New Cysteine Protease Inhibitors: Electrophilic (Het)arenes and Unexpected Prodrug Identification for the Trypanosoma Protease Rhodesain

 , , and
, , and
Abstract
:1. Introduction
2. Results
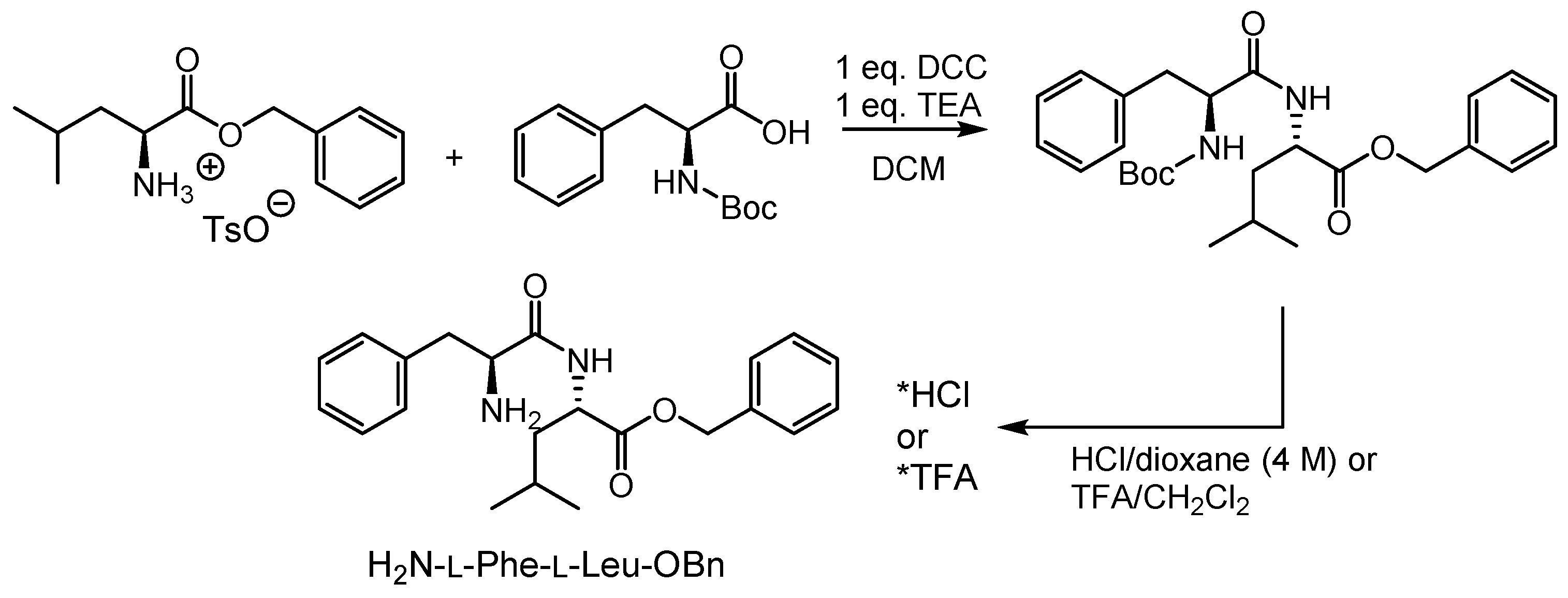
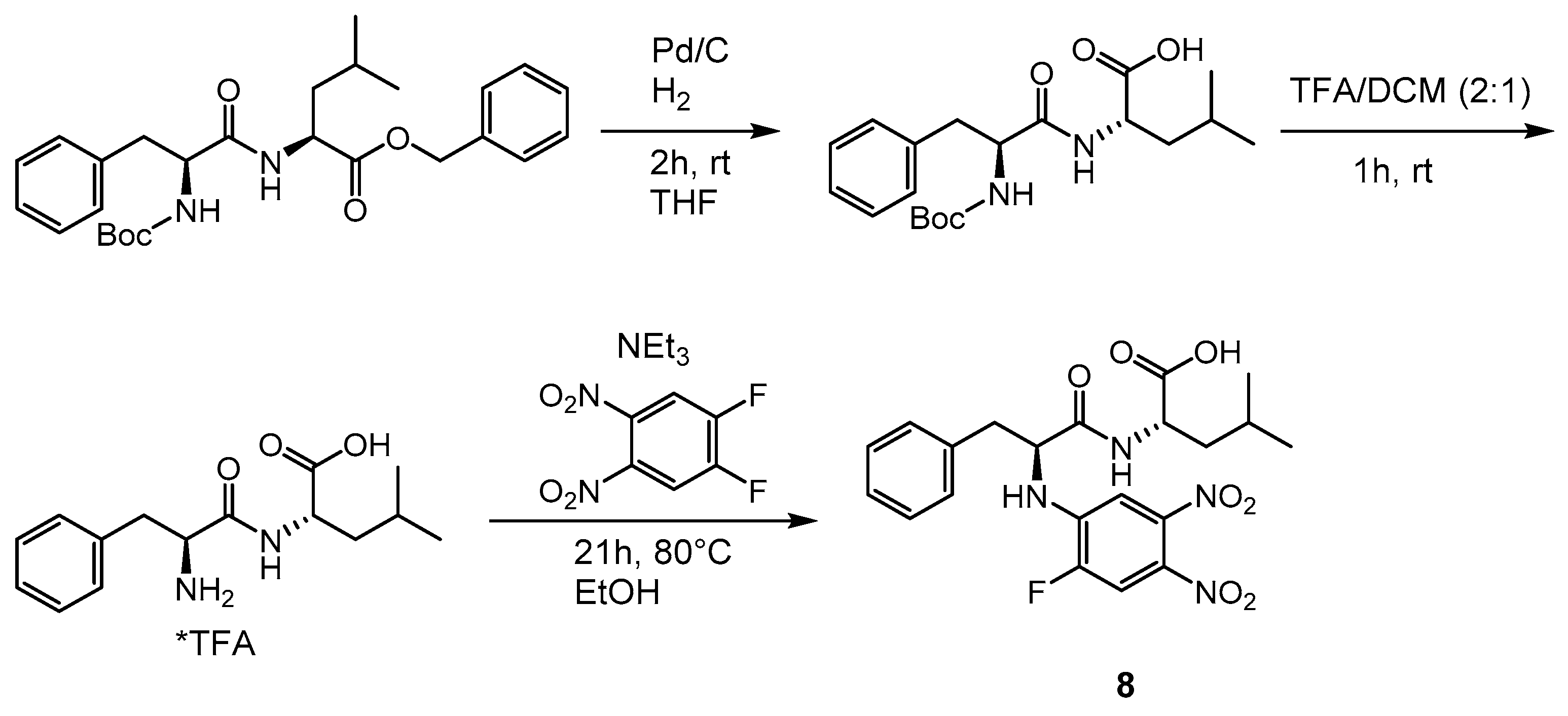
2.1. Syntheses
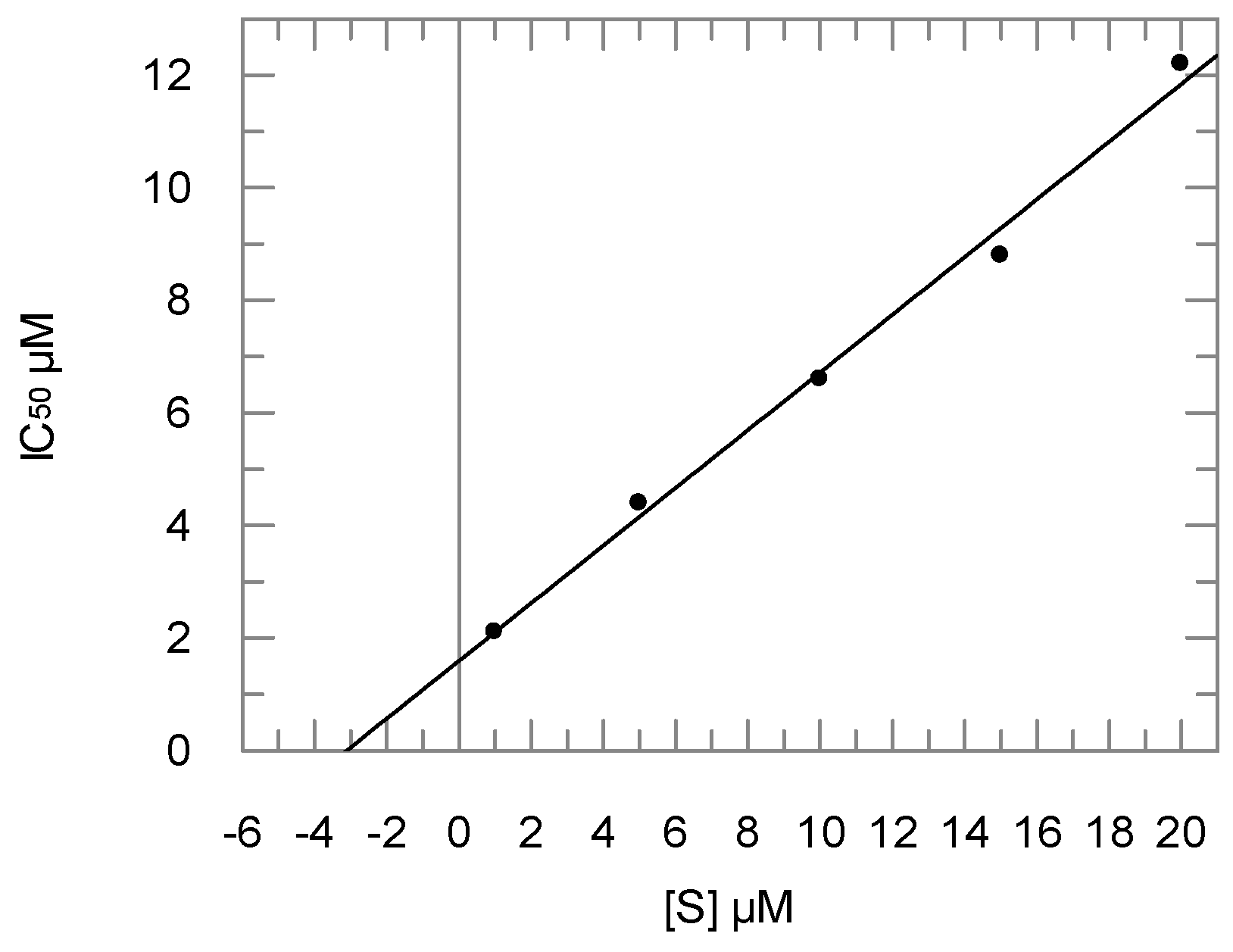
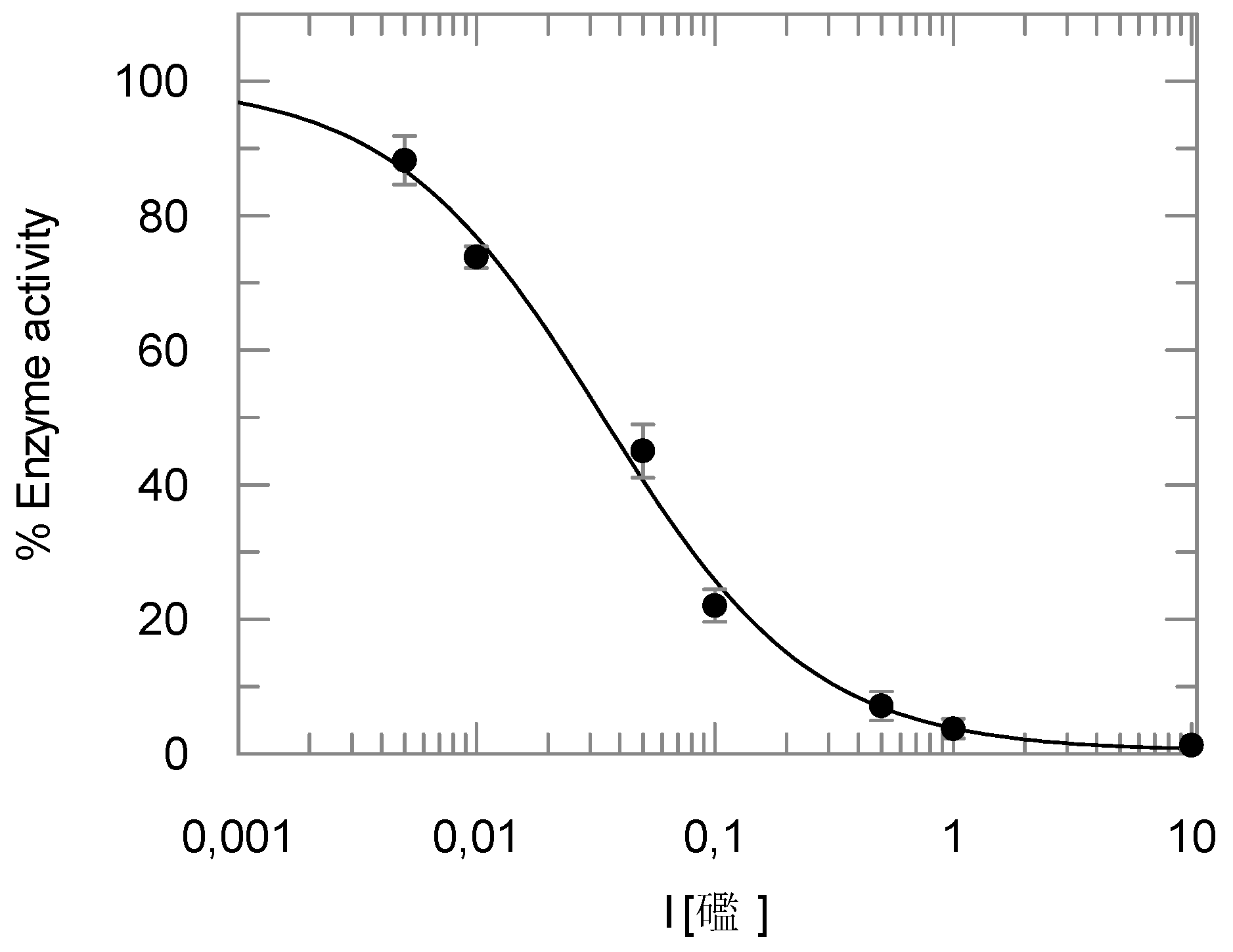
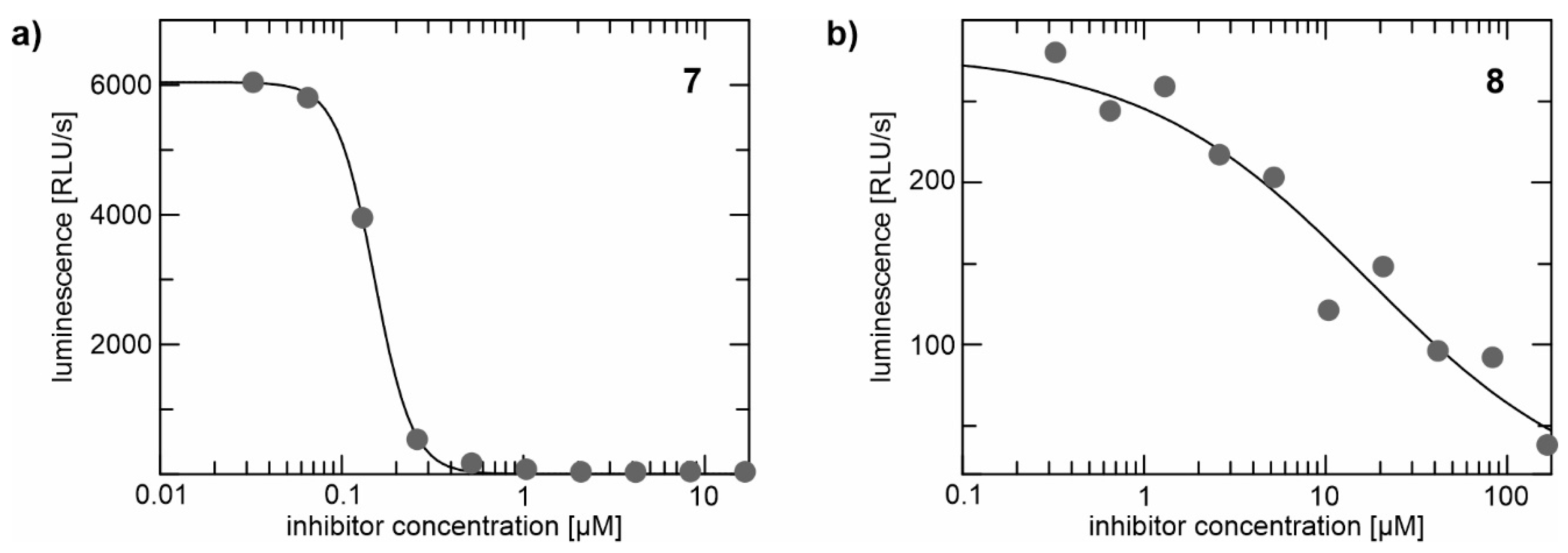
2.2. Enzyme Assays
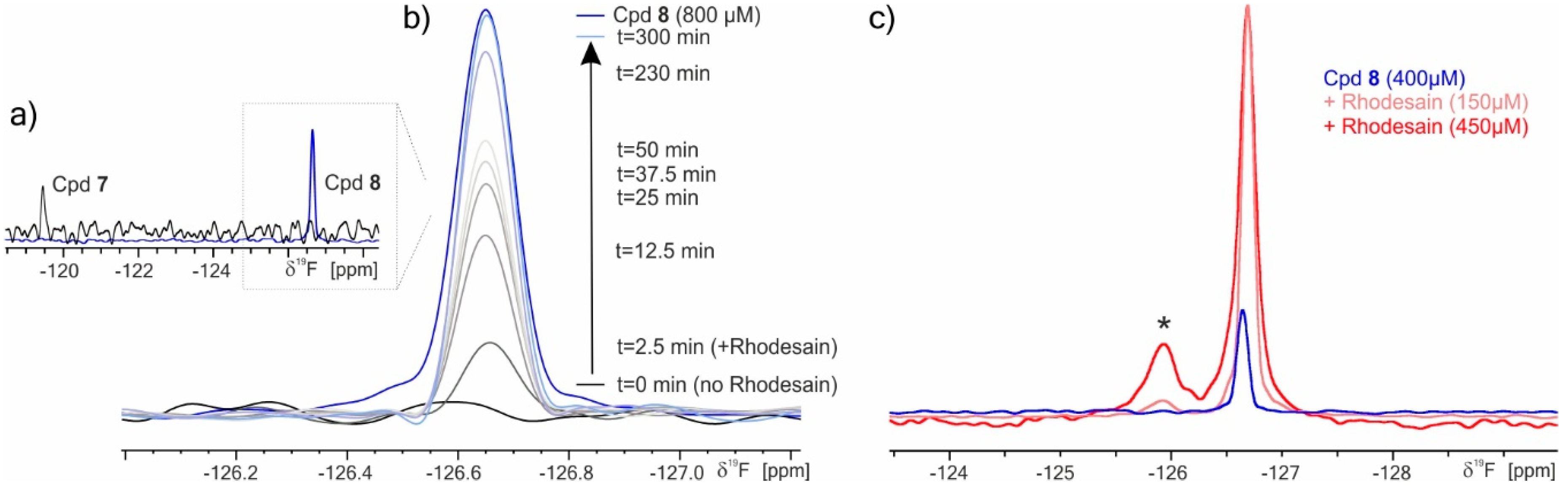
2.3. 19F NMR Spectroscopy
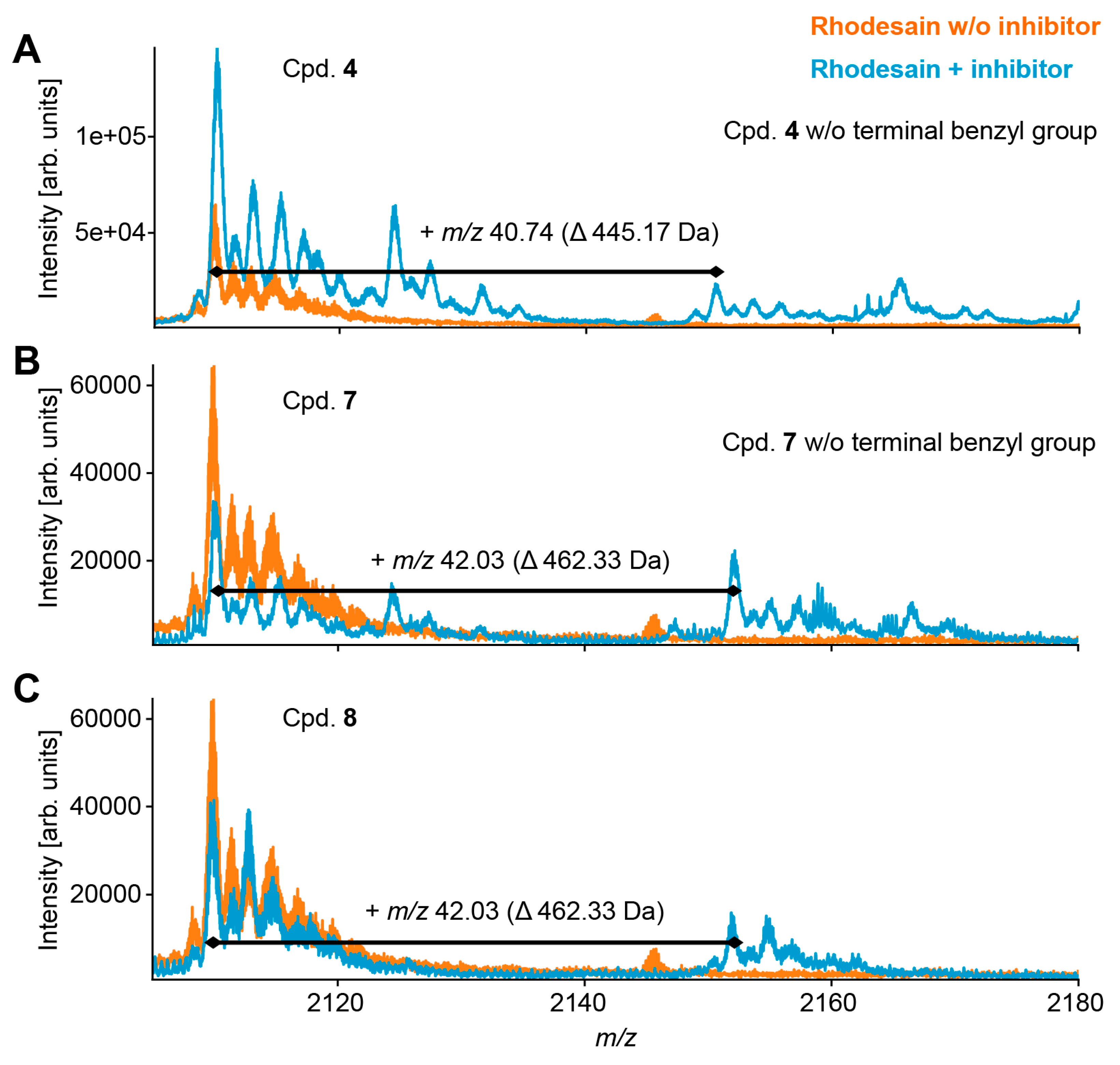
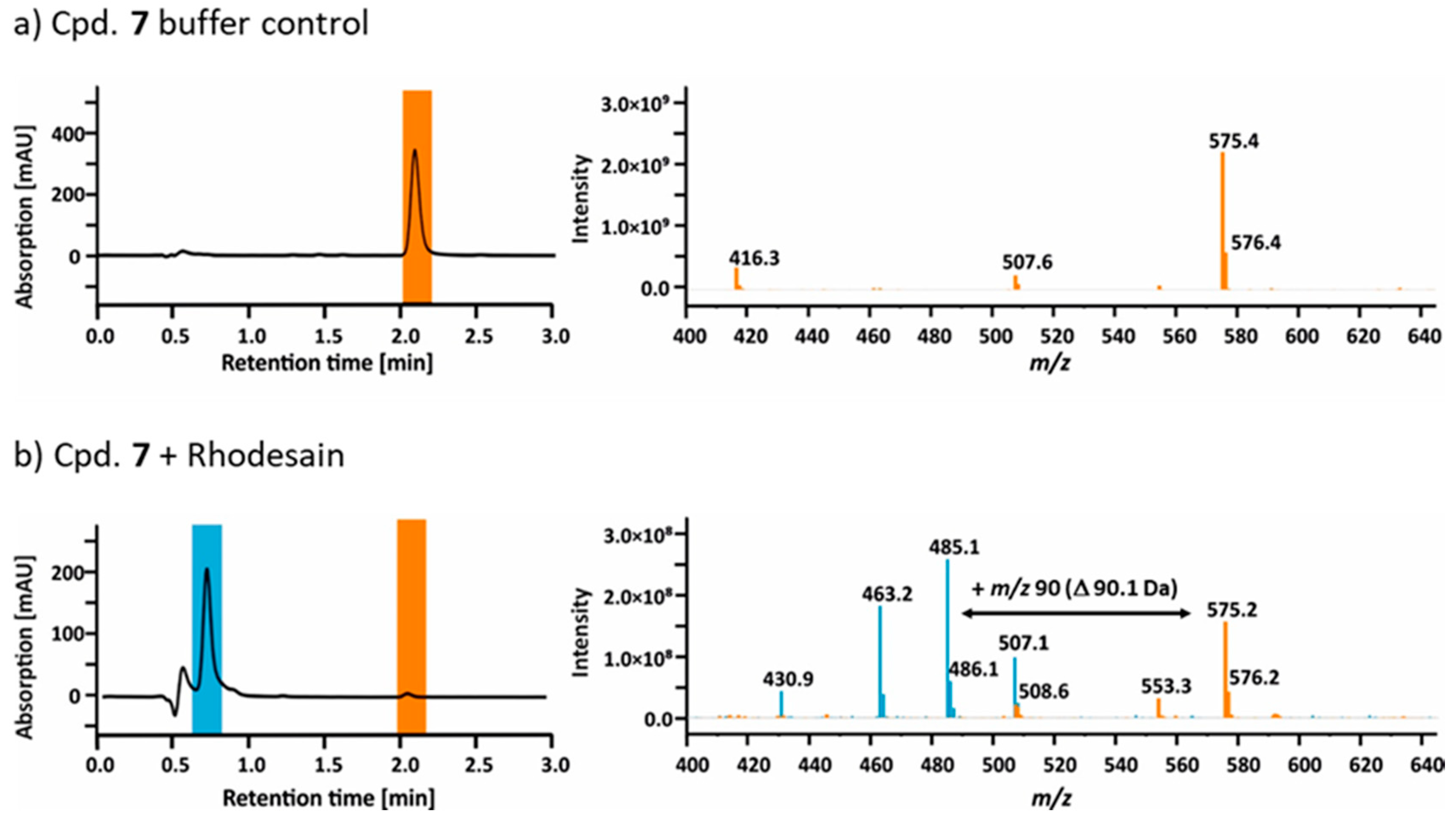
2.4. Mass Spectrometry
2.5. Hydrolysis of Ester 7 by Rhodesain
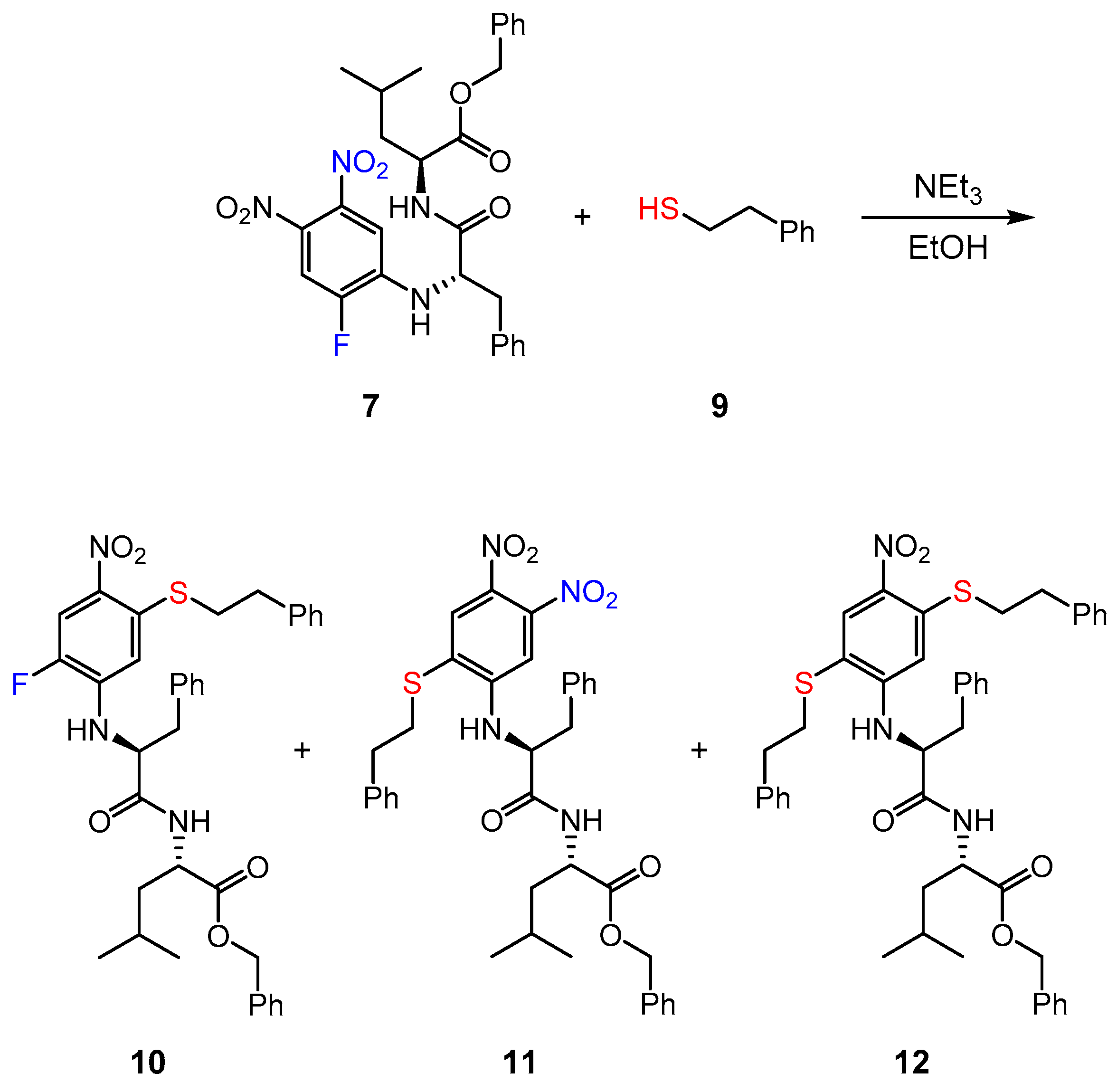
2.6. Reaction of Inhibitor 7 with a Low Molecular Weight Thiol
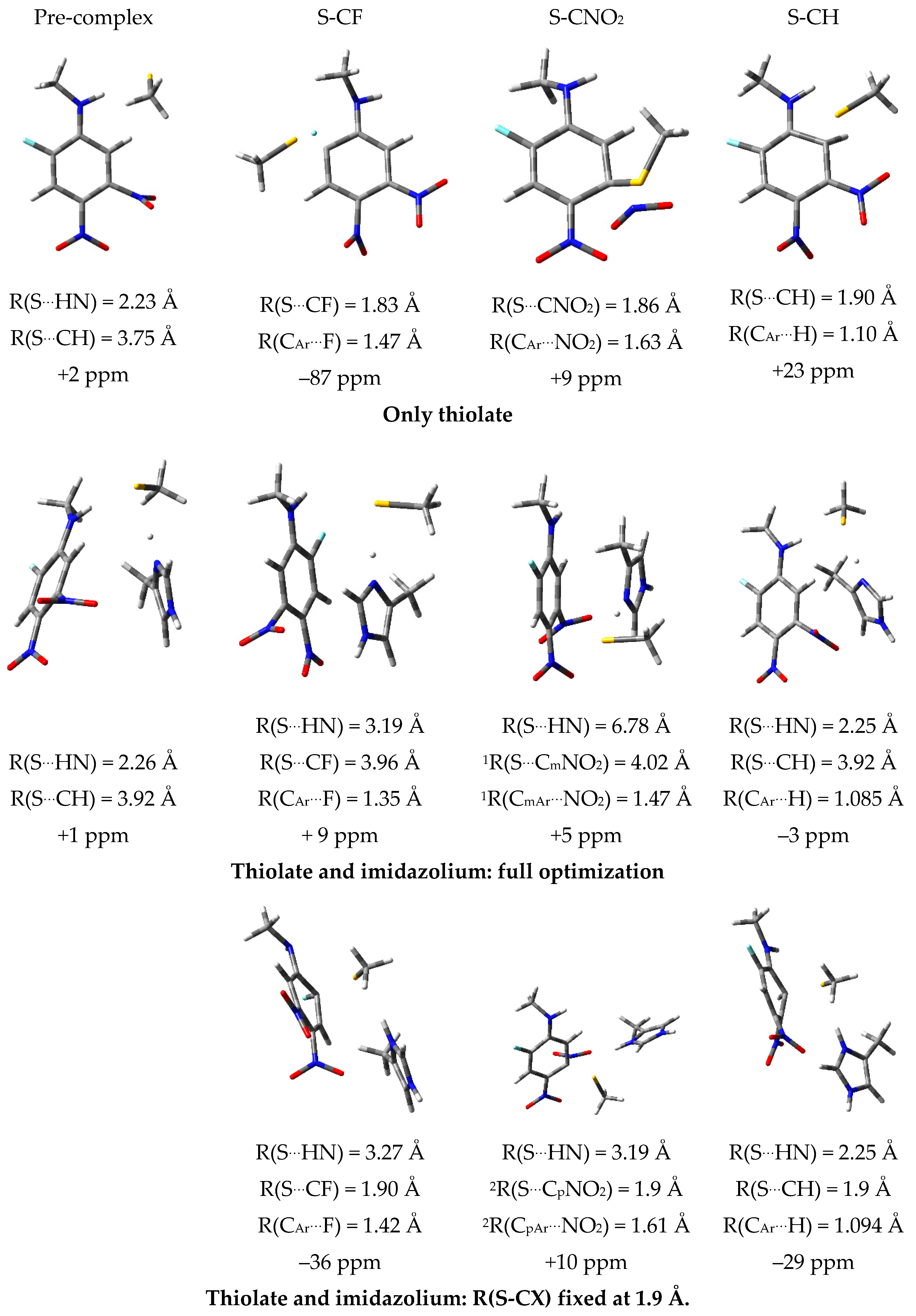
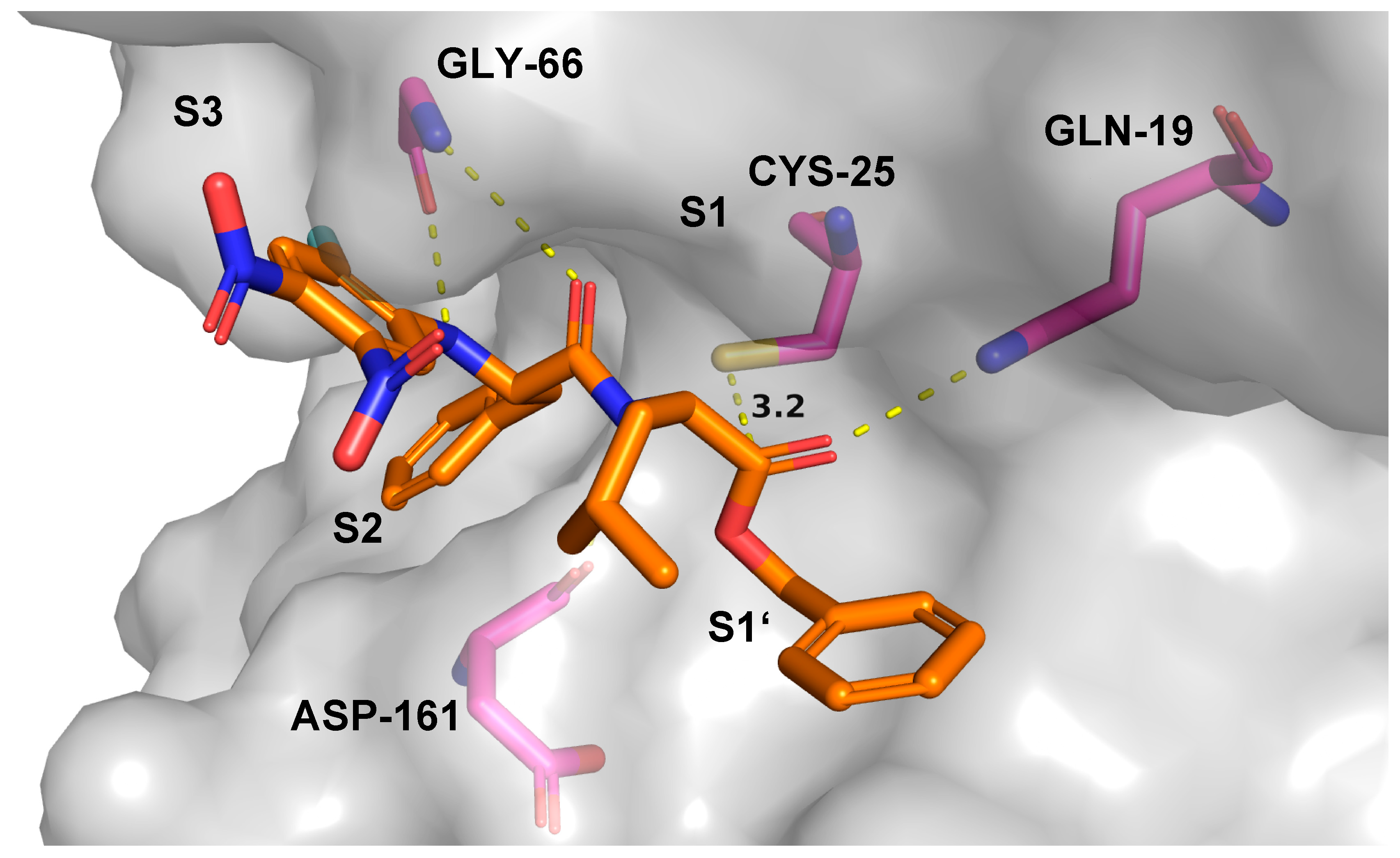
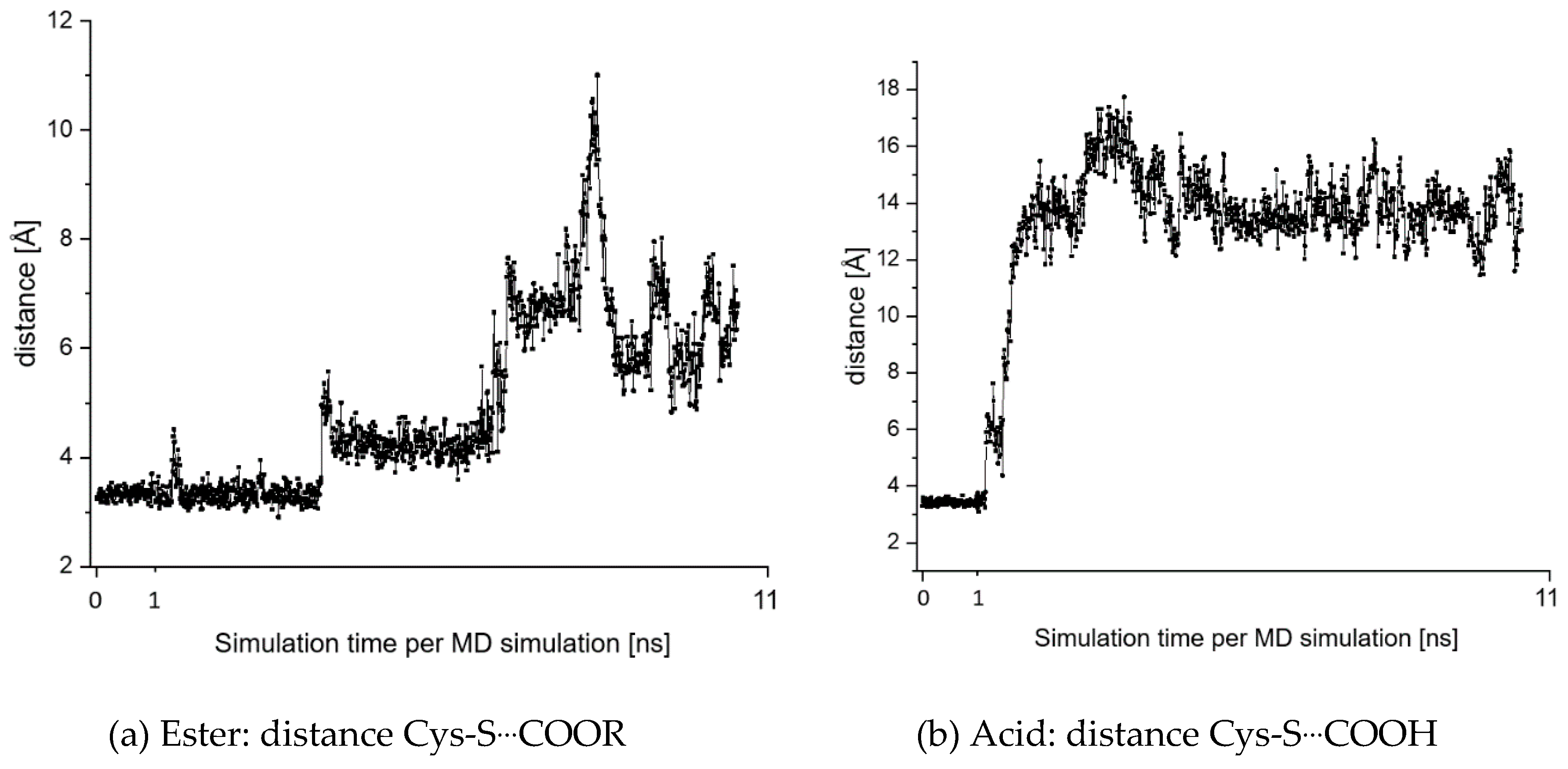
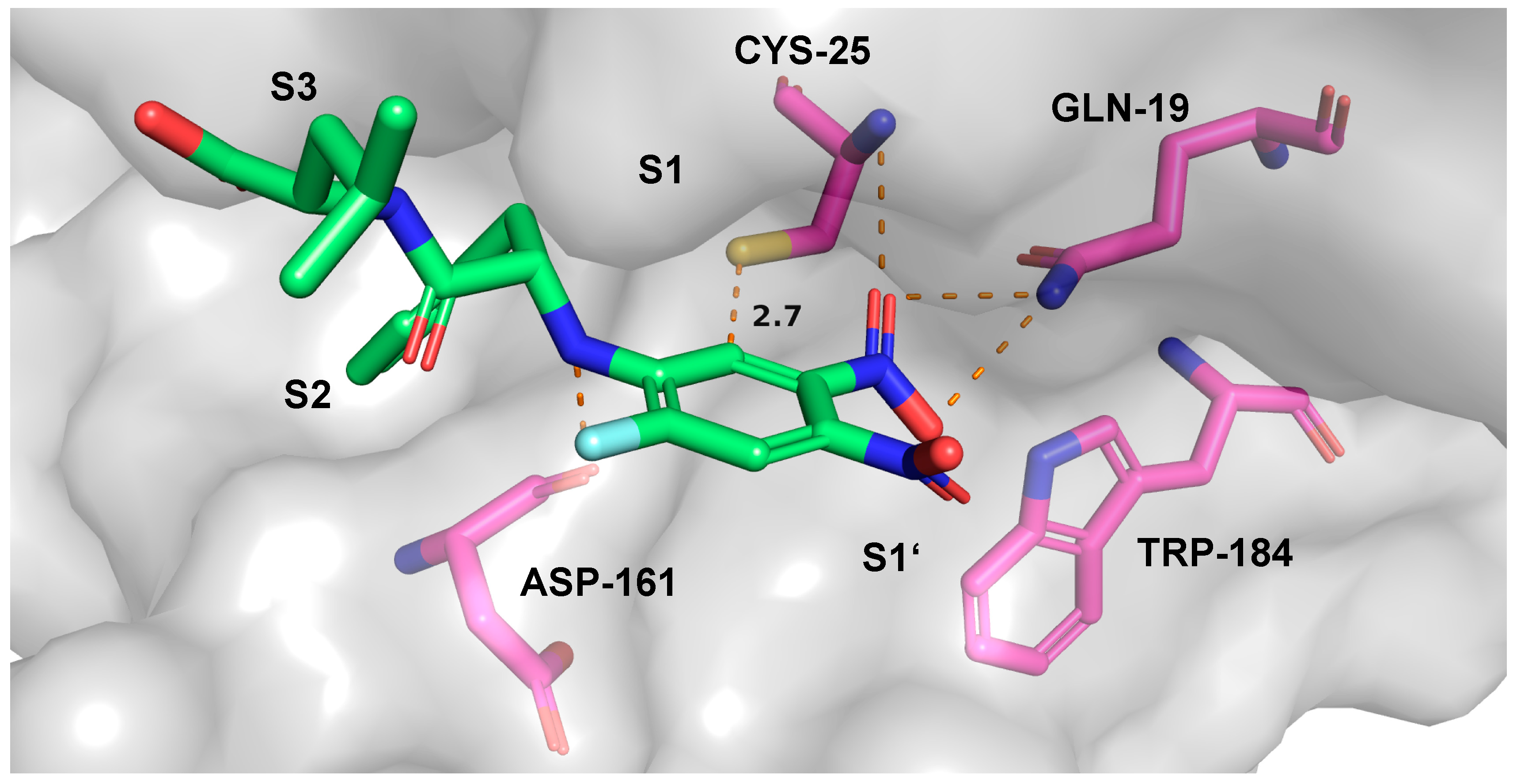
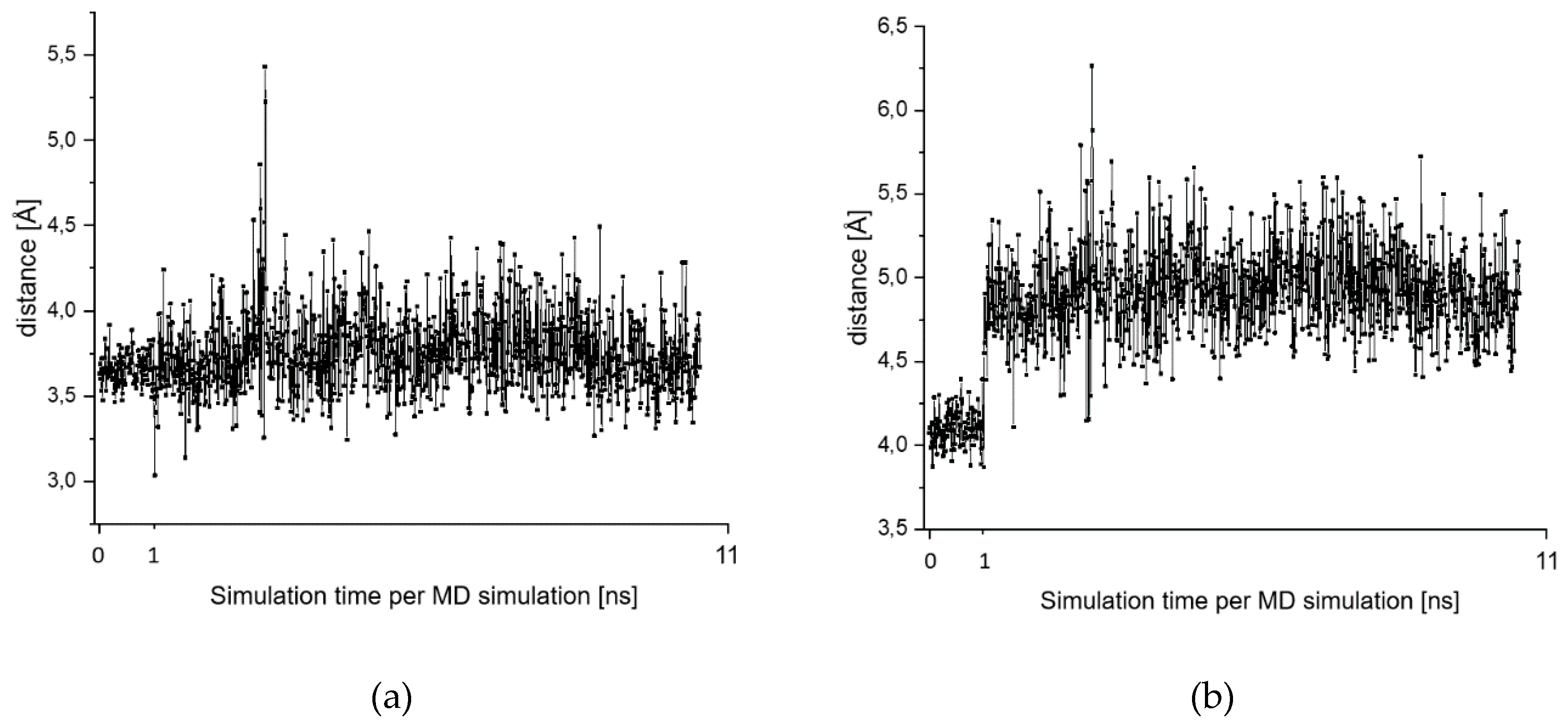
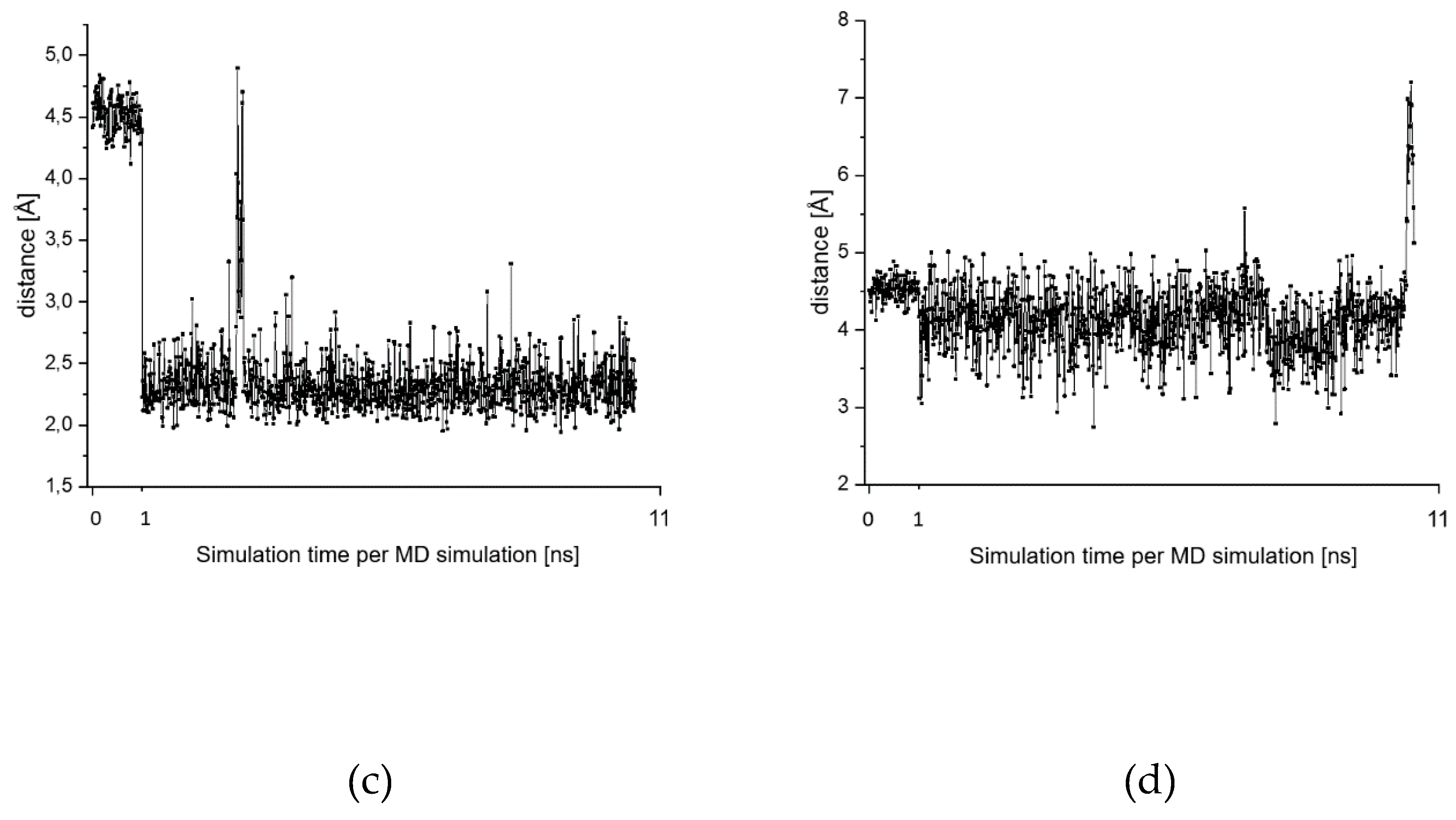
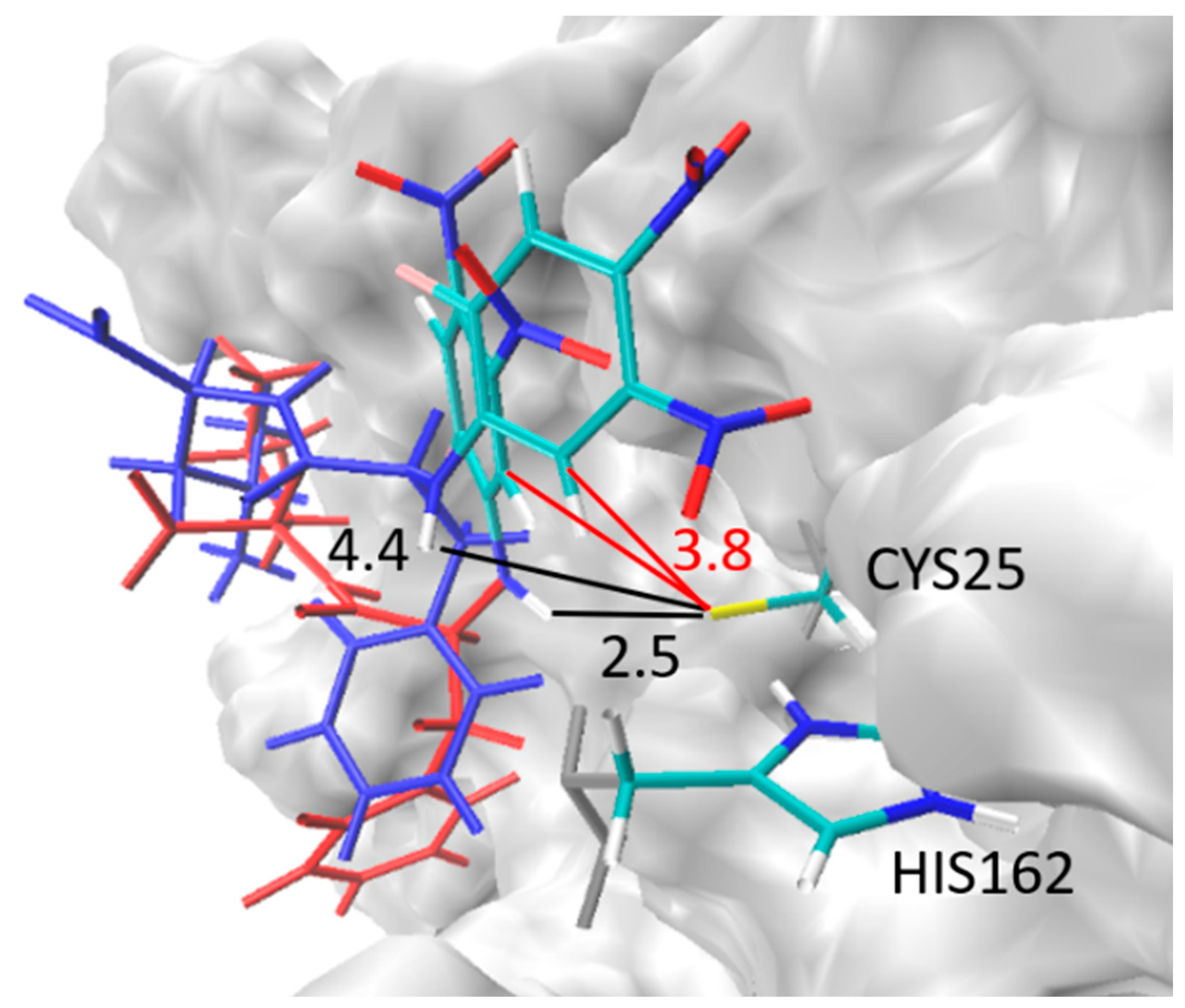
2.7. Theoretical Investigations of the Enzyme-Inhibitor Complex
2.8. T. b. brucei Cell Survival Assay
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Kluge, A.F. Targeted covalent drugs of the kinase family. Curr. Opin. Chem. Biol. 2010, 14, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Weerapana, E.; Cravatt, B.F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potashman, M.H.; Duggan, M.E. Covalent modifiers: An orthogonal approach to drug design. J. Med. Chem. 2009, 52, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Mah, R.; Thomas, J.R.; Shafer, C.M. Drug discovery considerations in the development of covalent inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 33–39. [Google Scholar] [CrossRef]
