
Muscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach

Abstract
:1. Long-Chain Fatty Acids
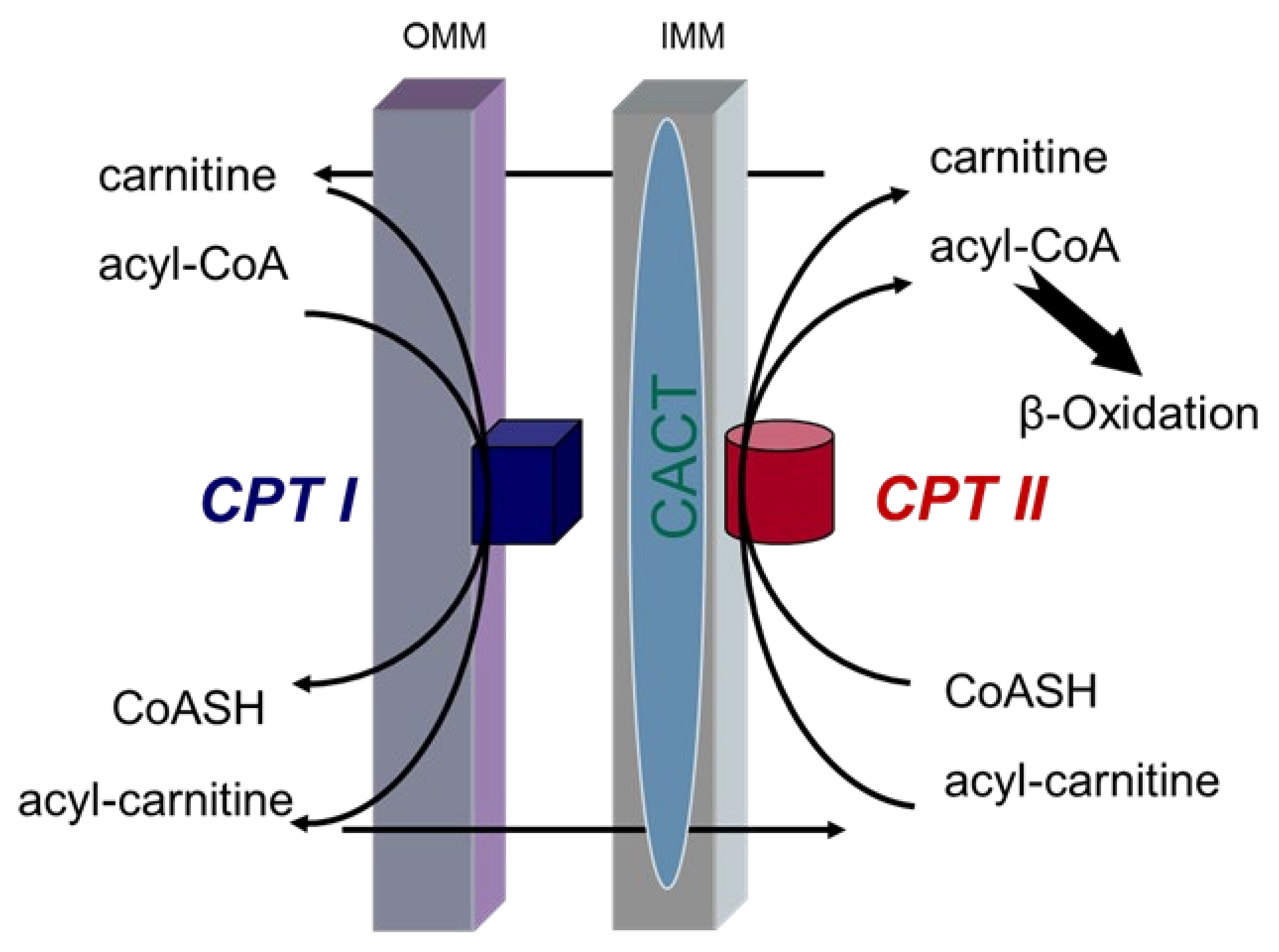
2. Long-Chain Fatty Acid Transport System
3. Carnitine Palmitoyltransferase II (CPT II) Deficiency
4. Three Phenotypes of CPT II Deficiency
5. Muscle Form of CPT II Deficiency
5.1. Clinical Presentations
5.2. Biochemical Features
5.3. Pathobiochemical Characteristics
5.4. Immunohistochemistry
5.5. Molecular Genetic Aspects
6. Recombinant Enzyme Studies
7. Fibroblast Growth Factor 21 (FGF-21) Mitochondrial Biomarker
8. Genotype-Phenotype Analysis
9. Manifesting Heterozygotes
10. Treatment
11. Conclusions and Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Demaugre, F.; Bonnefont, J.P.; Cepanec, C.; Scholte, J.; Saudubray, J.M.; Leroux, J.P. Immunoquantitative analysis of human carnitine palmitoyltransferase I and II defects. Pediatr. Res. 1990, 27, 497–500. [Google Scholar] [CrossRef] [Green Version]
- Dutta-Roy, A.K.; Kahn, N.N.; Sinha, A.K. Interaction of receptors for prostaglandin E1/prostacyclin and insulin in human erythrocytes and platelets. Life Sci. 1991, 49, 1129–1139. [Google Scholar] [CrossRef]
- Dutta-Roy, A.K. Insulin mediated processes in platelets, erythrocytes and monocytes/macrophages: Effects of essential fatty acid metabolism. Prostaglandins Leukot. Essent. Fatty Acids 1994, 51, 385–399. [Google Scholar] [CrossRef]
- Stubbs, C.D.; Smith, A.D. The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim. Biophys. Acta 1984, 779, 89–137. [Google Scholar] [CrossRef]
- Uauy, R.; Mena, P.; Valenzuela, A. Essential fatty acids as determinants of lipid requirements in infants, children and adults. Eur. J. Clin. Nutr. 1999, 53 (Suppl. 1), S66–S77. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef]
- Roe, C.; Ding, J. Mitochondrial fatty acid oxidation disorders. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume 8, pp. 2297–2326. ISBN 978-0-07-913035-8. [Google Scholar]
- Brosnan, J.T.; Kopec, B.; Fritz, I.B. The localization of carnitine palmitoyltransferase on the inner membrane of bovine liver mitochondria. J. Biol. Chem. 1973, 248, 4075–4082. [Google Scholar]
- McGarry, J.D.; Brown, N.F. The Mitochondrial Carnitine Palmitoyltransferase System—From Concept to Molecular Analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Fraser, F.; Corstorphine, C.G.; Zammit, V.A. Topology of carnitine palmitoyltransferase I in the mitochondrial outer membrane. Biochem. J. 1997, 323 Pt 3, 711–718. [Google Scholar] [CrossRef] [Green Version]
