Vibrational Dynamics of Crystalline 4-Phenylbenzaldehyde from INS Spectra and Periodic DFT Calculations

 ,
,  , and
, and
Abstract
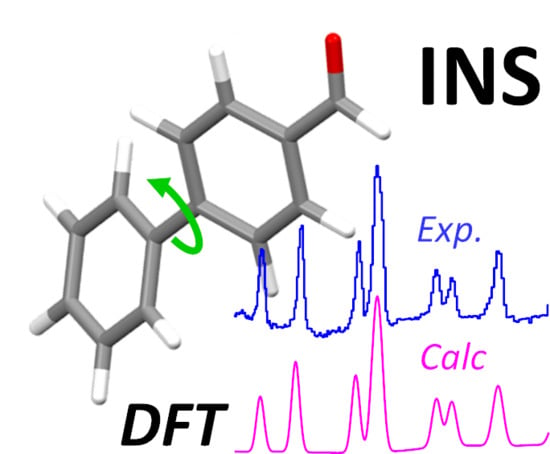
:
1. Introduction
2. Results and Discussion
2.1. Molecular Geometry and Intermolecular Interactions
2.2. Calculated vs. Experimental Spectra
2.3. Large Amplitude, Low Wavenumber Motions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Araujo, C.; Freire, C.S.R.; Nolasco, M.M.; Ribeiro-Claro, P.J.A.; Rudić, S.; Silvestre, A.J.D.; Vaz, P.D. Hydrogen Bond Dynamics of Cellulose through Inelastic Neutron Scattering Spectroscopy. Biomacromolecules 2018, 19, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Claro, P.J.A.; Vaz, P.D.; Nolasco, M.M.; Amado, A.M. Understanding the vibrational spectra of crystalline isoniazid: Raman, IR and INS spectroscopy and solid-state DFT study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 204, 452–459. [Google Scholar] [CrossRef]
- Parker, S.F.; Butler, I.R. Synthesis, Computational Studies, Inelastic Neutron Scattering, Infrared and Raman Spectroscopy of Ruthenocene. Eur. J. Inorg. Chem. 2019, 2019, 1142–1146. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Claro, P.J.; Vaz, P.D.; Nolasco, M.M.; Araujo, C.F.; Gil, F.P.S.C.; Amado, A.M. Vibrational dynamics of 4-fluorobenzaldehyde from periodic DFT calculations. Chem. Phys. Lett. X 2019, 2, 100006. [Google Scholar] [CrossRef]
- Bilski, P.; Druzbicki, K.; Jenczyk, J.; Mielcarek, J.; Wasicki, J. Molecular and Vibrational Dynamics in the Cholesterol-Lowering Agent Lovastatin: Solid-State NMR, Inelastic Neutron Scattering, and Periodic DFT Study. J. Phys. Chem. B 2017, 121, 2776–2787. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Ravindran, T.R.; Chandra, S.; Sarguna, R.M.; Das, B.K.; Sairam, T.N.; Sivasubramanian, V.; Thirmal, C.; Murugavel, P. Vibrational spectroscopic and computational studies on diisopropylammonium bromide. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 184, 211–219. [Google Scholar] [CrossRef]
- Ding, L.; Fan, W.-H.; Chen, X.; Chen, Z.-Y.; Song, C. Terahertz spectroscopy and solid-state density functional theory calculations of structural isomers: Nicotinic acid, isonicotinic acid and 2-picolinic acid. Mod. Phys. Lett. B 2017, 31. [Google Scholar] [CrossRef]
- Drużbicki, K.; Mielcarek, J.; Kiwilsza, A.; Toupet, L.; Collet, E.; Pajzderska, A.; Wąsicki, J. Computationally Assisted (Solid-State Density Functional Theory) Structural (X-ray) and Vibrational Spectroscopy (FT-IR, FT-RS, TDs-THz) Characterization of the Cardiovascular Drug Lacidipine. Cryst. Growth Des. 2015, 15, 2817–2830. [Google Scholar] [CrossRef]
- Bec, K.B.; Grabska, J.; Czarnecki, M.A.; Huck, C.W.; Wojcik, M.J.; Nakajima, T.; Ozaki, Y. IR Spectra of Crystalline Nucleobases: Combination of Periodic Harmonic Calculations with Anharmonic Corrections Based on Finite Models. J. Phys. Chem. B 2019, 123, 10001–10013. [Google Scholar] [CrossRef]
- Pawlukojć, A.; Hetmańczyk, Ł. INS, DFT and temperature dependent IR studies on dynamical properties of acetylcholine chloride. Vib. Spectrosc. 2016, 82, 37–43. [Google Scholar] [CrossRef]
- Drużbicki, K.; Mikuli, E.; Pałka, N.; Zalewski, S.; Ossowska-Chruściel, M.D. Polymorphism of Resorcinol Explored by Complementary Vibrational Spectroscopy (FT-RS, THz-TDS, INS) and First-Principles Solid-State Computations (Plane-Wave DFT). J. Phys. Chem. B 2015, 119, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Parker, S.F. Structure and vibrational spectroscopy of methanesulfonic acid. R. Soc. Open Sci. 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deringer, V.L.; George, J.; Dronskowski, R.; Englert, U. Plane-Wave Density Functional Theory Meets Molecular Crystals: Thermal Ellipsoids and Intermolecular Interactions. Acc. Chem. Res. 2017, 50, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rerat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WILEY Interdiscip. Rev. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual 2017; University of Torino: Torino, Italy, 2017. [Google Scholar]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeitschrift Fur Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Pawlukojć, A.; Hołderna-Natkaniec, K.; Bator, G.; Natkaniec, I. L-glutamine: Dynamical properties investigation by means of INS, IR, RAMAN, 1H NMR and DFT techniques. Chem. Phys. 2014, 443, 17–25. [Google Scholar] [CrossRef]