- Baillie, T.A. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 10, 96–114. [Google Scholar] [CrossRef]
- Johansson, M.H. Reversible Michael additions: Covalent inhibitors and prodrugs. Mini Rev. Med. Chem. 2012, 12, 1330. [Google Scholar]
- Palmer, J.T.; Rasnick, D.; Klaus, J.L.; Broemme, D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 1995, 38, 3193–3196. [Google Scholar] [CrossRef]
- Ettari, R.; Nizi, E.; Di Francesco, M.E.; Dude, M.-A.; Pradel, G.; Vicik, R.; Schirmeister, T.; Micale, N.; Grasso, S.; Zappala, M. Development of peptidomimetics with a vinyl sulfone warhead as irreversible falcipain-2 inhibitors. J. Med. Chem. 2008, 51, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Bonaccorso, C.; Micale, N.; Heindl, C.; Schirmeister, T.; Calabro, M.L.; Grasso, S.; Zappala, M. Development of novel peptidomimetics containing a vinyl sulfone moiety as proteasome inhibitors. ChemMedChem 2011, 6, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Cosconati, S.; Amendola, G.; Chouchene, K.; Wagner, A.; Hellmich, U.A.; Ulrich, K.; Krauth-Siegel, R.L.; Wich, P.R.; Schmid, I.; et al. Development of novel peptide-based Michael acceptors targeting rhodesain and falcipain-2 for the treatment of Neglected Tropical Diseases (NTDs). J. Med. Chem. 2017, 60, 6911–6923. [Google Scholar]