- Morillas, M.; López-Viñas, E.; Valencia, A.; Serra, D.; Gómez-Puertas, P.; Hegardt, F.G.; Asins, G. Structural model of carnitine palmitoyltransferase I based on the carnitine acetyltransferase crystal. Biochem. J. 2004, 379, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Eaton, S.; Bartlett, K.; Pourfarzam, M. Mammalian mitochondrial beta-oxidation. Biochem. J. 1996, 320, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Console, L.; Giangregorio, N.; Indiveri, C.; Tonazzi, A. Carnitine/acylcarnitine translocase and carnitine palmitoyltransferase 2 form a complex in the inner mitochondrial membrane. Mol. Cell. Biochem. 2014, 394, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerner, J.; Hoppel, C. Fatty acid import into mitochondria. Biochim. Biophys. Acta 2000, 1486, 1–17. [Google Scholar] [CrossRef]
- Vladutiu, G.D.; Quackenbush, E.J.; Hainline, B.E.; Albers, S.; Smail, D.S.; Bennett, M.J. Lethal neonatal and severe late infantile forms of carnitine palmitoyltransferase II deficiency associated with compound heterozygosity for different protein truncation mutations. J. Pediatr. 2002, 141, 734–736. [Google Scholar] [CrossRef]
- Sigauke, E.; Rakheja, D.; Kitson, K.; Bennett, M.J. Carnitine palmitoyltransferase II deficiency: A clinical, biochemical, and molecular review. Lab. Investig. J. Tech. Methods Pathol. 2003, 83, 1543–1554. [Google Scholar] [CrossRef] [Green Version]
- Pierce, M.R.; Pridjian, G.; Morrison, S.; Pickoff, A.S. Fatal carnitine palmitoyltransferase II deficiency in a newborn: New phenotypic features. Clin. Pediatr. 1999, 38, 13–20. [Google Scholar] [CrossRef]
- Boemer, F.; Deberg, M.; Schoos, R.; Caberg, J.-H.; Gaillez, S.; Dugauquier, C.; Delbecque, K.; François, A.; Maton, P.; Demonceau, N.; et al. Diagnostic pitfall in antenatal manifestations of CPT II deficiency. Clin. Genet. 2016, 89, 193–197. [Google Scholar] [CrossRef]
- Thuillier, L.; Sevin, C.; Demaugre, F.; Brivet, M.; Rabier, D.; Droin, V.; Aupetit, J.; Abadi, N.; Kamoun, P.; Saudubray, J.M.; et al. Genotype/phenotype correlation in carnitine palmitoyl transferase II deficiency: Lessons from a compound heterozygous patient. Neuromuscul. Disord. 2000, 10, 200–205. [Google Scholar] [CrossRef]
- Isackson, P.J.; Bennett, M.J.; Vladutiu, G.D. Identification of 16 new disease-causing mutations in the CPT2 gene resulting in carnitine palmitoyltransferase II deficiency. Mol. Genet. Metab. 2006, 89, 323–331. [Google Scholar] [CrossRef]
- Semba, S.; Yasujima, H.; Takano, T.; Yokozaki, H. Autopsy case of the neonatal form of carnitine palmitoyltransferase-II deficiency triggered by a novel disease-causing mutation del1737C. Pathol. Int. 2008, 58, 436–441. [Google Scholar] [CrossRef]
- Smeets, R.J.P.; Smeitink, J.A.M.; Semmekrot, B.A.; Scholte, H.R.; Wanders, R.J.A.; van den Heuvel, L.P.W.J. A novel splice site mutation in neonatal carnitine palmitoyl transferase II deficiency. J. Hum. Genet. 2003, 48, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Wieser, T. Carnitine Palmitoyltransferase II Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Demaugre, F.; Bonnefont, J.P.; Colonna, M.; Cepanec, C.; Leroux, J.P.; Saudubray, J.M. Infantile form of carnitine palmitoyltransferase II deficiency with hepatomuscular symptoms and sudden death. Physiopathological approach to carnitine palmitoyltransferase II deficiencies. J. Clin. Investig. 