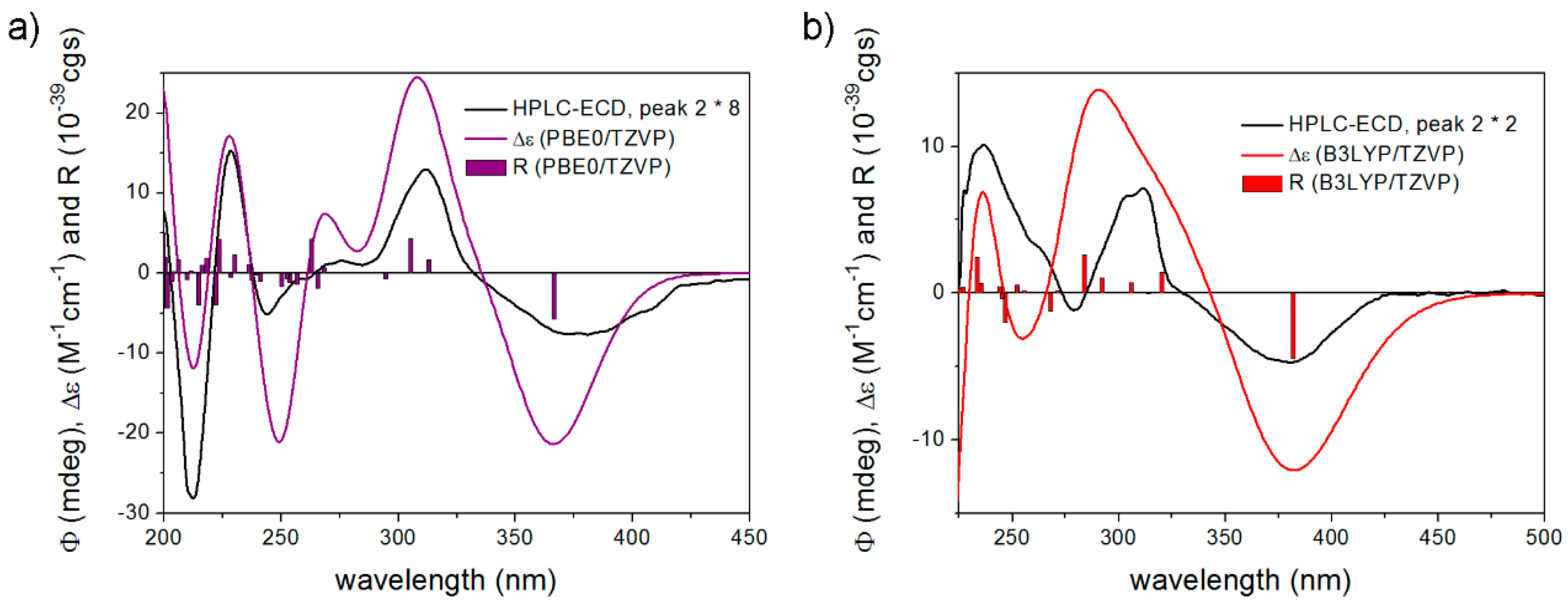
Melting points were determined on a Kofler hot-stage apparatus (Wagner and Munz, Munich, Germany) and are uncorrected. The reactions were monitored by thin layer chromatography (TLC). TLC plates were visualized under a UV lamp and developed by phosphomolybdic acid solution. The NMR spectra were recorded on Bruker-AMX 400 (1H: 400 MHz; 13C: 100 MHz, Bruker, Karlsruhe, Germany; Billerica, MA, USA) and Bruker Aspect 3000 (1H: 360 MHz, 13C: 90 MHz, Bruker, Karlsruhe, Germany; Billerica, MA, USA) spectrometers using TMS and the solvent peak as internal standard. Chemical shifts were reported as δ in ppm and 3JH,H coupling constants in Hz. Chiral HPLC separation of 10a and 11a were performed on a Jasco HPLC system with Chiralpak IC column (5 μm, 150 × 4.6 mm, hexan/propan-2-ol 1:1 eluent, 1 mLmin−1 flow rate, Daicel Chemical Industries Ltd., Tokyo, Japan) and HPLC-ECD spectra were recorded in stopped-flow mode on a JASCO J-810 electronic circular dichroism spectropolarimeter (JASCO Inc. Tokyo, Japan) equipped with a 10 mm HPLC flow cell. ECD ellipticity (ϕ) values were not corrected for concentration. For an HPLC-ECD spectrum, three consecutive scans were recorded and averaged with 2 nm bandwidth, 1 s response, and standard sensitivity. The HPLC-ECD spectrum of the eluent recorded in the same way was used as background. The concentration of the injected sample was set so that the HT value did not exceed 500 V in the HT channel down to 230 nm. IR spectra were recorded on a JASCO FT/IR-4100 spectrometer (JASCO Inc. Tokyo, Japan) and absorption bands are presented in cm−1. Electrospay Quadrupole Time-of-Flight HRMS measurements were performed with a MicroTOF-Q type QqTOF MS instrument equipped with an ESI source from Bruker (Bruker Daltoniks, Bremen, Germany).
3.2. Bioassay on AChE Inhibitory Activity
The AChE assay was performed using the method of Ellman et al. with slight modification [
51]. The cortex of Sprague–Dawley rat (400
–500 g, obtained from the Animal Center of Shanghai Institute of Materia Medica) was homogenized in cold sodium phosphate buffer (75 mM, pH 7.4) as the AChE source. All the experimental procedures were approved by the Animal Care and Use Committee of Shanghai Institute of Materia Medica (#2012-02-ZHY-32 and 2015-03-ZHY-64). The assay solution consisted of 50 μL of 0.1 M phosphate buffer, 50 μL of 0.2% 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, Sigma), 109 μL of deionized water, 10 μL of rat cortex homogenate, and 30 μL of 2 mM acetylthiocholine iodide (Sigma) as the substrate of the AChE enzymatic reaction. An appropriate concentration of compound (0.1 mM to 5 mM; 1 μL) and the assay solution were incubated for 20 min at room temperature and the reaction was terminated with 50 μL of 3% (
w/
v) sodium dodecylsulfate (SDS). The production of the yellow anion of 5-thio-2-nitrobenzoic acid was measured with a microplate reader (DTX 880, Beckman Coulter, Brea, CA, USA) at 450 nm. The inhibition percentage caused by the presence of test compound was calculated, and the IC
50 was defined as the concentration of the compound that reduced 50% of the enzymatic activity without inhibitor. The bioassay on neuroprotective activity was carried out according to the reference 25.
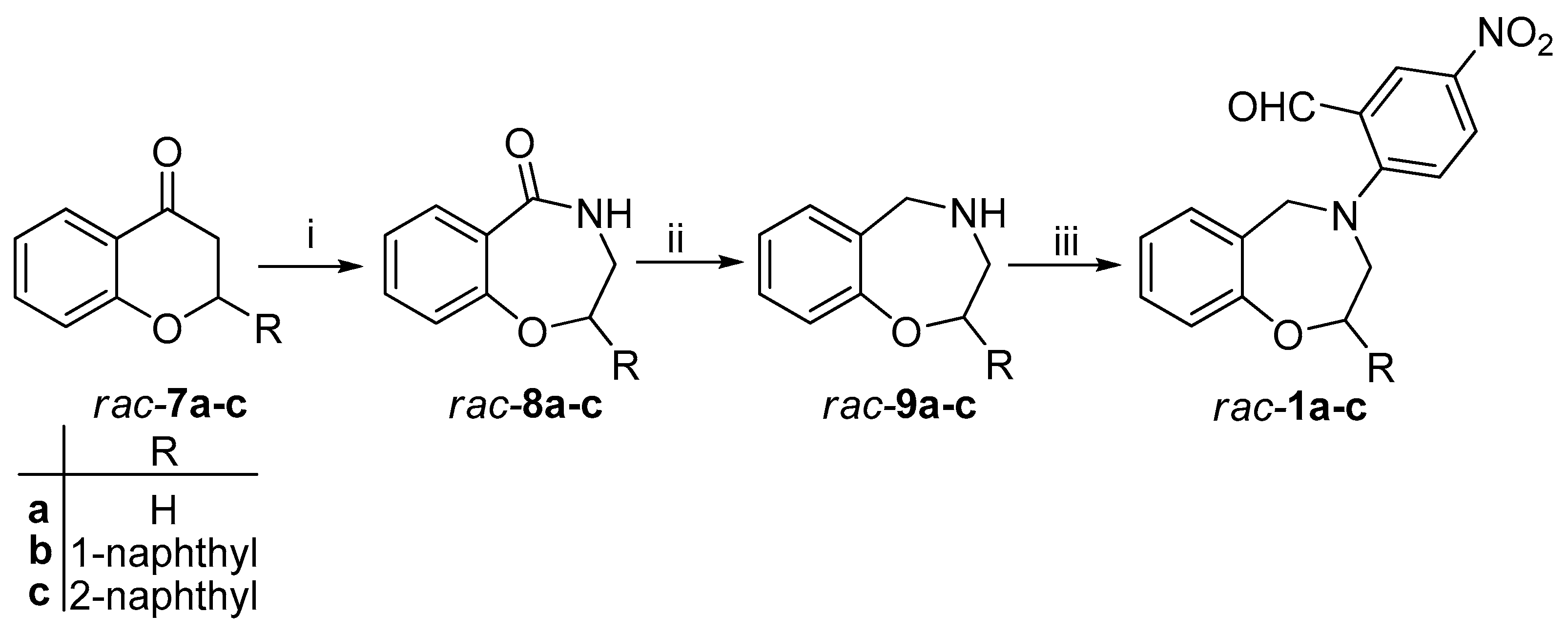
3,4-Dihydro-1,4-benzoxazepin-5(2H)-one (
rac-
8a): To a stirred solution of chroman-4-one (
7a) (10.0 g, 67.49 mmol) in water and sulfuric acid 1:1 (100 mL), sodium azide (6.6 g, 101.5 mmol) was added in two equal portions at 0 °C and stirred at room temperature. The reaction was monitored by TLC,visualized under a UV lamp and developed by phosphomolybdic acid solution. After 5 h, the reaction mixture was neutralized with sodium hydroxide solution and the aqueous layer was extracted with dichloromethane (5 × 50 mL). The combined organic layers were washed with water (50 mL), dried over MgSO
4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography on silica (EtOAc/hexane 2:5) to afford the
rac-
8a product as white solid [8.60 g, 52.71 mmol, 78%, mp 109–110 °C (lit. [
52] mp 115–116 °C)].