- Breuning, A.; Degel, B.; Schulz, F.; Büchold, C.; Stempka, M.; Machon, U.; Gelhaus, C.; Leippe, M.; Leyh, M.; Kisker, C.; et al. Michael acceptor based antiplasmodial and antitrypanosomal cysteine protease inhibitors with unusual amino acids. J. Med. Chem. 2010, 53, 1951–1963. [Google Scholar] [CrossRef] [PubMed]
- Machon, U.; Büchold, C.; Stempka, M.; Schirmeister, T.; Gelhaus, C.; Leippe, M.; Gut, J.; Rosenthal, P.J.; Kisker, C.; Leyh, M.; et al. On-bead screening of a combinatorial fumaric acid derived peptide library yields antiplasmodial cysteine protease inhibitors with unusual peptide sequences. J. Med. Chem. 2009, 52, 5662–5672. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Micale, N.; Schirmeister, T.; Gelhaus, C.; Leippe, M.; Nizi, E.; Di Francesco, M.E.; Grasso, S.; Zappala, M. Novel peptidomimetics containing a vinyl ester moiety as highly potent and selective falcipain-2 inhibitors. J. Med. Chem. 2009, 52, 2157–2160. [Google Scholar] [CrossRef] [PubMed]
- Schirmeister, T.; Kesselring, J.; Jung, S.; Schneider, T.H.; Weickert, A.; Becker, J.; Lee, W.; Bamberger, D.; Wich, P.R.; Distler, U.; et al. Quantum chemical-based protocol for the rational design of covalent inhibitors. J. Am. Chem. Soc. 2016, 138, 8332–8335. [Google Scholar] [CrossRef]
- Ehmke, V.; Quinsaat, J.E.; Rivera-Fuentes, P.; Heindl, C.; Schirmeister, T.; Diederich, F. Tuning and predicting biological affinity: Aryl nitriles as cysteine protease inhibitors. Org. Biomol. Chem. 2012, 10, 5764–5768. [Google Scholar] [CrossRef]