- Kearley, G.J. A review of the analysis of molecular vibrations using INS. Nucl. Instruments Methods Phys. Res. Sect. A.. 1995, 354, 53–58. [Google Scholar] [CrossRef]
- Verdal, N.; Hudson, B.S. Inelastic neutron scattering and periodic DFT studies of crystalline aromatic materials: Biphenylene—A Mills-Nixon molecule. Chem. Phys. Lett. 2007, 434, 241–244. [Google Scholar] [CrossRef]
- Vaz, P.D.; Nolasco, M.; Fonseca, N.; Amado, A.M.; Amorim Da Costa, A.M.; Félix, V.; Drew, M.G.B.; Goodfellow, B.J.; Ribeiro-Claro, P.J.A. C-H⋯O hydrogen bonding in 4-phenyl-benzaldehyde: A comprehensive crystallographic, spectroscopic and computational study. Phys. Chem. Chem. Phys. 2005, 7, 3027–3034. [Google Scholar] [CrossRef]
- Srishailam, K.; Reddy, B.V.; Rao, G.R. Investigation of torsional potentials, hindered rotation, molecular structure and vibrational properties of some biphenyl carboxaldehydes using spectroscopic techniques and density functional formalism. J. Mol. Struct. 2019, 1196, 139–161. [Google Scholar] [CrossRef]
- Nolasco, M.M.; Amado, A.M.; Ribeiro-Claro, P.J.A. Computationally-assisted approach to the vibrational spectra of molecular crystals: Study of hydrogen-bonding and pseudo-polymorphism. ChemPhysChem 2006, 7, 2150–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, N.F.C.; Nolasco, M.M.; Ribeiro-Claro, P.J.A. Effects of hydrogen-bond and cooperativity in the vibrational spectra of Luminol. Vib. Spectrosc. 2013, 64, 119–125. [Google Scholar] [CrossRef]
- Sardo, M.; Amado, A.M.; Ribeiro-Claro, P.J.A. Hydrogen bonding in nitrofurantoin polymorphs: A computation-assisted spectroscopic study. J. Raman Spectrosc. 2009, 40, 1956–1965. [Google Scholar] [CrossRef]
- Groner, P. Barrier, version 22; University of Missouri: Kansas City, MO, USA, 2019. [Google Scholar]
- Godunov, I.A.; Bataev, V.A.; Abramenkov, A.V.; Pupyshev, V.I. The Barriers to Internal Rotation of Benzaldehyde and Benzoyl Fluoride: “Reconciliation{’’} Between Theory and Experiment. J. Phys. Chem. A 2014, 118, 10159–10165. [Google Scholar] [CrossRef]
- Araujo, C.F.; Nolasco, M.M.; Ribeiro-Claro, P.J.A.; Rudić, S.; Silvestre, A.J.D.; Vaz, P.D.; Sousa, A.F. Inside PEF: Chain Conformation and Dynamics in Crystalline and Amorphous Domains. Macromolecules 2018, 51, 3515–3526. [Google Scholar] [CrossRef]
- Kurapov, P.B.; Klyuev, N.A.; Tyulin, V.I. Potential function of inner rotation of diphenyl. Theor. Exp. Chem. 1988, 24, 190–197. [Google Scholar] [CrossRef]
- Tylli, H.; Konschin, H. Raman-spectroscopic study of low frequency vibrations in anisole and anisole-d3. J. Mol. Struct. 1977, 42, 7–12. [Google Scholar] [CrossRef]
- Chiang, W.Y.; Laane, J. Fluorescence-spectra and torsional potential functions for trans-stilbene in its S0 and S1 (pi,pi*) electronic states. J. Chem. Phys. 1994, 100, 8755–8767. [Google Scholar] [CrossRef]
- Chiang, W.Y.; Laane, J. Fluorescence-spectra and torsional potential energy functions for 4-methoxy-trans-stilbene in its S0 and S1 (pi,pi*) electronic states. J. Phys. Chem. 1995, 99, 11823–11829. [Google Scholar] [CrossRef]
- Vaz, P.D.; Nolasco, M.M.; Gil, F.P.S.C.; Ribeiro-Claro, P.J.A.; Tomkinson, J. Hydrogen-bond dynamics of C-H···O interactions: The chloroform···acetone case. Chem. A Eur. J. 2010, 16, 9010–9017. [Google Scholar] [CrossRef] [PubMed]
- Vaz, P.D.; Nolasco, M.M.; Ribeiro-Claro, P.J.A. Intermolecular C-H center dot center dot center dot O interactions in cyclopentanone: An inelastic neutron scattering study. Chem. Phys. 2013, 427, 117–123. [Google Scholar] [CrossRef]
- Li, J.C.; Ross, D.K. Evidence for two kinds of hydrogen bond in ice. Nature 1993, 365, 327–329. [Google Scholar] [CrossRef]
- Corsaro, C.; Crupi, V.; Longo, F.; Majolino, D.; Venuti, V.; Wanderlingh, U. Mobility of water in Linde type A synthetic zeolites: An inelastic neutron scattering study. J. Phys. -Condens. Matter 2005, 17, 7925–7934. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 155114. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Cuesta, A.J. aCLIMAX 4.0.1, The new version of the software for analyzing and interpreting INS spectra. Comput. Phys. Commun. 2004, 157, 226–238. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D; Jmol Development Team: Minneapolis, MN, USA, 2016; Available online: http://jmol.sourceforge.net/ (accessed on 24 February 2020).