1H NMR (400 MHz, CDCl
3): δ = 3.50 (dd,
J = 9.6 Hz and 5.2 Hz, 3-H, 2 H), 4.39 (m,
J = 9.6 Hz and 5.2 Hz, 2-H, 2 H), 7.03 (d,
J = 8.0 Hz, 9-H, 1 H), 7.10 (m, 7-H, 1 H), 7.4 (m, 8-H, 1 H), 7.9 (dd,
J = 8.0 Hz and 1.6 Hz, 6-H, 1 H), 8.2 (br s, 4-H, 1 H).
13C NMR (100 MHz, CDCl
3): δ = 41.3 (C-3), 73.2 (C-2), 121.3 (C-9), 122.8 (C-7), 123.9 (C-5a), 131.6 (C-6), 133.3 (C-8), 155.3 (C-9a), 170.9 (C-5). IR (KBr): 1661, 3060, 3183, 3287 cm
−1. HRMS-ESI (
m/
z): [M + Na]
+ calc’d for C
9H
9NO
2Na, 186.053; found: 186.053.
2,3,4,5-Tetrahydro-1,4-benzoxazepine (
rac-
9a): To a stirred solution of
rac-
8a (4.0 g, 24.51 mmol) in dry THF (60 mL), 2.0 M lithium aluminium hydride solution in THF (5 mL, 0.38 g, 10.02 mmol) was added dropwise and the mixture was refluxed for 3 h. After cooling to room temperature, ethyl-acetate (5 mL), methanol (5 mL) and water (50 mL) were added and the mixture was concentrated under reduced pressure. The residue was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with water (20 mL), dried over MgSO
4, filtered and concentrated under reduced pressure. The
rac-
9a product was isolated as yellow oil (2.92 g, 80%) [
53].
1H NMR (400 MHz, CDCl
3): δ = 2.17 (s, 4-H, 1 H), 3.15 (m, 3-H, 2 H), 3.90 (s, 5-H, 2 H), 3.96 (m, 2-H, 2 H), 6.92–7.00 (m, 7-H and 9-H, 2 H), 7.08–7.20 (m, 6-H and 8-H, 2 H).
13C NMR (100 MHz, CDCl
3): δ = 52.0 (C-5), 52.8 (C-3), 74.8 (C-2), 120.8 (C-9), 123.2 (C-7), 128.1 (C-6), 129.1 (C-8), 134.6 (C-5a), 159.8 (C-9a). IR (KBr): 766, 1226, 1488, 2858, 2932, 3312 cm
−1. HRMS-ESI (
m/
z): [M + Na]
+ calc’d for C
9H
11NONa, 172.074; found: 186.074.
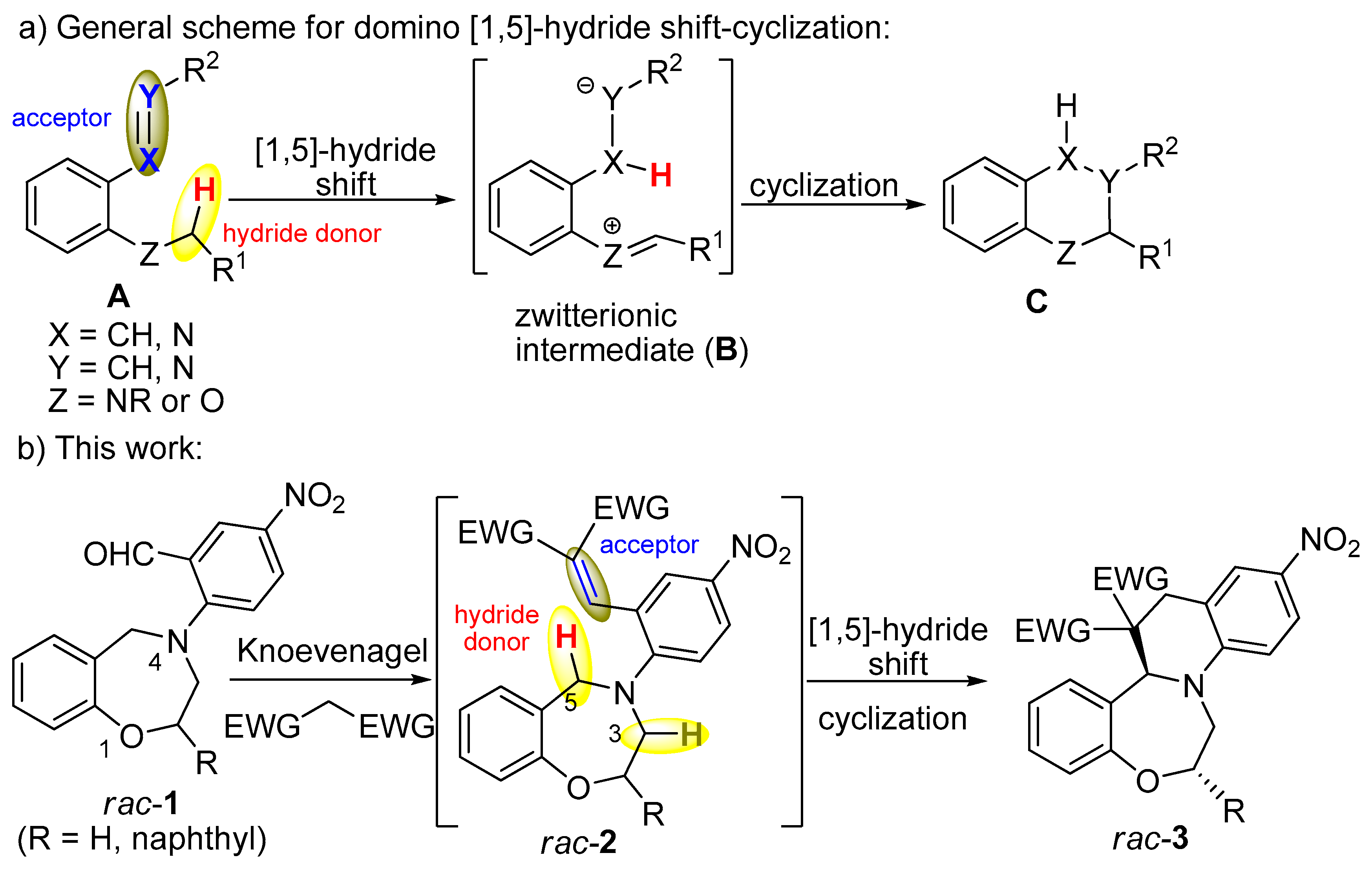
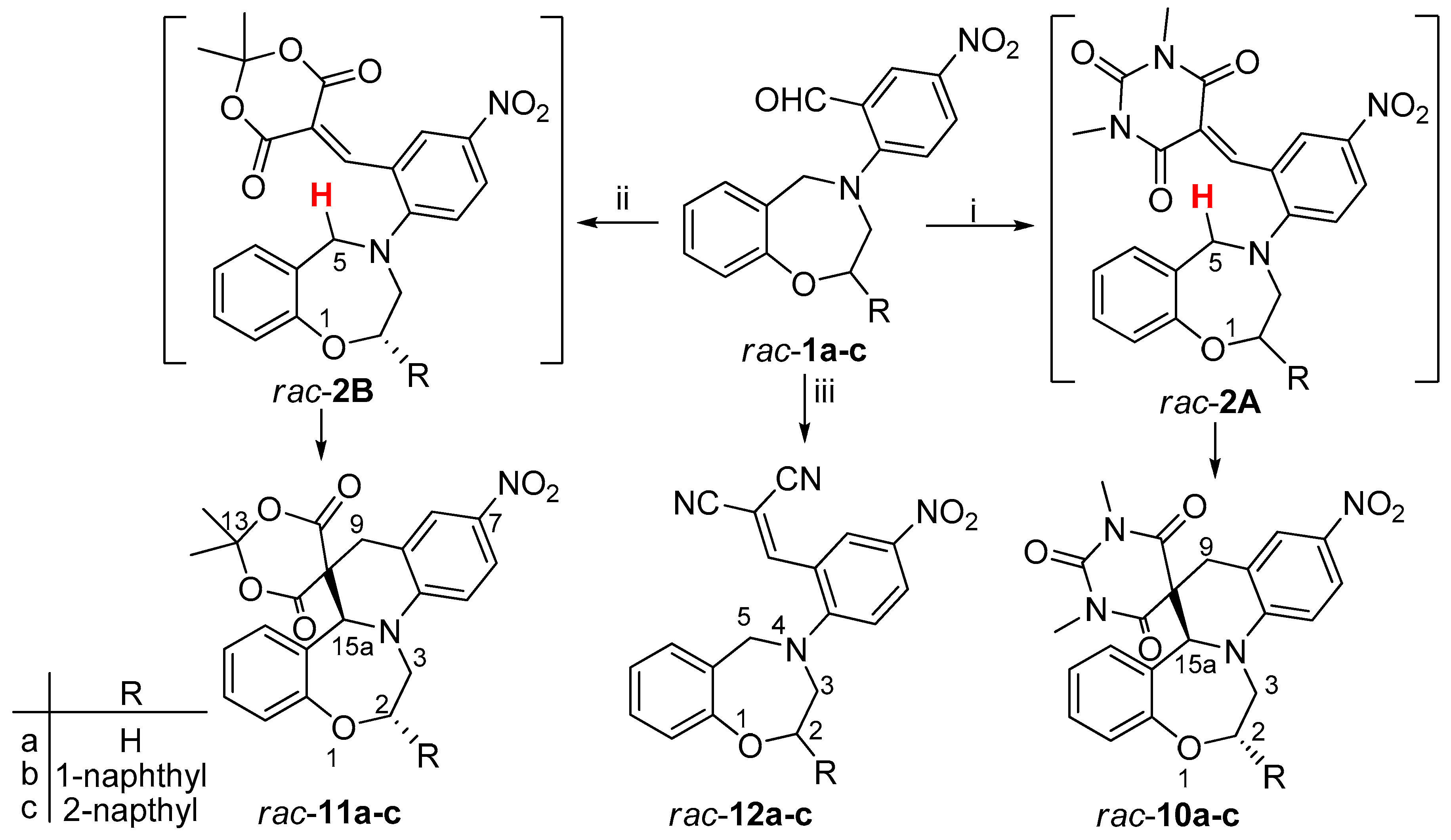
2-(2,3-Dihydro-1,4-benzoxazepin-4(5H)-yl)-5-nitrobenzaldehyde (rac-1a): To the stirred solution of rac-9a (1.50 g, 9.19 mmol) in dry toluene (25 mL), anhydrous K2CO3 (1.85 g, 13.39 mmol) and 2-fluoro-5-nitrobenzaldehyde (2.04 g, 12.06 mmol) were added and the mixture was refluxed for 5 h. After cooling to room temperature, the K2CO3 was filtered off and toluene was removed under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl-acetate/hexane 4:1) to give rac-1a as yellow solid (2.61 g, 95%, mp 101–102 °C). 1H NMR (400 MHz CDCl3): δ = 3.79 (m, J = 4.8 Hz, 3-H, 2 H), 4.35 (m, J = 4.8 Hz, 2-H, 1 H), 4.73 (s, 5-H, 2 H), 6.97 (d, J = 7.6 Hz, 9-H, 1 H), 7.01 (d, J = 9.2 Hz, 6′-H, 1 H), 7.09 (m, 7-H, 1 H), 7.23–7.27 (m, 6-H and 8-H, 2 H), 8.13 (dd, J = 9.2 Hz and 2.8 Hz, 5′-H, 1 H), 8.61 (d, J = 2.8 Hz, 3′-H, 1 H), 9.99 (s, 1H, -CHO). 13C-NMR (100 MHz, CDCl3): δ = 55.9 (C-3), 58.21 (C-5), 70.34 (C-2), 117.3 (C-9), 120.4 (C-6′), 123.4 (C-7), 124.0 (C-6a), 126.7 (C-2′), 129.0 (C-3′), 129.3* (C-8), 129.4* (C-6), 129.8* (C-5′), 139.7 (C-4′), 156.7 (C-9a), 158.7 (C-1′), 188.4 (-CHO). *exchangable signals. IR (KBr): 761, 1330, 1491, 1600, 1684 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C16H14N2O4Na, 321.085; found: 321.085.
