; Nitek, W.; Wyrzuc, K.; Kieć-Kononowicz, K.; Handzlik, J. The 5-aromatic hydantoin-3-acetate derivatives as inhibitors of the tumour multidrug resistance efflux pump P-glycoprotein (ABCB1): Synthesis, crystallographic and biological studies. Bioorg. Med. Chem. 2016, 24, 2815–2822. [Google Scholar] [CrossRef] [Green Version]
- Domínguez-Álvarez, E.; Gajdács, M.; Spengler, G.; Palop, J.A.; Marć, M.A.; Kieć-Kononowicz, K.; Amaral, L.; Molnár, J.; Jacob, C.; Handzlik, J.; et al. Identification of selenocompounds with promising properties to reverse cancer multidrug resistance. Bioorg. Med. Chem Lett. 2016, 26, 2821–2824. [Google Scholar] [CrossRef] [Green Version]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Sobiło, A.; Olejarz, A.; Kucwaj-Brysz, K.; Satała, G.; Bojarski, A.J.; Wesołowska, A.; et al. In the search for a lead structure among series of potent and selective hydantoin 5-HT7R agents: The drug-likeness in vitro study. Chem. Biol. Drug Des. 2017, 90, 1295–1306. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Kucwaj-Brysz, K.; Wesołowska, A.; Kieć-Kononowicz, K.; Handzlik, J. MF-8, a novel promising arylpiperazine-hydantoin based 5-HT7 receptor antagonist: In vitro drug-likeness studies and in vivo pharmacological evaluation. Bioorg. Med. Chem. Lett. 2018, 28, 878–883. [Google Scholar] [CrossRef]
- Latacz, G.; Hogendorf, A.S.; Hogendorf, A.; Lubelska, A.; Wierońska, J.M.; Woźniak, M.; Cieślik, P.; Kieć-Kononowicz, K.; Handzlik, J.; Bojarski, A.J. Search for a 5-CT alternative. In vitro and in vivo evaluation of novel pharmacological tools: 3-(1-alkyl-1 H-imidazol-5-yl)-1 H-indole-5-carboxamides, low-basicity 5-HT7 receptor agonists. MedChemComm 2019, 9, 1882–1890. [Google Scholar] [CrossRef]
- Vilar, S.; Sobarzo-Sanchez, E.; Uriarte, E. In silico prediction of P-glycoprotein binding: Insights from molecular docking studies. Curr. Med. Chem. 2019, 26, 1746–1760. [Google Scholar] [CrossRef]
- Chufan, E.E.; Sim, H.M.; Ambudkar, S.V. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): Recent biochemical and structural studies. Adv. Cancer Res. 2015, 125, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, R.; Singh, U.; Patil, A.; Khomane, K.S.; Bagul, P.; Bansal, A.K.; Sangamwar, A.T. In silico model for P-glycoprotein substrate prediction: Insights from molecular dynamics and in vitro studies. J. Comput. Aided Mol. Des. 2013, 27, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Pajeva, I.K.; Hanl, M.; Wiese, M. Protein contacts and ligand binding in the inward-facing model of human P-glycoprotein. ChemMedChem 2013, 8, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, F.; Vasanthanathan, P.; Ecker, G.F. Ligand and structure-based classification models for prediction of P-glycoprotein inhibitors. J. Chem. Inf. Model. 2014, 54, 218–229. [Google Scholar] [CrossRef]
- Dolghih, E.; Bryant, C.; Renslo, A.R.; Jacobson, M.P. Predicting binding to p-glycoprotein by flexible receptor docking. PLoS Comput. Biol. 2011, 7, e1002083. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site. J. Biol. Chem. 2003, 278, 20449–20452. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Clarke, D.M. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 2001, 276, 14972–14979. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Clarke, D.M. Mutational analysis of ABC proteins. Arch. Biochem. Biophys. 2008, 476, 51–64. [Google Scholar] [CrossRef]