1991, 87, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahyaoui, R.; Espinosa, M.G.; Gómez, C.; Dayaldasani, A.; Rueda, I.; Roldán, A.; Ugarte, M.; Lastra, G.; Pérez, V. Neonatal carnitine palmitoyltransferase II deficiency associated with Dandy-Walker syndrome and sudden death. Mol. Genet. Metab. 2011, 104, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Bouchireb, K.; Teychene, A.-M.; Rigal, O.; de Lonlay, P.; Valayannopoulos, V.; Gaudelus, J.; Sellier, N.; Bonnefont, J.P.; Brivet, M.; de Pontual, L. Post-mortem MRI reveals CPT2 deficiency after sudden infant death. Eur. J. Pediatr. 2010, 169, 1561–1563. [Google Scholar] [CrossRef]
- Vladutiu, G.D.; Bennett, M.J.; Fisher, N.M.; Smail, D.; Boriack, R.; Leddy, J.; Pendergast, D.R. Phenotypic variability among first-degree relatives with carnitine palmitoyltransferase II deficiency. Muscle Nerve 2002, 26, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Deschauer, M.; Zierz, S. Carnitine palmitoyltransferase II (CPT II) deficiency: Genotype-phenotype analysis of 50 patients. J. Neurol. Sci. 2014, 338, 107–111. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; DiMauro, P.M.M. Muscle Carnitine Palmityltransferase Deficiency and Myoglobinuria. Science 1973, 182, 929–931. [Google Scholar] [CrossRef]
- Corti, S.; Bordoni, A.; Ronchi, D.; Musumeci, O.; Aguennouz, M.; Toscano, A.; Lamperti, C.; Bresolin, N.; Comi, G.P. Clinical features and new molecular findings in Carnitine Palmitoyltransferase II (CPT II) deficiency. J. Neurol. Sci. 2008, 266, 97–103. [Google Scholar] [CrossRef]
- Deschauer, M.; Wieser, T.; Zierz, S. Muscle Carnitine Palmitoyltransferase II Deficiency: Clinical and Molecular Genetic Features and Diagnostic Aspects. Arch. Neurol. 2005, 62, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.R.; Deschauer, M.; Zierz, S. Phenotype of carnitine palmitoyltransferase II (CPT II) deficiency: A questionnaire-based survey. J. Clin. Neurosci. 2019, 59, 32–36. [Google Scholar] [CrossRef]
- Kaneoka, H.; Uesugi, N.; Moriguchi, A.; Hirose, S.; Takayanagi, M.; Yamaguchi, S.; Shigematsu, Y.; Yasuno, T.; Sasatomi, Y.; Saito, T. Carnitine palmitoyltransferase II deficiency due to a novel gene variant in a patient with rhabdomyolysis and ARF. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2005, 45, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.-P.; Demaugre, F.; Prip-Buus, C.; Saudubray, J.-M.; Brivet, M.; Abadi, N.; Thuillier, L. Carnitine Palmitoyltransferase Deficiencies. Mol. Genet. Metab. 1999, 68, 424–440. [Google Scholar] [CrossRef]