2-(2,3-Dihydro-1,4-benzoxazepin-4(5H)-yl)-5-nitrobenzylidene]propanedinitrile (rac-12a): To a stirred solution of rac-1a (100 mg, 0.34 mmol) in chloroform (10 mL), anhydrous MgSO4 (150 mg, 1.24 mmol) and malononitrile (130 mg, 1.97 mmol) were added and the mixture was refluxed for 8 h. After cooling to room temperature, the MgSO4 was filtered off and chloroform was removed under reduced pressure. Water (10 mL) and dichloromethane (20 mL) were added, the two layers were separated and the aqueous phase was washed with dichloromethane (2 × 10 mL). The combined organic layers were washed with concentrated NaHCO3 solution, dried over MgSO4, filtered and concentrated under reduced pressure. The oily product was crystallized with ether to give rac-12a as brown solid (72 mg, 62%, mp 246–248 °C). 1H NMR (400 MHz CDCl3): δ = 3.75 (m, 3-H, 2 H), 4.34 (m, 2-H, 2 H), 4.45 (s, 5-H, 2 H), 7.07 (d, J = 8.0 Hz, 6-H, 1 H), 7.15–7.20 (m, 3′-H,7-H, 9-H, 3 H), 7.26-7.33 (m, 8-H, 1 H), 7.73 (s, 7′-H, 1 H), 8.2 (dd, J = 9.2 Hz and 2.4 Hz, 4′-H, 1 H), 8.84 (d, J = 2.4 Hz, 6′-H, 1 H). 13C NMR (100 MHz, CDCl3): δ = 56.8 (C-3), 59.1 (C-5), 70.6 (C-2), 84.1 (C-2”),111.8 (CN), 112.9 (CN), 118.8 (C-9), 120.8 (C-3′), 121.5 (C-1′), 124.1 (C-7),125.9 (C-6′), 127.6 (C-5a), 128.9 (C-4′), 129.4 (C-6), 129.8 (C-8), 140.9 (C-5′), 156.9 (C-7′), 158.2 (C-9a), 159.2 (C-2′). IR (KBr): 781, 1509, 1570, 2232, 2925 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C19H14N4O3Na, 369.096; found: 369.096.
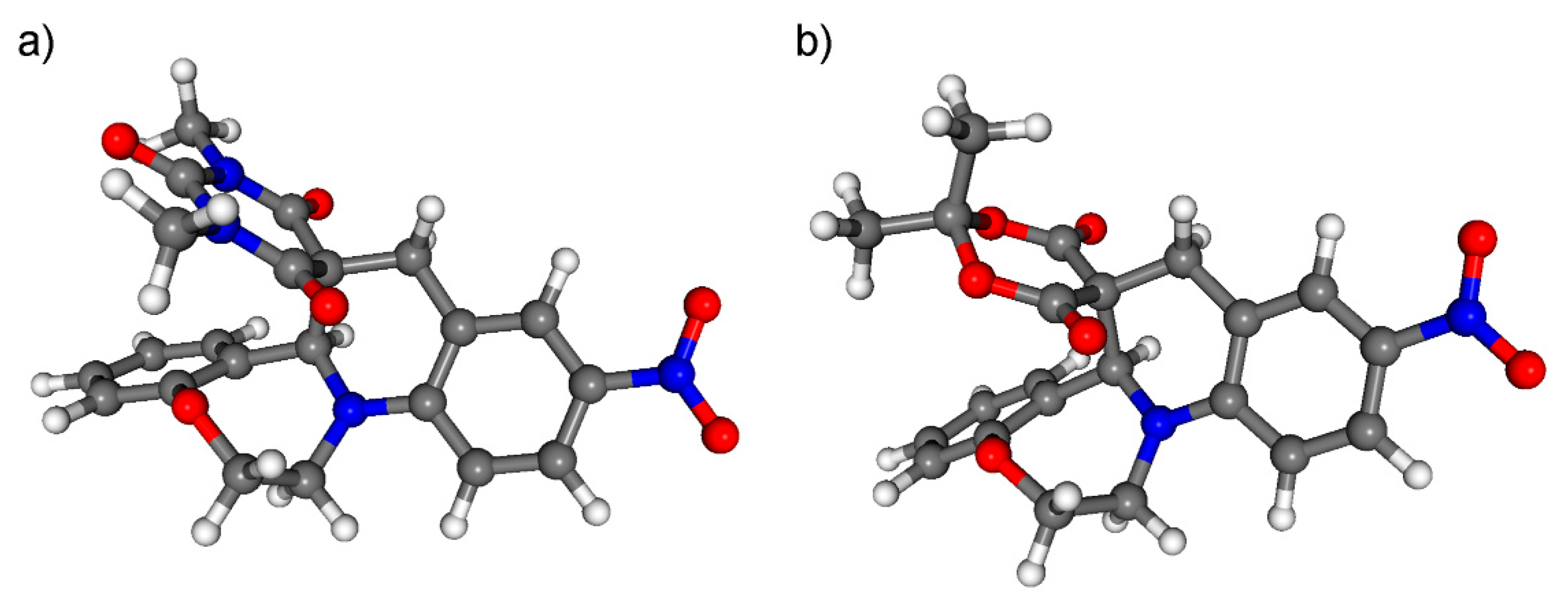
1,3-Dimethyl-11’-nitro-1’,2’-dihydro-2H,7b’H,9’H-spiro[pyrimidine-5,8’-quinolino[1,2-d][1,4]benzoxazepine]-2,4,6(1H,3H)-trione (rac-10a): To a stirred solution of rac-1a (100 mg, 0.34 mmol) in chloroform (10 mL), anhydrous MgSO4 (150 mg, 1.25 mmol) and 1,3-dimetylbarbituric acid (80 mg, 0.51 mmol) were added and the mixture was refluxed for 6 h. After cooling to room temperature, the MgSO4 was filtered off and chloroform was removed under reduced pressure. Water (10 mL) and dichloromethane (20 mL) were added and two layers were separated. The aqueous phase was extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with concentrated NaHCO3 solution, dried over MgSO4, filtered and concentrated under reduced pressure. The oily product was crystallized with ether to give rac-10a as yellow solid (139 mg, 95%, mp 247–250 °C). 1H NMR (400 MHz DMSO-d6): δ = 2.76 (s, N-CH3, 3 H), 2.89 (s, N-CH3, 3 H), 3.20 (d, J = 17.2 Hz, 9-Ha, 1 H), 3.48 (m, 3-Ha, 1 H), 3.71–3.76 (overlapping m, 9-Hb and 3-Hb, 2 H), 3.99–4.05 (m, 2-Ha, 1 H), 4.22 (m, J = 3.6 Hz and 16.4 Hz, 2-Hb, 1 H), 4.82 (s, 15-Ha, 1 H), 6.98–7.02 (m, 16-H, 19-H, 2 H), 7.10 (d, J = 9.2 Hz, 5-H, 1 H), 7.17 (m, 17-H, 1 H), 7.39 (m, 18-H, 1 H), 7.97 (dd, J = 9.2 Hz and 2.8 Hz, 6-H, 1 H), 8.02 (bs, 8-H, 1 H). 13C NMR (100 MHz, DMSO-d6): δ = 28.0 (N-CH3), 28.1 (N-CH3), 33.6 (C-9), 42.8 (C-3), 51.3 (C-10), 69.8 (C-2), 70.5 (C-15a), 109.9 (C-5), 121.9 (C-15b), 122.4 (C-6), 123.1 (C-8), 124.4 (C-17, C-19), 127.2 (C-8a), 129.7 (C-18), 131.7 (C-16), 136.1 (C-7), 148.4 (C-13), 149.9 (C-19a), 152.9 (C-4a), 166.8 (C-15), 168.8 (C-11). IR (KBr): 1320, 1680, 2924 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C22H20N4O6Na, 459.128; found: 459.128.