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X-ray (A) | X-ray (B) | CASTEP(A) | CASTEP(B) | G09 | |
|---|---|---|---|---|---|
| Bond length/pm | |||||
| C7=O | 118.1 | 118.7 | 122.7 | 122.8 | 120.6 |
| C4-C8 | 149.2 | 145.5 | 147.8 | 147.8 | 147.7 |
| C1-C7 | 148.7 | 148.4 | 146.8 | 146.8 | 147.4 |
| Dihedral angle/° | |||||
| C2C1-C7O | −1.95 | −2.06 | −2.16 | −2.26 | −0.10 |
| C3C4-C8C9 | 31.9 | 31.6 | 32.6 | 31.0 | 38.9 |
| Distance/pm | |||||
| C9(H)···O | 341 | 361 | 333.7 | 348.1 | - |
| π...π (C1···C4’) 1 | 414.7 | 411.1 | 418.5 | 418.1 | - |
| CASTEP 1 | Observed | Approx. Description |
|---|---|---|
| 3104 | 3030 | νC-H |
| 1379 | 1384 | βC7-H |
| 1298 | 1312 | βC-H |
| 1170 | 1164 | βC-H |
| 1106 | 1108 | βC-H |
| 1077 | 1075 | βC-H |
| 1030 | 1032 | βC-H |
| 994 | 994 | γC7-H |
| 975 | 973 | γC-H |
| 953 | 951 | γC-H |
| 926 | 929 | γC-H |
| 845 | 855 | γC-H |
| 833 | 832 | νC-C |
| 762 | 762 | γC-H |
| 720 | 715 | δring |
| 702 | 702 | δring |
| 638 | 641 | αring |
| 622 | 623 | βC=O |
| 609 | 610 | αring |
| 545 | 545 | δring |
| 493 | 493 | αring |
| 476 | 475 | δring |
| 408 | 408 | δring |
| 385 | 388 | βC8-C4 |
| 317 | 324 | γC8-C4 |
| 278 | 281 | αring |
| 225 | 229 | γC-CHO |
| 210-201 | 212-207 | βC-CHO |
| 153 | 152-162 | τ-CHO |
| 130 | 136 | βC4-C8 |
| 109-117 | 118-128 | τ-C6H5 |
| 89 | 101 | Libration (Ia) |
| 67 | 86 | Librations (Ic, Ib) |
| 48 | 70 | Translation (b,c) |
| 37 | 45 | Translation(a) |
| System | V4/cm−1 | Ref. | |
|---|---|---|---|
| 4-phenylbenzaldehyde | INS, crystal | 2600 ± 80 | This work |
| Biphenyl | Raman, crystal | 2650–200 | [29] |
| Anisole | Raman, crystal | 4033 | [30] |
| Trans-Stilbene | Fluorescence, supersonic jet | 1550 | [31] |
| 4-Methoxy-stilbene | Fluorescence, supersonic jet | 1430 | [32] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolasco, M.M.; Araujo, C.F.; Vaz, P.D.; Amado, A.M.; Ribeiro-Claro, P. Vibrational Dynamics of Crystalline 4-Phenylbenzaldehyde from INS Spectra and Periodic DFT Calculations. Molecules 2020, 25, 1374. https://doi.org/10.3390/molecules25061374
Nolasco MM, Araujo CF, Vaz PD, Amado AM, Ribeiro-Claro P. Vibrational Dynamics of Crystalline 4-Phenylbenzaldehyde from INS Spectra and Periodic DFT Calculations. Molecules. 2020; 25(6):1374. https://doi.org/10.3390/molecules25061374
Chicago/Turabian StyleNolasco, Mariela M., Catarina F. Araujo, Pedro D. Vaz, Ana M. Amado, and Paulo Ribeiro-Claro. 2020. "Vibrational Dynamics of Crystalline 4-Phenylbenzaldehyde from INS Spectra and Periodic DFT Calculations" Molecules 25, no. 6: 1374. https://doi.org/10.3390/molecules25061374