- Gempel, K.; Kottlors, M.; Jaksch, M.; Gerbitz, K.D.; Bauer, M.F. Adult carnitine palmitoyltransferase II deficiency: Detection of characteristic carnitine esters in serum by tandem mass spectrometry. J. Inherit. Metab. Dis. 1999, 22, 941–942. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; von Praun, C.; Baumkötter, J.; Lehnert, W.; Ensenauer, R.; Gerbitz, K.-D.; Bauer, M.F. “Adult” form of muscular carnitine palmitoyltransferase II deficiency: Manifestation in a 2-year-old child. Eur. J. Pediatr. 2001, 160, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, H.; Klar, A.; Korn-Lubetzki, I.; Wanders, R.J.; Elpeleg, O.N. Muscular carnitine palmitoyltransferase II deficiency in infancy. Pediatr. Neurol. 2000, 22, 148–150. [Google Scholar] [CrossRef]
- Fanin, M.; Anichini, A.; Cassandrini, D.; Fiorillo, C.; Scapolan, S.; Minetti, C.; Cassanello, M.; Donati, M.A.; Siciliano, G.; D’Amico, A.; et al. Allelic and phenotypic heterogeneity in 49 Italian patients with the muscle form of CPT-II deficiency. Clin. Genet. 2012, 82, 232–239. [Google Scholar] [CrossRef]
- Anichini, A.; Fanin, M.; Vianey-Saban, C.; Cassandrini, D.; Fiorillo, C.; Bruno, C.; Angelini, C. Genotype-phenotype correlations in a large series of patients with muscle type CPT II deficiency. Neurol. Res. 2011, 33, 24–32. [Google Scholar] [CrossRef]
- Reuschenbach, C.; Zierz, S. Mutant carnitine palmitoyltransferase associated with myoadenylate deaminase deficiency in skeletal muscle. J. Pediatr. 1988, 112, 600–603. [Google Scholar] [CrossRef]
- Ørngreen, M.C.; Dunø, M.; Ejstrup, R.; Christensen, E.; Schwartz, M.; Sacchetti, M.; Vissing, J. Fuel utilization in subjects with carnitine palmitoyltransferase 2 gene mutations: CPT2 Gene Mutations and Fuel Use. Ann. Neurol. 2005, 57, 60–66. [Google Scholar] [CrossRef]
- Martín, M.A.; Rubio, J.C.; De Bustos, F.; Del Hoyo, P.; Campos, Y.; García, A.; Börnstein, B.; Cabello, A.; Arenas, J. Molecular analysis in Spanish patients with muscle carnitine palmitoyltransferase deficiency. Muscle Nerve 1999, 22, 941–943. [Google Scholar] [CrossRef]
- DiMauro, S.; Papadimitriou, A. Carnitine palmitoyltransferase deficiency. In Myology; Engel, A.G., Banker, B.Q., Eds.; McGraw-Hill: New York, NY, USA, 1986; Volume 2, pp. 1697–1708. [Google Scholar]
- Gieron, M.A.; Korthals, J.K. Carnitine palmityltransferase deficiency with permanent weakness. Pediatr. Neurol. 1987, 3, 51–53. [Google Scholar] [CrossRef]
- Wieser, T.; Deschauer, M.; Olek, K.; Hermann, T.; Zierz, S. Carnitine palmitoyltransferase II deficiency: Molecular and biochemical analysis of 32 patients. Neurology 2003, 60, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- Deschauer, M.; Morgenroth, A.; Joshi, P.R.; Gläser, D.; Chinnery, P.F.; Aasly, J.; Schreiber, H.; Knape, M.; Zierz, S.; Vorgerd, M. Analysis of spectrum and frequencies of mutations in McArdle disease. Identification of 13 novel mutations. J. Neurol. 2007, 254, 797–802. [Google Scholar] [CrossRef]