2,2-Dimethyl-11’-nitro-1’,2’-dihydro-7b’H,9’H-spiro[1,3-dioxane-5,8’-quinolino[1,2-d][1,4]benzoxazepine]-4,6-dione (rac-11a): To the stirred solution of rac-1a (100 mg, 0.34 mmol) in chloroform (10 mL), anhydrous MgSO4 (150 mg, 1.25 mmol) and Meldrum’s acid (98 mg, 0.68 mmol) were added and the mixture was refluxed for 6 h. After cooling to room temperature, the MgSO4 was filtered off and chloroform was removed under reduced pressure. Water (10 mL) and dichloromethane (20 mL) were added and layers were separated. The aqueous phase was washed with dichloromethane (2 × 10 mL) and the combined organic layers were washed with concentrated NaHCO3 solution, dried over MgSO4, filtered and concentrated under reduced pressure. The oily product was crystallized with ether to give rac-11a as yellow solid (123 mg, 87%, mp 236–238 °C). 1H NMR (360 MHz, DMSO-d6): δ = 0.94 (s, CH3, 3 H), 1.58 (s, CH3, 3 H), 3.57 (m, 3-Ha, 9-H, 3 H), 3.71 (m, 2-Ha, 1 H), 4.03 (m, 3-Hb, 1 H), 4.20 (m, 2-Hb, 1 H), 5.01 (s, 15a-H, 1 H), 7.07-7.14 (m, H-16, H-17, H-19, 3 H), 7.21 (m, 18-H, 1 H), 7.45 (m, 6-H, 1 H), 8.00 (m, 5-H, 8-H, 2 H). 13C NMR (90 MHz, CDCl3): δ = 26.4 (CH3), 29.5 (CH3), 34.6 (C-9), 43.2 (C-3), 49.3 (C-10), 68.9 (C-2), 69.7 (C-15a), 105.2 (C-13), 110.0 (C-19), 119.9 (C-8a), 122.8 (C-5), 123.7 (C-6), 124.6 (C-8), 124.7 (C-17), 130.4 (C-18), 131.5 (C-16), 135.9 (C-7), 148.3 (C-19a), 153.6 (C-4a), 163.6 (C-15), 167.7 (C-11). IR (KBr): 1262, 1313, 1736, 2924 cm−1.
2-(Naphthalen-1-yl)-chroman-4-one (
rac-
7b): To the stirred solution of the corresponding chalcone derivative (4.3 g, 16mmol) in EtOH, 130 mL of cc. NaOAc solution was added and refluxed for 5 h. After cooling it down to room temperature, EtOH was removed under reduced pressure. After addition of water, the aqueous layer was extracted with dichloromethane (5 × 50 mL). The combined organic layers were dried over MgSO
4, filtered and concentrated under reduced pressure. The oily crude product was crystallized with cold diisopropyl-ether affording yellow crystals (3.14 g, 73%, mp 74–76 °C) [
54].
1H-NMR (360 MHz, CDCl
3): δ = 3.05 (dd,
J = 14.4 and 3.6 Hz, 1 H, 3-H
a), 3.24 (dd,
J = 14.4 and 10.8 Hz, 1 H, 3-H
b), 6.19 (dd,
J = 10.8 Hz and 3.6 Hz, 1 H, 2-H), 7.07-7.52 (m, 2 H, 6′-H, 7′-H), 7.53–7.57 (m, 4 H, 3′-H, 4′-H, 5′-H, 8′-H), 7.75 (d, 1 H, 8-H), 7.89 (m, 2 H, 2′-H, 6′-H), 8.00 (m, 2 H, 5-H, 7-H).
13C-NMR (90 MHz, CDCl
3): δ = 43.9 (C-3), 76.8 (C-2), 118.2 (C-8), 121.1 (C-4a), 121.7 (C-6), 122.8 (C-8′), 123.8 (C-3′), 125.3 (C-6′), 125.9 (C-2′), 126.6 (C-7′), 127.1 (C-5), 129.1 (C-4′), 129.3 (C-5′), 130.1 (C-8a’), 133.8 (C-4a’), 134.1 (C-1), 136.2 (C-7), 161.7 (C-8a), 192.2 (C-4). IR (KBr): 1222, 1302, 1606, 1684, 3050 cm
−1.
2-(Naphthalen-2-yl)-chroman-4-one (
rac-
7c): To a stirred solution of the chalcone derivative (4.3 g, 16 mmol) in EtOH, 130 mL of cc. NaOAc solution was added and refluxed for 5 h. After cooling it down to room temperature, EtOH was removed under reduced pressure. The residue was taken in water and extracted with dichloromethane (5 × 50 mL). The combined organic layers were dried over MgSO
4, filtered and concentrated under reduced pressure. The oily crude product was crystallized with cold diisopropyl-ether affording yellow crystals (3.27 g, 76%, mp 110–112 °C) [
54].
1H-NMR (360 MHz, CDCl
3): δ = 2.90 (dd,
J = 16.5 Hz and 2.5 Hz, 1 H, 3-H
a) 3.09 (dd,
J = 16.5 Hz and 13.0 Hz, 1 H, 3-H
b). 5.56 (dd,
J=13.0 Hz and 2.5 Hz, 1 H, 2-H). 7.01-7.07 (m, 2 H, 6′-H, 7′-H). 7.47–7.55 (m, 4 H, 3′-H, 4′-H, 5′-H, 8′-H). 7.82–7.92 (m, 5 H, 5-H, 1′-H, 5-H, 6-H, 7-H), 7.93 (d,
J = 7.6 Hz, 1 H, 8-H).
13C-NMR (90 MHz, CDCl
3): δ = 44.5 (C-3), 79.6 (C-2), 118.1 (C-8), 120.9 (C-4a), 121.6 (C-6), 123.6 (C-3′), 125.3 (C-4′), 126.4 (C-6′, C-7′), 127.0 (C-1′), 127.7 (C-5′), 128.1 (C-5), 128.7 (C-8′), 133.1 (C-8a’), 133.3 (C-4a’), 135.9 (C-2′), 136.2 (C-7), 161.4 (C-8a), 191.8 (C-4). IR (KBr): 1063, 1223, 1688, 3025 cm
−1.
2-(Naphthalen-1-yl)-3,4-dihydrobenzo[f][1,4]oxazepin-5(2H)-one (rac-8b): To the stirred soluton of rac-7b (3.0 g, 11 mmol) in acetic acid (30 mL), sodium azide (1.1 g, 17 mmol) was added in two parts at 0 °C, then 3.0 mL of cc. H2SO4 was added dropwise, and the reaction mixture was stirred at room temperature for 5 h. The reaction mixture was neutralized with sodium hydroxide solution and extracted with dichloromethane (5 × 50 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography on silica (hexane/EtOAc 4:1) to give the product as white powder (3.14 g, 73%, mp 74–76 °C). 1H-NMR (400 MHz, CDCl3): δ = 3.57–3.62 (m, 1 H, 3-Ha) 3.65–3.71 (m, 1 H, 3-Hb), 5.58 (m, 1 H, 2-H), 7.10 (d, J = 8.0 Hz 1 H, 9-H), 7.21 (m, 1 H, 7-H), 7.46-7.49 (m, 4 H, 2-H’, 3-H’, 6-H, 8-H), 7.62 (s, 1 H, 4-H), 7.81–7.85 (m, 5 H, 4′-H, 5′-H, 6′-H, 7′-H, 8′-H), 13C-NMR (100 MHz, CDCl3): δ = 46.2 (C-3), 85.9 (C-2), 122.5 (C-9), 123.8 (C-7), 124.0 (C-8′), 125.5 (C-2′), 125.9 (C-5a), 126.4 (C-6′), 126.5 (C-3′), 127.7 (C-7′), 128.2 (C-4′), 128.5 (C-5′), 131.0 (C-6), 133.1 (C-8a’), 133.3 (C-4a’), 133.4 (C-8), 154.6 (C-9a), 171.2 (C-5), IR (KBr): 1024, 1217, 1462, 1604, 1655, 3062, 3212, 3283 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C19H19NO2Na, 312.100; found: 312.099.