- Zeino, M.; Saeed, M.E.; Kadioglu, O.; Efferth, T. The ability of molecular docking to unravel the controversy and challenges related to P-glycoprotein-a well-known, yet poorly understood drug transporter. Invest. New Drugs 2014, 32, 618–625. [Google Scholar] [CrossRef]
- Syed, S.B.; Arya, H.; Fu, I.H.; Yeh, T.K.; Periyasamy, L.; Hsieh, H.P.; Coumar, M.S. Targeting P-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972. [Google Scholar] [CrossRef] [Green Version]
- Pimthon, J.; Dechaanontasup, R.; Ratanapiphop, C.; Phromprasert, C. Homology modeling and substrate binding studies of human P-glycoprotein. Pharm. Sci. Asia 2017, 44, 96–107. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, M.L. P-glycoprotein inhibition for optimal drug delivery. Drug Target. Insights 2013, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Maludziński, P.; Szymańska, E.; Kieć-Kononowicz, K. The lipophilicity estimation of 5-arylidene derivatives of (2-thio) hydantoin with antimycobacterial activity. Biomed. Chromatogr. 2007, 21, 291–298. [Google Scholar] [CrossRef]
- Szymańska, E.; Kieć-Kononowicz, K. Antimycobacterial activity of 5-arylidene derivatives of hydantoin. Farmaco 2002, 57, 355–362. [Google Scholar] [CrossRef]
- Matys, A.; Podlewska, S.; Witek, K.; Witek, J.; Bojarski, A.J.; Schabikowski, J.; Otrębska-Machaj, E.; Latacz, G.; Szymańska, E.; Kieć-Kononowicz, K.; et al. Imidazolidine-4-one derivatives in the search for novel chemosensitizers of Staphyloccocus aureus MRSA: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2015, 101, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Cornwell, M.M.; Pastan, I.; Gottesman, M.M. Certain calcium-channel blockers bind specifically to multidrug-resistant human KB carcinoma membrane vesicles and inhibit drug binding to P-glycoprotein. J. Biol. Chem. 1987, 262, 2166–2170. [Google Scholar]
- Takács, D.; Csonka, Á.; Horváth, Á.; Windt, T.; Gajdács, M.; Riedl, Z.; Hajós, G.; Amaral, L.; Molnár, J.; Spengler, G. Reversal of ABCB1-related multidrug resistance of colonic adenocarcinoma cells by phenothiazines. Anticancer Res. 2015, 35, 3245–3251. [Google Scholar]
- Żesławska, E.; Kinces, A.; Unger, V.; Tóth, V.; Spengler, G.; Nitek, W.; Tejchman, W. Exocyclic sulfur and selenoorganic compounds towards their anticancer effects: Crystallographic and biological studies. Anticancer Res. 2018, 38, 4577–4584. [Google Scholar] [CrossRef]
- Pei, J.; Kim, B.H.; Tang, M.; Grishin, N.V. PROMALS web server for accurate multiple protein sequence alignments. Nucleic Acids Res. 2007, 35, 649–652. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-1: Protein Preparation Wizard; Schrödinger LLC: New York, NY, USA, 2018.
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2010, 27, 343–350. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modeling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Tusnády, G.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Maestro, LigPrep, Macromodel; Schrödinger LLC: New York, NY, USA, 2018.
- Schrödinger Release 2018-1: Induced Fit. Docking Protocol, Glide, Prime; Schrödinger LLC: New York, NY, USA, 2018.
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger LLC: New York, NY, USA, 2018.
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Cpd | Group | R | n | Amine |
| 7 | A |  | --- |  |
| 8 | A |  | --- |  |
| 9 | A |  | --- |  |
| 10 | A |  | --- |  |
| 11 | A |  | --- |  |
| 12 | A |  | --- |  |
| 13 | A |  | --- |  |
| 14 | B |  | 2 | --- |
| 15 | B |  | 3 | --- |
| 16 | B |  | 3 | --- |
| 17 | B |  | 3 | --- |