- Martín, M.A.; Rubio, J.C.; Buchbinder, J.; Fernández-Hojas, R.; del Hoyo, P.; Teijeira, S.; Gámez, J.; Navarro, C.; Fernández, J.M.; Cabello, A.; et al. Molecular heterogeneity of myophosphorylase deficiency (McArdle’s disease): A genotype-phenotype correlation study. Ann. Neurol. 2001, 50, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Blanc, P.L.; Carrier, H.; Thomas, L.; Chavaillon, J.M.; Robert, D. Acute rhabdomyolysis with carnitine-palmityl-transferase deficiency. Intensive Care Med. 1982, 8, 307. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar]
- Taroni, F.; Verderio, E.; Fiorucci, S.; Cavadini, P.; Finocchiaro, G.; Uziel, G.; Lamantea, E.; Gellera, C.; DiDonato, S. Molecular characterization of inherited carnitine palmitoyltransferase II deficiency. Proc. Natl. Acad. Sci. USA 1992, 89, 8429–8433. [Google Scholar] [CrossRef] [Green Version]
- Demaugre, F.; Bonnefont, J.-P.; Mitchell, G.; Nguyen-Hoang, N.; Pelet, A.; Rimoldi, M.; Donato, S.D.; Saudubray, J.-M. Hepatic and Muscular Presentations of Carnitine Palmitoyl Transferase Deficiency: Two Distinct Entities. Pediatr. Res. 1988, 24, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Taroni, F.; Verderio, E.; Dworzak, F.; Willems, P.J.; Cavadini, P.; DiDonato, S. Identification of a common mutation in the carnitine palmitoyltransferase II gene in familial recurrent myoglobinuria patients. Nat. Genet. 1993, 4, 314–320. [Google Scholar] [CrossRef]
- Zierz, S.; Engel, A.G. Regulatory properties of a mutant carnitine palmitoyltransferase in human skeletal muscle. Eur. J. Biochem. 1985, 149, 207–214. [Google Scholar] [CrossRef] [PubMed]
- de Sain-van der Velden, M.G.M.; Diekman, E.F.; Jans, J.J.; van der Ham, M.; Prinsen, B.H.C.M.T.; Visser, G.; Verhoeven-Duif, N.M. Differences between acylcarnitine profiles in plasma and bloodspots. Mol. Genet. Metab. 2013, 110, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Diekman, E.F.; van der Pol, W.L.; Nievelstein, R.A.J.; Houten, S.M.; Wijburg, F.A.; Visser, G. Muscle MRI in patients with long-chain fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2014, 37, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Laforêt, P.; Vianey-Saban, C. Disorders of muscle lipid metabolism: Diagnostic and therapeutic challenges. Neuromuscul. Disord. 2010, 20, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.C.; Nishino, I. State of the art in muscle lipid diseases. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2010, 29, 351–356. [Google Scholar]
- Lehmann, D.; Zierz, S. Normal protein content but abnormally inhibited enzyme activity in muscle carnitine palmitoyltransferase II deficiency. J. Neurol. Sci. 2014, 339, 183–188. [Google Scholar] [CrossRef]
- Joshi, P.R.; Young, P.; Deschauer, M.; Zierz, S. Expanding mutation spectrum in CPT II gene: Identification of four novel mutations. J. Neurol. 2013, 260, 1412–1414. [Google Scholar] [CrossRef]