- Ehmke, V.; Winkler, E.; Banner, D.W.; Haap, W.; Schweizer, W.B.; Rottmann, M.; Kaiser, M.; Freymond, C.; Brun, R.; Schirmeister, T.; et al. Optimization of triazine nitriles as rhodesain inhibitors: Structure-activity relationships, bioisosteric imidazopyridine nitriles, and X-ray crystal structure analysis with human cathepsin L. ChemMedChem 2013, 8, 967–975. [Google Scholar] [CrossRef]
- Schirmeister, T.; Schmitz, J.; Jung, S.; Schmenger, T.; Krauth-Siegel, R.L.; Gütschow, M. Evaluation of dipeptide nitriles as inhibitors of rhodesain, a major cysteine protease of Trypanosoma brucei. Bioorg. Med. Chem. Lett. 2016, 27, 45–50. [Google Scholar] [CrossRef]
- Giroud, M.; Ivkovic, J.; Martignoni, M.; Fleuti, M.; Trapp, N.; Haap, W.; Kuglstatter, A.; Benz, J.; Kuhn, B.; Schirmeister, T.; et al. Inhibition of the cysteine protease human cathepsin L by triazine nitriles: Amide⋅⋅⋅heteroarene π-stacking interactions and chalcogen bonding in the S3 pocket. ChemMedChem 2017, 12, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Lynas, J.F. Strategies for the inhibition of serine proteases. CMLS Cell. Mol. Life Sci. 2001, 58, 596–624. [Google Scholar] [CrossRef] [PubMed]
- Siklos, M.; BenAissa, M.; Thatcher, G.R.J. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettari, R.; Zappala, M.; Evers, A.; Gelhaus, C.; Leippe, M.; Schirmeister, T.; Micale, N.; Grasso, S. Synthesis of novel peptidomimetics as inhibitors of protozoan cysteine proteases falcipain-2 and rhodesain. Eur. J. Med. Chem. 2010, 45, 3228–3233. [Google Scholar] [CrossRef]
- Otto, H.-H.; Schirmeister, T. Cysteine proteases and their inhibitors. Chem. Rev. 1997, 97, 133–171. [Google Scholar] [CrossRef]