2-(Naphthalen-2-yl)-3,4-dihydrobenzo[f][1,4]oxazepin-5(2H)-one (rac-8c): To the stirred solution of rac-7c (3.0 g, 11 mmol) in acetic acid (30 mL), sodium azide (1.1 g, 17 mmol) was added in two parts at 0 °C, then 3.0 mL of cc. H2SO4 was added dropwise and stirred at room temperature for 5 h. The reaction mixture was neutralized with sodium hydroxide solution and the aqueous mixture was extracted with dichloromethane (5 × 50 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography on silica (hexane/EtOAc 4:1) to give the product as white powder (3.27 g, 76%, mp 110–112 °C). 1H-NMR (400 MHz, CDCl3) δ = 3.55–3.69 (m, 2 H, 3-H), 5.57 (br s, 1H, 2-H), 7.09 (d, J = 8.0 Hz, 1 H, 9-H), 7.18 (m, 1 H, 7-H), 7.47 (m, 4 H, 2′-H, 3′-H, 6-H, 8-H), 7.82-7.84 (m, 5 H 4′-H, 5′-H, 6′-H, 7′-H, 8′-H), 13C-NMR (100 MHz, CDCl3): δ = 46.2 (C-3), 85.9 (C-2), 122.5 (C-9), 123.8 (C-1′), 124.0 (C-9), 125.5 (C-7), 125.9 (C-5a), 126.4 (C-6′), 126.5 (C-7′), 127.7 (C-5′), 128.2 (C-4′), 128.5 (C-8′), 131.0 (C-6), 133.1 (C-8a’), 133.3 (C-4a’), 133.4 (C-8), 136.4 (C-2′), 154.6 (C-9a), 171.2 (C-5). IR (KBr): 1064, 1223, 1299, 1460, 1696, 2958 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C19H19NO2Na, 312.100; found: 312.099.
2-(Naphthalen-1-yl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepine (rac-9b): To the stirred solution of rac-8b (2.0 g, 6.9 mmol) in dry THF (10 mL), lithium aluminum hydride solution in hexane (2.0 M) was added dropwise (10 mL, 0.72 g, 20.04 mmol) and the mixture was refluxed for 1 h. After cooling to room temperature, 20 mL 4 M NaOH solution was added and the mixture was concentrated under reduced pressure. The residue was extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The product was isolated as brown oil (1.82 g, 91%). 1H-NMR (400 MHz, CDCl3): δ = 1.86 (br s, 1 H, 4-H) 3.24–3.45 (dd, J = 12.0 Hz and 8.0 Hz, 1 H, 3-Ha), 3.44 (d, J = 12.0 Hz, 1 H, 3-Ha), 3.97 (d, J = 16.0 Hz, 1 H, 5-Ha), 4.15 (d, J = 12.0 Hz, 1 H, 5-Hb), 4.80 (d, J = 8.0 Hz, 1 H, 2-H), 7.01–7.09 (m, 2 H, 7-H, 9-H), 7.16–7.21 (m, 2 H, 5′-H, 6-H), 7.46–7.52 (m, 3 H, 2′-H, 3′-H, 8-H), 7.82–7.88 (m, 4 H, 4′-H, 6′-H, 7′-H, 8′-H), 13C-NMR (100 MHz, CDCl3): δ = 52.6 (C-5), 59.0 (C-3), 86.5 (C-2), 121.7 (C-9), 123.8 (C-7), 124.1 (C-8′), 124.7 (C-6′), 126.0 (C-2′), 126.3 (C-3′), 127.8 (C-7), 128.0 (C-4), 128.2 (C-8), 128.5 (C-6), 129.1 (C-5′), 132.9 (C-5a), 133.3 (C-8a’), 135.7 (C-4a’), 137.9 (C-1′), 159.2 (C-9a), IR (KBr): 1047, 1232, 1483, 1599, 2899, 3241, 3315 cm−1.
2-(Naphthalen-2-yl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepine (rac-9c): To the stirred solution of rac-8c (2.0 g, 6.9 mmol) in dry THF (10 mL), lithium aluminum hydride solution in hexane (2.0 M) was added dropwise (10 mL, 0.72 g, 20.04 mmol) and the mixture was refluxed for 1 h. After cooling to room temperature, 20 mL 4 M NaOH solution was added and the mixture was concentrated under reduced pressure. The residue was extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The product was isolated as brown oil (1.88g, 94%). 1H-NMR (400 MHz, CDCl3): δ = 1.94 (br s, 1 H, 4-H). 3.25–3.47 (m, 1 H, 3-Ha), 3.45 (d, J = 16.0 Hz, 1 H, 3-Hb), 3.98 (d, J = 16.0 Hz, 1 H, 5-Ha), 4.16 (d, J = 16.0 Hz 1 H, 5-Hb), 4.81 (d, J = 8.0 Hz 1 H, 2-H), 7.02-7.10 (m, 2 H, 6-H, 8-H), 7.17–7.22 (m, 2 H, 7-H, 9-H), 7.45–7.52 (m, 3 H, 3′-H, 6′-H, 8′-H), 7.82-7.89 (m, 4 H, 1′-H, 4′-H, 5′-H, 7′-H), 13C-NMR (100 MHz, CDCl3): δ = 52.6 (C-5), 59.0 (C-3), 86.5 (C-2), 121.7 (C-9), 123.8 (C-7), 124.1 (C-3′), 124.7 (C-1′), 126.0 (C-6′), 126.3 (C-7′), 127.7 (C-4′), 128.1 (C-8), 128.2 (C-5′), 128.5 (C-8′), 129.1 (C-6), 132.9 (C-5a), 133.3 (C-8a’), 135.7 (C-4a’), 137.9 (C-2′), 159.2 (C-9a), IR (KBr): 1078, 1241, 1470, 2963, 3240, 3350 cm−1.
2-(Naphthalen-1-yl)-4-(4-nitrophenyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepine (rac-1b): To the stirred solution of rac-9b (1.30 g, 4.70 mmol) in dry toluene (20 mL), 2-chloro-5-nitrobenzaldehyde (1.75 g 9.4 mmol) and anhydrous K2CO3 (3.2 g 2.3 mmol) were added and the mixture was refluxed for 5 h. After cooling to room temperature, the K2CO3 was filtered off and toluene was removed under reduced pressure. The residue was purified by column chromatography on silica gel (hexane/EtOAc 4:1) affording the product as yellow solid (1.01 g, 78%, mp 179–181 °C). 1H-NMR (400 MHz, CDCl3): δ = 3.71 (dd, J = 14.0 Hz and 9.0 Hz, 1 H, 3-Ha) 4.12 (dd, J = 14.0 Hz and 4.0 Hz, 1 H, 3-Hb), 4.74 (d, J = 16.0 Hz,1 H, 5-Ha), 4.95 (d, J = 16.0 Hz, 1 H, 5-Hb), 5.45 (dd, J = 9.0 Hz and 4.0 Hz, 1 H, 2-H), 7.05 (m, 2 H, 7-H, 9-H), 7.15–7.17 (m, 1 H, 6”-H), 7.30-7.32 (m, 2 H, 6-H, 8-H), 7.49–7.52 (m, 3 H, 2′-H, 3′-H, 4′-H), 7.84–7.89 (m, 4 H, 5′-H, 6′-H, 7′-H, 8′-H), 8.13–8.16 (dd, J = 8.0 Hz and 2. 8 Hz, 1 H, 5”-H), 8.60 (d, J = 2.8 Hz, 1 H, 3”-H), 9.95 (s, 1 H, CHO), 13C-NMR (100 MHz, CDCl3): δ = 56.3 (C-5), 64.5 (C-3), 82.9 (C-2), 117.8 (C-9), 121.1 (C-6”), 123.6 (C-7), 124.0 (C-8′), 124.4 (C-5a), 125.1 (C-6′), 126.5 (C-2′), 126.6 (C-3′), 127.8 (C-7′), 128.1 (C-8), 128.7 (C-4′), 129.2 (C-3”), 129.3 (C-5′), 129.5 (C-6), 129.9 (C-5”), 133.1 (C-8a’), 133.2 (C-4a’), 135.5 (C-4”), 140.0 (C-1′), 156.8 (C-9a), 158.3 (C-1”), 188.4 (CHO). IR (KBr): 1489, 1600, 1684, 2750, 2917, 3053 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C26H20N2O4Na, 447.132; found: 447.132.
2-(Naphthalen-2-yl)-4-(4-nitrophenyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepine (rac-1c): To the stirred solution of rac-9c (1.30 g, 4.70 mmol) in dry toluene (20 mL), 2-chloro-5-nitrobenzaldehyde (1.75 g, 9.4 mmol) and anhydrous K2CO3 (3.2 g, 2.3 mmol) were added and the mixture was refluxed for 5 h. After cooling to room temperature, the K2CO3 was filtered off and toluene was removed under reduced pressure. The residue was purified by column chromatography on silica gel (hexan/EtOAc 4:1) to give the product as yellow solid (1.05 g, 81%, mp 124–126 °C). 1H-NMR (400 MHz, CDCl3): δ = 3.70 (m, 1 H, 3-Ha), 4.12 (d, J = 12.0 Hz, 1 H, 3-Hb), 4.73 (d, J = 16.0 Hz, 1H, 5-Ha), 4.95 (d, J = 16.0 Hz, 1 H, 5-Hb), 5.45 (d, J = 12.0 Hz, 1 H, 2-H), 7.02–7.16 (m, 3 H, 5′-H, 6”-H, 8′-H), 7.25–7.32 (m, 2 H, 7-H, 9-H), 7.79–7.51 (m, 3 H, 6′-H, 7′-H, 8′-H), 7.83–7.89 (m, 4 H, 1′-H, 3′-H, 4′-H, 6′-H), 8.13 (dd, J = 8.0 Hz, 1 H, 5”-H), 8.59 (d, J = 2.8 Hz, 1H, C-3”), 9.94 (s, 1 H, CHO), 13C-NMR (100 MHz, CDCl3): δ = 56.3 (C-5), 64.5 (C-3), 82.9 (C-2), 117.7 (C-9), 121.1 (C-6”), 123.6 (C-7), 123.9 (C-3′), 124.4 (C-5a), 125.1 (C-1′), 126.4 (C-6′), 126.5 (C-6′), 126.6 (C-7′), 127.7 (C-2′), 127.8 (C-4′), 127.9 (C-8), 128.6 (C-3”), 129.1 (C-5′), 129.2 (C-8′), 129.4 (C-6), 129.8 (C-5”), 133.1 (C-8a’), 133.2 (C-4a’), 135.5 (C-4”), 139.9 (C-2′’), 156.8 (C-9a), 158.3 (C-1”), 188.4 (CHO), IR (KBr): 1238, 1326, 1503, 1598, 1680, 2918, 3058 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C26H20N2O4Na, 447.132; found: 447.132.