- Lehmann, D.; Motlagh, L.; Robaa, D.; Zierz, S. Muscle Carnitine Palmitoyltransferase II Deficiency: A Review of Enzymatic Controversy and Clinical Features. Int. J. Mol. Sci. 2017, 18, 82. [Google Scholar] [CrossRef] [Green Version]
- Motlagh, L.; Golbik, R.; Sippl, W.; Zierz, S. Stabilization of the thermolabile variant S113L of carnitine palmitoyltransferase II. Neurol. Genet. 2016, 2, e53. [Google Scholar] [CrossRef] [Green Version]
- Bank, W.J.; DiMauro, S.; Bonilla, E.; Capuzzi, D.M.; Rowland, L.P. A disorder of muscle lipid metabolism and myoglobinuria. Absence of carnitine palmityl transferase. N. Engl. J. Med. 1975, 292, 443–449. [Google Scholar] [CrossRef]
- Reza, M.J.; Kar, N.C.; Pearson, C.M.; Kark, R.A. Recurrent myoglobinuria due to muscle carnitine palmityl transferase deficiency. Ann. Intern. Med. 1978, 88, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Freddo, L.; Battistella, P.; Bresolin, N.; Pierobon-Bormioli, S.; Armani, M.; Vergani, L. Carnitine palmityl transferase deficiency: Clinical variability, carrier detection, and autosomal-recessive inheritance. Neurology 1981, 31, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, K.Y.; Hoppel, C.L.; Romine, J.S.; Sipe, J.C.; Gross, S.R.; Higginbottom, P.A. Partial deficiency of muscle carnitine palmitoyltransferase with normal ketone production. N. Engl. J. Med. 1978, 298, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Ionasescu, V.; Hug, G.; Hoppel, C. Combined partial deficiency of muscle carnitine palmitoyltransferase and carnitine with autosomal dominant inheritance. J. Neurol. Neurosurg. Psychiatry 1980, 43, 679–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layzer, R.B.; Havel, R.J.; McIlroy, M.B. Partial deficiency of carnitine palmityltransferase: Physiologic and biochemical consequences. Neurology 1980, 30, 627–633. [Google Scholar] [CrossRef]
- Vladutiu, G.D.; Saponara, I.; Conroy, J.M.; Grier, R.E.; Brady, L.; Brady, P. Immunoquantitation of carnitine palmitoyl transferase in skeletal muscle of 31 patients. Neuromuscul. Disord. 1992, 2, 249–259. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shaw, K.; Cooper, D.N. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr. Protoc. Bioinforma. 2012, 39, 1–20. [Google Scholar] [CrossRef]
- Motlagh, L.; Golbik, R.; Sippl, W.; Zierz, S. Malony-CoA inhibits the S113L variant of carnitine-palmitoyltransferase II. Biochim. Biophys. Acta 2016, 1861, 34–40. [Google Scholar] [CrossRef]
- Olpin, S.E.; Afifi, A.; Clark, S.; Manning, N.J.; Bonham, J.R.; Dalton, A.; Leonard, J.V.; Land, J.M.; Andresen, B.S.; Morris, A.A.; et al. Mutation and biochemical analysis in carnitine palmitoyltransferase type II (CPT II) deficiency. J. Inherit. Metab. Dis. 2003, 26, 543–557. [Google Scholar] [CrossRef]
- Scholle, L.M.; Lehmann, D.; Deschauer, M.; Kraya, T.; Zierz, S. FGF-21 as a Potential Biomarker for Mitochondrial Diseases. Curr. Med. Chem. 2018, 25, 2070–2081. [Google Scholar] [CrossRef]