- Powers, J.C.; Asgian, J.L.; Ekici, O.D.; James, K.E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002, 102, 4639–4750. [Google Scholar] [CrossRef]
- Schirmeister, T.; Klockow, A. Cysteine protease inhibitors containing small rings. Mini Rev. Med. Chem. 2003, 3, 585–596. [Google Scholar] [CrossRef]
- Vicik, R.; Helten, H.; Schirmeister, T.; Engels, B. Rational design of aziridine containing cysteine protease inhibitors with improved potency - Studies on inhibition mechanism. ChemMedChem 2006, 1, 1021–1028. [Google Scholar] [CrossRef]
- Park, T.; Yang, C.; Yu, Z. Specific inhibitors of lysozyme and peptidases inhibit the growth of the rumen protozoan Entodinium caudatum without decreasing feed digestion or fermentation in vitro. J. Appl. Microbiol. 2019, 127, 670–682. [Google Scholar] [CrossRef]
- Hartmann, K.; Mielczarek, P.; Silberring, J. Synthesis of the novel covalent cysteine proteases inhibitor with iodacetic functional group. Molecules 2020, 25, 813. [Google Scholar] [CrossRef] [Green Version]
- Shannon, D.A.; Banerjee, R.; Webster, E.R.; Bak, D.W.; Wang, C.; Weerapana, E. Investigating the proteome reactivity and selectivity of aryl halides. J. Am. Chem. Soc. 2014, 136, 3330–3333. [Google Scholar] [CrossRef] [PubMed]
- Leesnitzer, L.M.; Parks, D.J.; Bledsoe, R.K.; Cobb, J.E.; Collins, J.L.; Consler, T.G.; Davis, R.G.; Hull-Ryde, E.A.; Lenhard, J.M.; Patel, L.; et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry 2002, 41, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Laufer, S.A. Emerging and re-emerging warheads for Targeted Covalent Inhibitors: Applications in medicinal chemistry and chemical biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Spokoyny, A.M.; Zou, Y.; Ling, J.J.; Yu, H.; Lin, Y.