| 18 | B |  | 3 | --- |
| 19 | B |  | 3 | --- |
| 20 | B |  | 3 | --- |
| Effects on Rhodamine 123 Accumulation | ||||
|---|---|---|---|---|
| Concentration 2 µM | Concentration 20 µM | |||
| Cpd | FAR | FAR Quotient (%) | FAR | FAR Quotient (%) |
| 7 1 | 0.955 | 7.31 | 0.927 | 7.09 |
| 8 2 | 1.359 | 8.49 | 2.409 | 15.04 |
| 9 1 | 0.766 | 5.86 | 1.645 | 12.58 |
| 10 2 | 1.108 | 6.92 | 1.992 | 12.44 |
| 11 2 | 1.203 | 7.51 | 23.419 | 146.22 |
| 12 1 | 1.161 | 8.88 | 6.045 | 46.23 |
| 13 2 | 0.968 | 6.04 | 2.073 | 12.94 |
| 14 1 | 0.814 | 6.22 | 0.847 | 6.48 |
| 15 1 | 0.887 | 6.78 | 2.221 | 16.98 |
| 16 1 | 0.927 | 7.09 | 2.426 | 18.55 |
| 17 2 | 0.929 | 5.80 | 2.301 | 14.37 |
| 18 1 | 1.038 | 7.94 | 18.095 | 138.37 |
| 19 2 | 1.272 | 7.94 | 1.615 | 10.08 |
| 20 2 | 0.896 | 5.59 | 1.237 | 7.72 |
| VER | - | - | 13.077 1/16.016 2 | 100.00 |
| DMSO | 0.672 1/0.664 2 (V/V%) | 5.141/4.15 2 | - | - |
| Cpd | Mouse T-Lymphoma Cells | ||||
|---|---|---|---|---|---|
| PAR | MDR | ||||
| IC50 (µM) | SD ± | IC50 (µM) | SD ± | SI | |
| 7 | 53.52 | 2.37 | 54.17 | 1.42 | 0.99 |
| 8 | 23.97 | 2.09 | 46.42 | 3.37 | 0.52 |
| 9 | 12.82 | 1.35 | 18.11 | 2.53 | 0.71 |
| 10 | 53.96 | 3.80 | 63.76 | 0.30 | 0.85 |
| 11 | 39.06 | 2.36 | 45.50 | 0.60 | 0.86 |
| 12 | 15.82 | 1.15 | 30.80 | 1.56 | 0.51 |
| 13 | 82.22 | 3.08 | >100 | - | ≤0.82 |
| 14 | 36.85 | 1.12 | 57.37 | 5.74 | 0.64 |
| 15 | 17.48 | 0.53 | 20.76 | 0.91 | 0.84 |
| 16 | 16.00 | 0.98 | 31.29 | 2.11 | 0.51 |
| 17 | 21.49 | 0.45 | 23.41 | 1.21 | 0.92 |
| 18 | 15.96 | 0.68 | 27.75 | 1.29 | 0.58 |
| 19 | 28.59 | 0.60 | >100 | - | ≤0.29 |
| 20 | 19.60 | 2.35 | 24.88 | 0.10 | 0.79 |
| DOX | 0.7 | 0.56 | 2.14 | 0.76 | 0.33 |
| DMSO | >1% | - | >1% | - | - |
| Cpd | Mouse T-Lymphoma Cells | ||||
|---|---|---|---|---|---|
| PAR | MDR | ||||
| IC50 (µM) | SD ± | IC50 (µM) | SD ± | SI | |
| 7 | 17.60 | 0.87 | 26.33 | 0.32 | 0.67 |
| 8 | 14.77 | 1.41 | 25.98 | 1.17 | 0.57 |
| 9 | 7.78 | 0.31 | 16.53 | 1.10 | 0.47 |
| 10 | 26.42 | 1.43 | 22.45 | 0.94 | 1.18 |
| 11 | 24.77 | 0.89 | 25.60 | 0.26 | 0.97 |
| 12 | 12.70 | 0.08 | 16.20 | 0.30 | 0.78 |
| 13 | 17.37 | 0.17 | 48.58 | 0.21 | 0.36 |
| 14 | 24.93 | 0.77 | 27.86 | 0.95 | 0.89 |
| 15 | 10.26 | 0.32 | 13.64 | 0.22 | 0.75 |
| 16 | 11.39 | 0.38 | 12.16 | 0.04 | 0.94 |
| 17 | 12.17 | 1.07 | 14.25 | 0.58 | 0.85 |
| 18 | 13.38 | 0.20 | 10.82 | 0.61 | 1.24 |
| 19 | 14.39 | 0.90 | 43.82 | 4.29 | 0.33 |
| 20 | 14.78 | 0.41 | 17.31 | 0.70 | 0.85 |
| DOX | 0.28 | 0.06 | 1.75 | 0.38 | 0.16 |
| DMSO | >1% | - | >1% | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczor, A.; Nové, M.; Kincses, A.; Spengler, G.; Szymańska, E.; Latacz, G.; Handzlik, J. Search for ABCB1 Modulators Among 2-Amine-5-Arylideneimidazolones as a New Perspective to Overcome Cancer Multidrug Resistance. Molecules 2020, 25, 2258. https://doi.org/10.3390/molecules25092258
Kaczor A, Nové M, Kincses A, Spengler G, Szymańska E, Latacz G, Handzlik J. Search for ABCB1 Modulators Among 2-Amine-5-Arylideneimidazolones as a New Perspective to Overcome Cancer Multidrug Resistance. Molecules. 2020; 25(9):2258. https://doi.org/10.3390/molecules25092258
Chicago/Turabian StyleKaczor, Aneta, Márta Nové, Annamária Kincses, Gabriella Spengler, Ewa Szymańska, Gniewomir Latacz, and Jadwiga Handzlik. 2020. "Search for ABCB1 Modulators Among 2-Amine-5-Arylideneimidazolones as a New Perspective to Overcome Cancer Multidrug Resistance" Molecules 25, no. 9: 2258. https://doi.org/10.3390/molecules25092258