1,3-Dimethyl-2’-(naphthalen-1-yl)-11’-nitro-1’,2’-dihydro-2H,7b’H,9’H-spiro[pyrimidine-5,8’-quinolino[1,2-d][1,4]benzoxazepine]-2,4,6(1H,3H)-trione (rac-10b): To the stirred solution of rac-1b (100 mg, 0.23 mmol) in chloroform (5 mL), anhydrous MgSO4 (150 mg, 1.25 mmol) and barbituric acid (38 mg, 0.29 mmol) were added and the mixture was refluxed for 24 h. After cooling to room temperature, the MgSO4 was filtered off and chloroform was removed under reduced pressure. The residue was purified by column chromatography on silica gel (hexane/EtOAc 1:1) affording the product as yellow solid (36 mg, 36%, mp 289–291 °C) 1H-NMR (400 MHz, DMF-d7): δ = 2.71 (s, 3 H, CH3), 2.98 (s, 3 H, CH3), 3.31 (d, J = 16.0 Hz, 1 H, 9-Ha), 3.83 (d, J = 16.0 Hz, 1H, 9-Hb), 4.10 (m, 1 H, 3-Ha), 4.61 (d, J = 16.0 Hz, 1 H, 3-Hb), 4.88 (s, 1 H, 15a-H), 5.79 (m, 1 H, 2-H), 6.09 (m, 1 H, 5-H), 7.00–7.04 (m, 5 H, Ph-H), 7.26–7.33 (m, 3 H, Ph-H), 7.48–7.61 (m, 2 H, Ph-H), 7.85–7.93 (m, 2 H, Ph-H), 8.03 (s, 1H, 8′-H), 8.13 (d, J = 8.0 Hz, 1 H, 6-H). 13C-NMR (100 MHz, DMF-d7): δ = 29.0 (CH3) 29.1 (CH3), 46.7 (C-9), 53.1 (C-15a), 72.2 (C-3), 76.6 (C-10), 111.6 (C-2), 105.5 (C-13), 123.7 (C-19), 124.2 (C-5), 124.6 (C-17), 124.7 (C-15b), 124.8 (C-6) 125.7 (C-8), 125.8 (C-8′), 124.8 (C-2′), 127.1 (C-6′), 127.8 (C-3′), 128.8 (C-7′), 129.9 (C-18), 130.4 (C-4′), 131.2 (C-5′),132.3 (C-16) 132.7 (C-8a’), 133.7 (C-4a’), 135.1 (C-7), 138.3 (C-1′), 149.9 (C-13), 151.4 (C-19a), 152.4 (C-4a), 168.6 (C-15), 170.3 (C-11). IR (KBr): 1322, 1601, 1683, 1698, 2853, 2923 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C32H26N4O6Na, 585.175; found: 585.175.
1,3-Dimethyl-2’-(naphthalen-2-yl)-11’-nitro-1’,2’-dihydro-2H,7b’H,9’H-spiro[pyrimidine-5,8’-quinolino[1,2-d][1,4]benzoxazepine]-2,4,6(1H,3H)-trione (rac-10c): To the stirred solution of rac-1c (100 mg, 0.23 mmol) in chloroform (5 mL), anhydrous MgSO4 (150 mg, 1.25 mmol) and 1,3-dimethylbarbituric acid (38 mg, 0.29 mmol) were added and the mixture was refluxed for 24 h. After cooling to room temperature, the MgSO4 was filtered off and the chloroform was removed under reduced pressure. The residue was crystallized with cold diisopropyl-ether affording the product as yellow solid (52 mg, 52%, mp 312–314 °C) The sample could not be dissolved in NMR solvents. IR (KBr): 1320, 1507, 1680, 1746, 2953, 3057 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C32H26N4O6Na, 585.175; found: 585.175.
2,2-Dimethyl-2’-(naphthalen-1-yl)-11’-nitro-1’,2’-dihydro-7b’H,9’H-spiro[1,3-dioxane-5,8’-quinolino[1,2-d][1,4]benzoxazepine]-4,6-dione (rac-11b): To the stirred solution of rac-1b (100 mg, 0.23 mmol) in chloroform (5 mL), anhydrous MgSO4 (150 mg, 1.25 mmol) and Meldrum’s acid (43 mg, 0.29 mmol) were added and the mixture was refluxed for 24 h. After cooling to room temperature, the MgSO4 was filtered off and the chloroform was removed under reduced pressure. The residue was purified by column chromatography on silica gel (hexane/EtOAc 2:1) affording the product as yellow solid (50 mg, 50%, mp 160–162 °C). 1H-NMR (400 MHz, CDCl3): δ = 1.05 (s, 3 H, CH3), 1.65 (s, 3 H, CH3), 3.24 (d, J = 16.0 Hz, 1 H, 9-Ha), 3.90 (d, J = 16.0 Hz, 1 H, 9-Hb), 4.19 (m, 1 H, 3-Ha), 4.33 (m, 1 H, 3-Hb), 5.08 (s, 1 H, 15a-H), 6.08 (d, J = 8.0 Hz, 1 H, 2-H), 6.55 (d, J = 8.0 Hz, 1 H, 5-H), 6.88(d, J = 8.0 Hz, 1 H, 17-H), 7.02 (m, 1 H, Ar-H), 7.21–7.34 (m, 4 H, Ar-H), 7.57–7.62 (m, 2 H, Ar-H), 7.89 (m, 1 H, 16-H), 8.10 (m, 2 H, 6-H, 8-H), 8.31 (m, 1 H, 18-H). 13C-NMR (100 MHz, CDCl3): δ = 26.9 (CH3) 30.4 (CH3), 35.9 (C-9), 46.5 (C-3), 49.9 (C-10), 70.6 (C-15a), 74.8 (C-2), 105.5 (C-13), 109.9 (C-19), 119.5 (C-5), 123.1 (C-17), 123.6 (C-6), 124.3 (C-8), 124.4 (C-8′), 124.8 (C-2′), 125.1 (C-8a), 125.5 (C-6′), 126.1 (C-3′), 126.9 (C-15b), 126.9 (C-7′), 128.7 (C-18), 129.5 (C-4′), 130.8 (C-16), 131.3 (C-5′), 131.5 (C-8a’), 132.5 (C-4a’), 133.8 (C-7), 137.9 (C-1′), 147.8 (C-19a), 152.2 (C-4a), 164.4 (C-15), 168.2 (C-11). IR (KBr): 1195, 1509, 1603, 1737, 1775, 2930, 3056 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C32H20N2O7Na, 573.164; found: 573.163.
2’,2’-Dimethyl-2-(naphtalene-2-yl)11-nitro-1,2,7b,9-tetrahydrospiro[benzo[6,7][1,4]oxazepine[4,5-a]quinoline-8,5’-[1,3]dioxane]-4’,6’-dione (rac-11c): To the stirred solution of rac-1c (100 mg, 0.23 mmol) in chloroform (5 mL), anhydrous MgSO4 (150 mg, 1.25 mmol) and Meldrum’s acid (43 mg, 0.29 mmol) were added and the mixture was refluxed for 24 h. After cooling to room temperature, the MgSO4 was filtered off and the chloroform was removed under reduced pressure. The residue was purified by column chromatography on silica gel (hexane/EtOAc 2:1) affording the product as yellow solid (98 mg, 98%, mp 188–190 °C). 1H-NMR (400 MHz, CDCl3): δ = 1.06 (s, 3 H, CH3) 1.62 (s, 3 H, CH3), 3.18 (d, J = 16.0 Hz, 1 H, 9-Ha), 3.88 (d, J = 16.0 Hz, 1 H, 9-Hb), 4.09 (m, 1 H, 3-Ha), 4.29 (dd, J = 16.0 Hz and 4.0 Hz, 1 H, 3-Hb), 5.04 (s, 1H, 15a-H), 5.42 (dd, J = 12.0 Hz and 4.0 Hz, 1 H, 2-H), 6.81 and 6.89 (2 × d, J = 8.0 Hz, 2 H, 5-H and 19-H), 7.15-7.24 (m, 4 H, Ar-H), 7.26-7.32 (m, 3 H, Ar-H), 7.34–7.58 (m, 3 H, Ar-H), 8.12 (m, 2 H, 1′-H, 8-H). 13C-NMR (100 MHz): δ = 27.0 (CH3), 30.5 (CH3), 35.7 (C-9), 47.0 (C-3), 50.1 (C-10), 70.6 (C-15a), 80.1 (C-2), 105.05 (C-13), 110.1 (C-19) 119.5 (C-5), 124.3 (C-17), 124.9 (C-3′), 125,1 (C-1′), 125.2 (C-6), 125.5 (C-8), 126.4 (C-8a), 126.5 (C-6′), 126.6 (C-7′), 126.8 (C-15b), 127.6 (C-4′), 128.1 (C-5′), 128.4 (C-8), 131.0 (C-8′), 131.4 (C-16), 132.9 (C-8a’), 133.3 (C-4a’), 134.6 (C-7), 137.9 (C-2′), 147.7 (C-19a), 152.2 (C-4a), 164.5 (C-15), 168.2 (C-11). IR (KBr): 1201, 1320, 1507, 1736, 1773, 3057 cm−1. HRMS-ESI (m/z): [M + Na]+ calc’d for C32H20N2O7Na, 573.164; found: 573.163.