- Suomalainen, A. Biomarkers for mitochondrial respiratory chain disorders. J. Inherit. Metab. Dis. 2011, 34, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Koester, A.; Flier, J.S.; Kharitonenkov, A.; Maratos-Flier, E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology 2009, 150, 4931–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koene, S.; de Laat, P.; van Tienoven, D.H.; Vriens, D.; Brandt, A.M.; Sweep, F.C.G.J.; Rodenburg, R.J.T.; Donders, A.R.T.; Janssen, M.C.H.; Smeitink, J.A.M. Serum FGF21 levels in adult m.3243A>G carriers: Clinical implications. Neurology 2014, 83, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Warfel, J.D.; Wicks, S.E.; Ghosh, S.; Salbaum, J.M.; Burk, D.; Dubuisson, O.S.; Mendoza, T.M.; Zhang, J.; Noland, R.C.; et al. Impaired Mitochondrial Fat Oxidation Induces FGF21 in Muscle. Cell Rep. 2016, 15, 1686–1699. [Google Scholar] [CrossRef] [Green Version]
- Motlagh Scholle, L.; Lehmann, D.; Joshi, P.R.; Zierz, S. Normal FGF-21-Serum Levels in Patients with Carnitine Palmitoyltransferase II (CPT II) Deficiency. Int. J. Mol. Sci. 2019, 20, 1400. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivelä, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428. [Google Scholar] [CrossRef]
- Pereyra, A.S.; Hasek, L.Y.; Harris, K.L.; Berman, A.G.; Damen, F.W.; Goergen, C.J.; Ellis, J.M. Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J. Biol. Chem. 2017, 292, 18443–18456. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Choi, J.; Selen Alpergin, E.S.; Zhao, L.; Hartung, T.; Scafidi, S.; Riddle, R.C.; Wolfgang, M.J. Loss of Hepatic Mitochondrial Long-Chain Fatty Acid Oxidation Confers Resistance to Diet-Induced Obesity and Glucose Intolerance. Cell Rep. 2017, 20, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Choi, J.; Scafidi, S.; Wolfgang, M.J. Hepatic Fatty Acid Oxidation Restrains Systemic Catabolism during Starvation. Cell Rep. 2016, 16, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Ellis, J.M.; Wolfgang, M.J. Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell Rep. 2015, 10, 266–279. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Hurtado, E.; Lee, J.; Choi, J.; Wolfgang, M.J. Fatty acid oxidation is required for active and quiescent brown adipose tissue maintenance and thermogenic programing. Mol. Metab. 2018, 7, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Liang, C.; Edema-Hildebrand, F.; Riley, C.; Needham, M.; Sue, C.M. Fibroblast growth factor 21 is a sensitive biomarker of mitochondrial disease. Neurology 2013, 81, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Durka-Kęsy, M.; Stępień, A.; Tomczykiewicz, K.; Fidziańska, A.; Niebrój-Dobosz, I.; Pastuszak, Z. Myopathy in the course of carnitine palmitoyltransferase II deficiency. Neurol. Neurochir. Pol. 2012, 46, 600–602. [Google Scholar]