-S.; Pentelute, B.L.J. A perfluoroaryl-cysteine SNAr chemistry approach to unprotected peptide stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. [Google Scholar] [CrossRef] [Green Version]
- Dolle, R.A.; Hoyer, D.; Rinker, J.M.; Ross, T.M.; Schmidt, S.J.; Helaszek, C.T.; Ator, M.A. 3-Chloro-4-carboxamido-6-arylpyridazines as non-peptide class of interleukin-1β converting enzyme inhibitor. Bioorg. Med. Chem. Lett. 1997, 7, 1003–1006. [Google Scholar] [CrossRef]
- Shie, J.-J.; Fang, J.-M.; Kuo, C.-J.; Kuo, T.-H.; Liang, P.-H.; Huang, H.-J.; Yang, W.-B.; Lin, C.-H.; Chen, J.-L.; Wu, Y.-T.; et al. Discovery of potent anilide inhibitors against the Severe Acute Respiratory Syndrome 3CL protease. J. Med. Chem. 2005, 48, 4469–4473. [Google Scholar] [CrossRef]
- Li, Z.; Brecher, M.; Deng, Y.-Q.; Zhang, J.; Sakamuru, S.; Liu, B.; Huang, R.; Koetzner, C.A.; Allen, C.A.; Jones, S.A.; et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017, 27, 1046–1064. [Google Scholar] [CrossRef] [Green Version]
- Olson, O.C. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef]
- Mohamed, M.M. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Andricopulo, A.D. Targeting cysteine proteases in trypanosomatid disease drug discovery. Pharmacol. Ther. 2017, 180, 49–61. [Google Scholar] [CrossRef]
- Ettari, R.; Previti, S.; Tamborini, L.; Cullia, G.; Grasso, S.; Zappala, M. The inhibition of cysteine proteases rhodesain and TbCatB: A valuable approach to treat Human African Trypanosomiasis. Mini Rev. Med. Chem. 2016, 16, 1374–1391. [Google Scholar] [CrossRef] [PubMed]