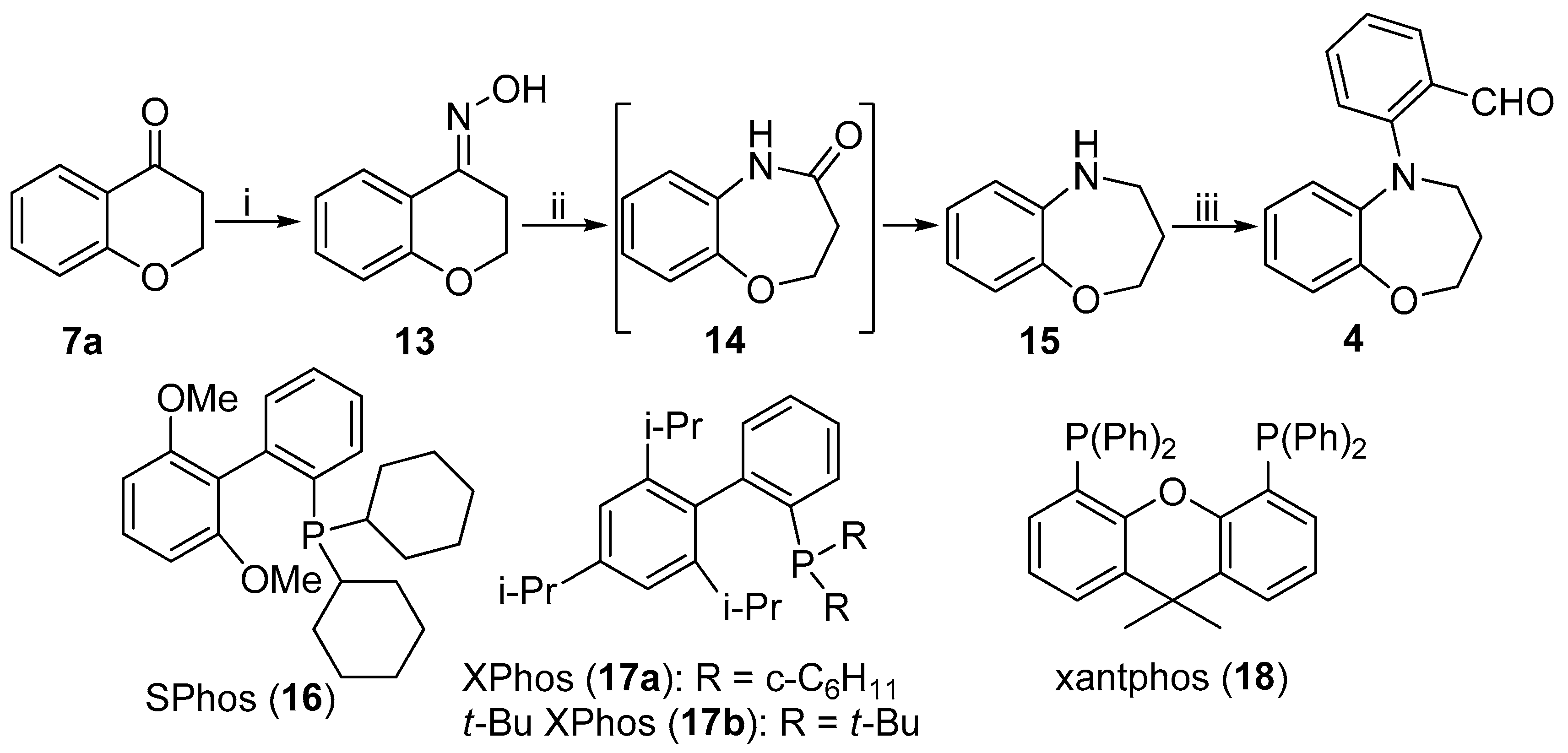
(4E)-N-Hydroxy-2,3-dihydro-4H-chromen-4-imine (13): Hydroxylamine hydrochloride (2.81 g, 40.44 mmol) was added to the stirred solution of chroman-4-one (7a, 3.0 g, 20.25 mmol) in pyridine (25 mL) and the mixture was stirred for 1.5 h. Cool water (100 mL) was added to the reaction mixture and the aqueous phase was extracted with ethyl-acetate (3 × 50 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (hexane/EtOAc 3:1) to give 13 as white solid (3.1 g, 94%, mp 140–141 °C). 1H NMR (360 MHz, CDCl3): δ = 3.01 (m, J = 6.1 Hz, 2 H, 3-H), 4.25 (m, J = 6.1 Hz, 2 H, 2-H), 6.89–6.96 (m, 2 H, 6-H, 8-H), 7.25 (m, 1 H, 7-H), 7.81 (dd, J = 9.7 Hz and 1.8 Hz, 1 H, 5-H), 9.39 (br s, 1 H, OH). 13C NMR (90 MHz, CDCl3): δ = 23.5 (C-3), 64.9 (C-2), 117.8 (C-8), 118.1 (C-5a), 121.5 (C-6), 123.9 (C-5), 131.2 (C-7), 149.9 (C-4), 156.7 (C-8a). IR (KBr): 1046, 1253, 1454, 1603, 3263 cm−1. HRMS-ESI (m/z): [M + H]+ calc’d for C9H9NO2H, 164.071; found: 164.071.
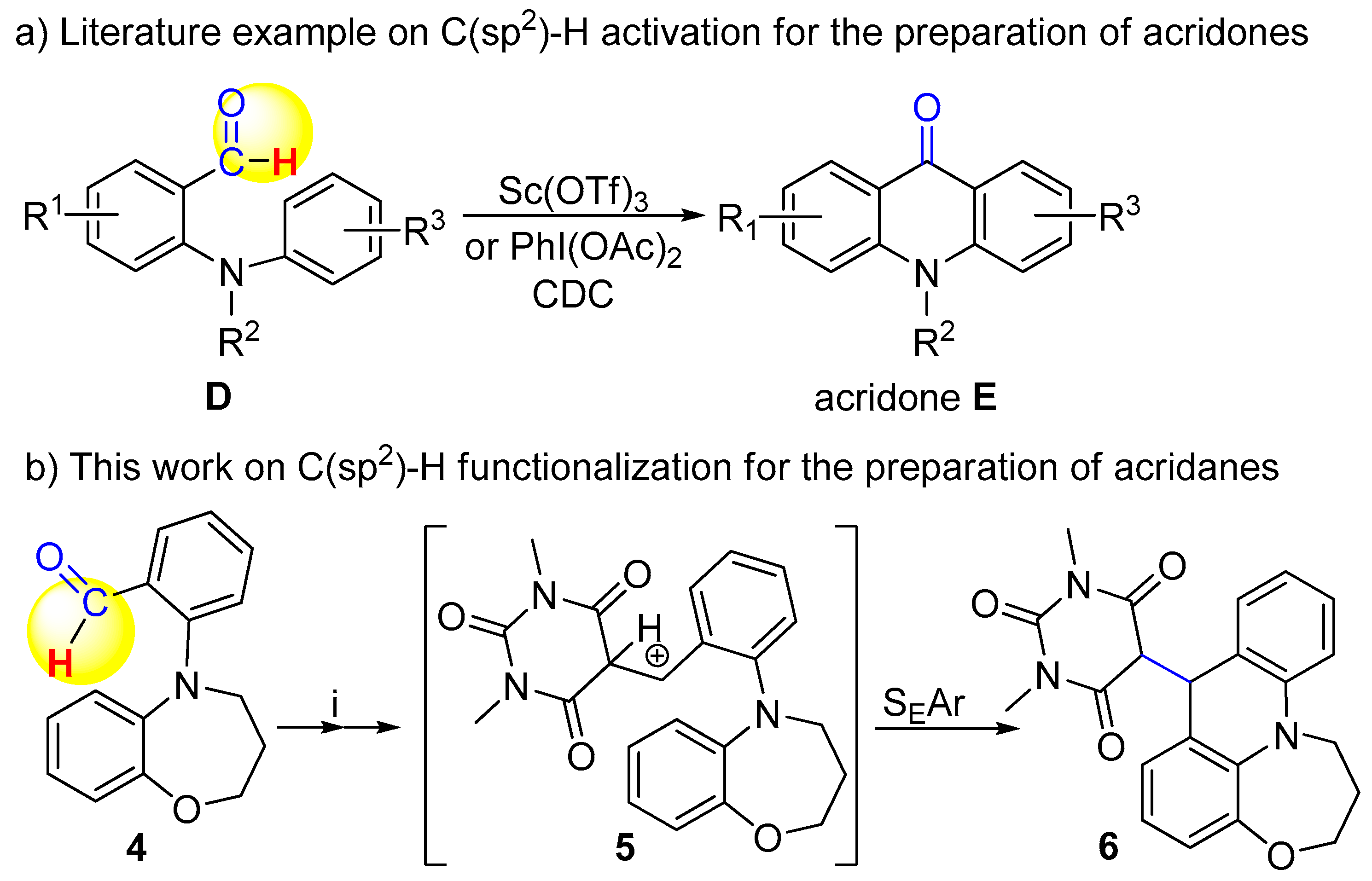
2,3-Dihydro-1,5-benzoxazepin-4(5H)-one (15): The solution of 13 (1.0 g, 6.13 mmol) in dry dichloromethane (60 mL) was cooled to 5 °C and added to DIBAL-H (40 mL, 1.0M in hexane) under argon atmosphere and the mixture was stirred for 24 h. The reaction mixture was diluted with ethyl-acetate (20 mL), and filtered through celite bench. The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure to afford 15 as brown solid (883 mg, 97%, mp 48–52 °C). 1H NMR (400 MHz, CDCl3): δ = 1.98 (m, J = 5.6 Hz, 2H, 3-H), 3.19 (m, J = 5.6 Hz, 2 H, 4-H), 3.57 (br s, 1 H, -NH), 4.07 (m, J = 5.6 Hz, 2 H, 2-H), 6.73 (dd, J = 8.8 Hz and 1.2 Hz, 1 H, 6-H), 6.76 (m, 1 H, 7-H), 6.85 (m, 1 H, 8-H), 6.96 (dd, J = 9.2 Hz and 1.2 Hz, 1 H, 9-H). 13C NMR (100 MHz, CDCl3): δ = 31.9 (C-3), 45.9 (C-4), 71.4 (C-2), 119.5 (C-9), 120.7 (C-6), 121.8 (C-8), 123.3 (C-7), 142.0 (C-6a), 150.1 (C-9a). IR (KBr): 1053, 1250, 1595, 2952, 3282. HRMS-ESI (m/z): [M + Na]+ calc’d for C9H11NONa, 172.074; found: 172.074.