- Shinohara, M.; Saitoh, M.; Takanashi, J.; Yamanouchi, H.; Kubota, M.; Goto, T.; Kikuchi, M.; Shiihara, T.; Yamanaka, G.; Mizuguchi, M. Carnitine palmitoyl transferase II polymorphism is associated with multiple syndromes of acute encephalopathy with various infectious diseases. Brain Dev. 2011, 33, 512–517. [Google Scholar] [CrossRef]
- Yamazaki, N.; Shinohara, Y.; Shima, A.; Yamanaka, Y.; Terada, H. Isolation and characterization of cDNA and genomic clones encoding human muscle type carnitine palmitoyltransferase I. Biochim. Biophys. Acta 1996, 1307, 157–161. [Google Scholar] [CrossRef]
- Yasuno, T.; Kaneoka, H.; Tokuyasu, T.; Aoki, J.; Yoshida, S.; Takayanagi, M.; Ohtake, A.; Kanazawa, M.; Ogawa, A.; Tojo, K.; et al. Mutations of carnitine palmitoyltransferase II (CPT II) in Japanese patients with CPT II deficiency. Clin. Genet. 2008, 73, 496–501. [Google Scholar] [CrossRef]
- Joshi, P.R.; Deschauer, M.; Zierz, S. Clinically symptomatic heterozygous carnitine palmitoyltransferase II (CPT II) deficiency. Wien. Klin. Wochenschr. 2012, 124, 851–854. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Pasquali, M.; Singh, R.; Matern, D.; Rinaldo, P.; di San Filippo, C.A.; Palmieri, F.; Longo, N. Response to therapy in carnitine/acylcarnitine translocase (CACT) deficiency due to a novel missense mutation. Am. J. Med. Genet. A. 2004, 126, 150–155. [Google Scholar] [CrossRef]
- Katsuya, H.; Misumi, M.; Ohtani, Y.; Miike, T. Postanesthetic acute renal failure due to carnitine palmityl transferase deficiency. Anesthesiology 1988, 68, 945–948. [Google Scholar] [CrossRef]
- Wieser, T.; Kraft, B.; Kress, H.G. No carnitine palmitoyltransferase deficiency in skeletal muscle in 18 malignant hyperthermia susceptible individuals. Neuromuscul. Disord. 2008, 18, 471–474. [Google Scholar] [CrossRef]
- Roe, C.R.; Yang, B.-Z.; Brunengraber, H.; Roe, D.S.; Wallace, M.; Garritson, B.K. Carnitine palmitoyltransferase II deficiency: Successful anaplerotic diet therapy. Neurology 2008, 71, 260–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnefont, J.-P.; Bastin, J.; Behin, A.; Djouadi, F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N. Engl. J. Med. 2009, 360, 838–840. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exon 1 | Exon 4 |
|
|
| Exon 2 | |
| |
| Exon 3 | |
| |
| Exon 5 | |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, P.R.; Zierz, S. Muscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach. Molecules 2020, 25, 1784. https://doi.org/10.3390/molecules25081784
Joshi PR, Zierz S. Muscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach. Molecules. 2020; 25(8):1784. https://doi.org/10.3390/molecules25081784
Chicago/Turabian StyleJoshi, Pushpa Raj, and Stephan Zierz. 2020. "Muscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach" Molecules 25, no. 8: 1784. https://doi.org/10.3390/molecules25081784