- Vicik, R.; Busemann, M.; Gelhaus, C.; Stiefl, N.; Scheiber, J.; Schmitz, W.; Schulz, F.; Mladenovic, M.; Engels, B.; Leippe, M.; et al. Aziridide-based inhibitors of cathepsin L: Synthesis, inhibition activity, and docking studies. ChemMedChem 2006, 1, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Barthels, F.; Marincola, G.; Marciniak, T.; Konhäuser, M.; Hammerschmidt, S.; Bierlmeier, J.; Distler, U.; Wich, P.R.; Tenzer, S.; Schwarzer, D.; et al. Asymmetric disulfanylbenzamides as irreversible and selective inhibitors of Staphylococcus aureus sortase A. ChemMedChem 2020. accepted. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef]
- Dixon, M. The graphical determination of Km and Ki. Biochem. J. 1972, 129, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Nitsche, C.; Holloway, S.; Schirmeister, T.; Klein, C.D. Biochemistry and medicinal chemistry of the Dengue virus protease. Chem. Rev. 2014, 114, 11348–11381. [Google Scholar] [CrossRef]
- Millies, B.; Hammerstein, v.F.; Gellert, A.; Hammerschmidt, S.; Barthels, F.; Göppel, U.; Immerheiser, M.; Elgner, F.; Jung, N.; Basic, M.; et al. Proline-based allosteric inhibitors of Zika and Dengue virus NS2B/NS3 proteases. J. Med. Chem. 2019, 62, 11359–11382. [Google Scholar] [CrossRef]
- Barrett, A.J.; Woessner, J.F.; Rawlings, N.D. (Eds.) Handbook of Proteolytic Enzymes; Elsevier: London, UK, 2012; Volume 2, 984p, Available online: https://www.sciencedirect.com/book/9780120796113/handbook-of-proteolytic-enzymes (accessed on 23 March 2020).
- Kerr, I.D.; Lee, J.H.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 2009, 284, 25697–25703. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.v.R. Efficient diffuse function-augmented basis-sets for anion calculations. 3. The 3-21+G basis set for 1st-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Physics 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct.: Theochem 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Helten, H.; Schirmeister, T.; Engels, B. Theoretical studies about the influence of different ring substituents on the nucleophilic ring opening of three-membered heterocycles and possible implications for the mechanisms of cysteine protease inhibitors. J. Org. Chem. 2005, 70, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Helten, H.; Schirmeister, T.; Engels, B. Model calculations about the influence of protic environments on the alkylation step of epoxide, aziridine, and thiirane based cysteine protease inhibitors. J. Phys. Chem. A 2004, 108, 7691–7701. [Google Scholar] [CrossRef]
- Buback, V.; Mladenovic, M.; Engels, B.; Schirmeister, T. Rational design of improved aziridine-based inhibitors of cysteine proteases. J. Phys. Chem. B 2009, 113, 5282–5289. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom−Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6621. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Improved second-order Moller-Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Phys. Chem. 2003, 118, 9095–9102. [Google Scholar] [CrossRef]
- Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- LeadIT/FlexX. Version 2.3.2; BioSolveIT GmbH: St. Augustin, Germany, 2018. [Google Scholar]
- Mladenovic, M.; Junold, K.; Fink, R.F.; Thiel, W.; Schirmeister, T.; Engels, B. Atomistic insights into the inhibition of cysteine proteases: First QM/MM calculations clarifying the regiospecificity and the inhibition potency of epoxide- and aziridine-based inhibitors. J. Phys. Chem. B 2008, 112, 5458–5469. [Google Scholar] [CrossRef]
- Mladenovic, M.; Schirmeister, T.; Thiel, S.; Thiel, W.; Engels, B. The importance of the active site histidine for the activity of epoxide- or aziridine-based inhibitors of cysteine proteases. ChemMedChem 2007, 2, 120–128. [Google Scholar] [CrossRef]
- Mladenovic, M.; Ansorg, K.; Fink, R.F.; Thiel, W.; Schirmeister, T.; Engels, B. Atomistic Insights into the Inhibition of Cysteine Proteases: First QM/MM Calculations Clarifying the Stereoselectivity of Epoxide-Based Inhibitors. J. Phys. Chem. B 2008, 112, 11798–11808. [Google Scholar] [CrossRef]
- Wagner, A.; Le, T.A.; Brennich, M.; Klein, P.; Bader, N.; Diehl, E.; Paszek, D.; Weickhmann, A.K.; Dirdjaja, N.; Krauth-Siegel, R.L.; et al. Inhibitor-induced dimerization of an essential oxidoreductase from African trypanosomes. Angew. Chem. Int. Ed. 2019, 58, 3640–3644. [Google Scholar] [CrossRef] [PubMed]
- Vicik, R.; Hoerr, V.; Glaser, M.; Schultheis, M.; Hansell, E.; McKerrow, J.H.; Holzgrabe, U.; Caffrey, C.R.; Ponte-Sucre, A.; Moll, H.; et al. Aziridine-2,3-dicarboxylate inhibitors targeting the major cysteine protease of Trypanosoma brucei as lead trypanocidal agents. Bioorg. Med. Chem. Lett. 2006, 16, 2753–2757. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | Cath. L | Cath. B | Rhod. | DENV PR | Sortase A |
|---|---|---|---|---|---|
| % inh./Ki | % inh./Ki | % inh./Ki | % inh./Ki | % inh./Ki | |
| 1 | 57/nd | 16/nd | nd/nd | 19/nd | nd/nd |
| 2 | 56/nd | 33/nd | nd/nd | 24/nd | nd/nd |
| 3 | 60/nd | 22/nd | nd/nd | 28/nd | nd/nd |
| 4 | 99/0.66 | 20/nd | 75/0.29 | 10/nd | 11/nd |
| 5 | 8/nd | 10/nd | nd/nd | 11/nd | nd/nd |
| 6 | 50/nd | 21/nd | nd/nd | 12/nd | nd/nd |
| 7 | 98/1.6 | 63/4.4 | 45/nd | 31/nd | 16/nd |
| Structure | ωB97XD | MP2 | SCS-MP2 |
|---|---|---|---|
| Pre-complex | –8.8/–9.0/ | –12.3/–16.0/- | –10.3/–12.8/ |
| S-CH | +0.8/–11.5/–6.8 | –6.6/–16.0/–12.8 | –4.1/–12.9/–9.3 |
| S-CF | –3.3/–13.1/–3.1 | –13.2/–20.3/–12.5 | –11.1/–16.5/–10.1 |
| S-NO2 | –2.7/–9.3/+3.3 | –11.9/–17.0/–5.3 | –9.1/–13.3/–2.9 |
| Starting Structure | R(S-CNO2) | R(S-CH) | ΔE |
|---|---|---|---|
| MD 6 Frame 963 conformer with short Cys-S…H-N distance | 3.7 | 0.0 | |
| 3.3 | 1.4 | ||
| 3.1 | 1.7 | ||
| 2.9 | 2.5 | ||
| MD 6 Frame 1096 conformer with short Cys-S…H-N distance | 3.7 | 0.0 | |
| 3.3 | 0.7 | ||
| 3.1 | 0.8 | ||
| 2.9 | 2.6 | ||
| MD 6 Frame 542 conformer with short Cys-S…H-N distance | 4.9 | 0.0 | |
| 4.5 | 1.2 | ||
| 4.1 | 3.4 | ||
| 3.9 | 0.4 | ||
| 3.5 | 5.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, P.; Johe, P.; Wagner, A.; Jung, S.; Kühlborn, J.; Barthels, F.; Tenzer, S.; Distler, U.; Waigel, W.; Engels, B.; et al. New Cysteine Protease Inhibitors: Electrophilic (Het)arenes and Unexpected Prodrug Identification for the Trypanosoma Protease Rhodesain. Molecules 2020, 25, 1451. https://doi.org/10.3390/molecules25061451
Klein P, Johe P, Wagner A, Jung S, Kühlborn J, Barthels F, Tenzer S, Distler U, Waigel W, Engels B, et al. New Cysteine Protease Inhibitors: Electrophilic (Het)arenes and Unexpected Prodrug Identification for the Trypanosoma Protease Rhodesain. Molecules. 2020; 25(6):1451. https://doi.org/10.3390/molecules25061451
Chicago/Turabian StyleKlein, Philipp, Patrick Johe, Annika Wagner, Sascha Jung, Jonas Kühlborn, Fabian Barthels, Stefan Tenzer, Ute Distler, Waldemar Waigel, Bernd Engels, and et al. 2020. "New Cysteine Protease Inhibitors: Electrophilic (Het)arenes and Unexpected Prodrug Identification for the Trypanosoma Protease Rhodesain" Molecules 25, no. 6: 1451. https://doi.org/10.3390/molecules25061451