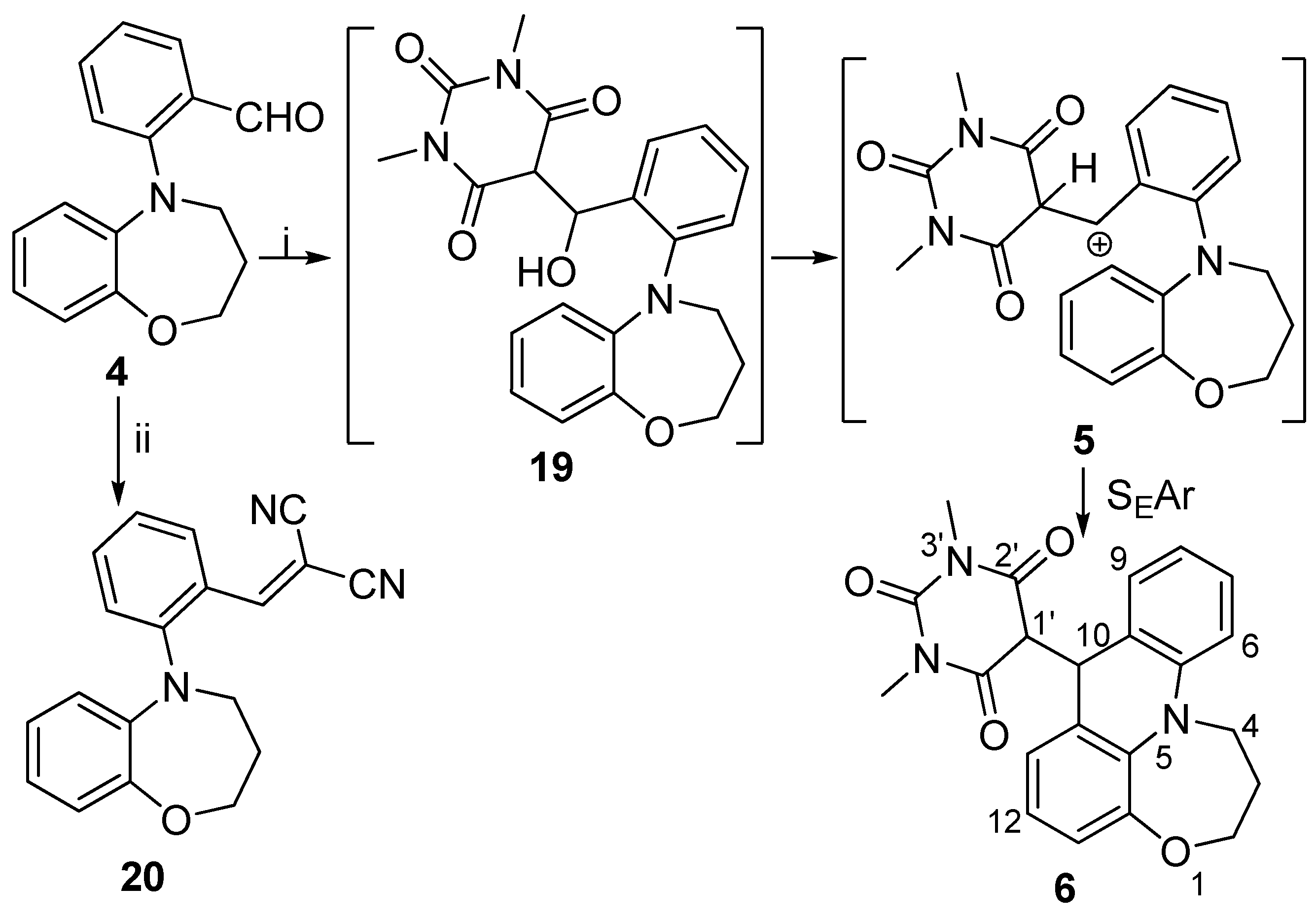
2-(3,4-Dihydro-1,5-benzoxazepin-5(2H)-yl)benzaldehyde (4): To the stirred solution of 15 (100 mg, 0.67 mmol) in dry 1,4-dioxane (4 mL), Pd(OAc)2 (11 mg, 0.05 mmol), 2-dicyclohexylphosphino-2′,6′-dimetoxybiphenyl (28 mg, 0.07 mmol), Cs2CO3 (328 mg, 1.00 mmol) and 2-bromobenzaldehyde (0.12 mL, 1.00 mmol) were added and the mixture was refluxed for 18 h under argon atmosphere. The reaction mixture was concentrated under reduced pressure and residue was purified by column chromatography on silica gel (hexane/EtOAc 8:1) to give 4 as yellow solid (97 mg, 87%, mp 89–92 °C). 1H NMR (400 MHz, CDCl3): δ = 2.07 (m, J = 5.6 Hz, 2H, 3-H), 3.83 (m, J = 5.6 Hz, 2 H, 4-H), 4.24 (m, J = 5.6 Hz, 2 H, 2-H), 6.52 (dd, J = 9.2 Hz and 1.2 Hz, 1 H, 9-H), 6.79 (m, 1H, 7-H), 6.88 (m, 1 H, 8-H), 7.01 (dd, J = 9.2 Hz and 1.2 Hz, 1 H, 6-H), 7.17 (m, J = 7.2 Hz, 1 H, 4′-H), 7.25 (d, J = 8.4 Hz, 1 H, 6′-H), 7.56 (m, 1 H, 5′-H), 7.81 (dd, J = 8.4 Hz and 1.2 Hz, 1 H, 3′-H), 10.14 (s, 1H, -CHO). 13C NMR (100 MHz, CDCl3): δ = 29.9 (C-3), 52.6 (C-4), 70.1 (C-2), 121.9 (C-9), 123.4 (C-6), 123.5 (C-6′), 123.6 (C-4′), 124.0 (C-8), 124.7 (C-7), 129.1 (C-3′), 129.9 (C-2′), 134.8 (C-5′), 142.6 (C-6a), 152.2 (C-9′), 152.3 (C-1′), 190.8 (CHO). IR (KBr): 1059, 1251, 1455, 1686. HRMS-ESI (m/z): [M + Na]+ calc’d for C16H15NO2Na, 276.100; found: 276.095.
[2-(3,4-Dihydro-1,5-benzoxazepin-5(2H)-yl)benzylidene]propanedinitrile (20): To the stirred solution of 4 (100 mg, 0.39 mmol) in dry toluene (3 mL), dimethylamine-hydrochloride (128 mg, 1.58 mmol) and malononitrile (156 mg, 2.37 mmol) were added and the mixture was stirred for 48 h. Water (10 mL) and dichloromethane (20 mL) were added and two layers were separated. The aqueous phase was extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with concentrated NaHCO3 solution (3 × 10 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The product was isolated as red solid (115 mg, 3.38 mmol, 96%, mp 118–121 °C).1H NMR (400 MHz, CDCl3): δ = 2.07 (m, 2 H, 3-H), 3.72 (m, 2 H, 4-H), 4.21 (m, 2 H, 2-H), 6.47 (d, J = 7.6 Hz, 1 H, 9-H), 6.87 (m, 1 H, 7-H), 7.01 (m, 1H, 8-H), 7.09 (m, 1H, 6-H), 7.16 (m, 1 H, 4′-H), 7.31 (d, J = 7.6 Hz, 1 H, 6′-H), 7.57 (t, J = 7.6 Hz, 1 H, 5′-H), 7.82 (m, 2 H, 3′-H, 10-H). 13C NMR (90 MHz, CDCl3): δ = 30.3 (C-3), 51.7 (C-4), 70.6 (C-2), 82.0 (C-11), 112.3 (-CN), 113.4 (-CN), 122.4 (C-9), 122.7 (C-6′), 123.1 (C-6), 124.1 (C-4′), 125.1 (C-8), 125.3 (C-7), 125.4 (C-2′), 129.5 (C-5′), 134.5 (C-3′), 142.6 (C-6a), 151.0 (C-9a), 153.7 (C-1′), 158.6 (C-10). IR (KBr): 1052, 1253, 1455, 2230.
5-(2,3-Dihydro-1H,8H-[1,4]oxazepino[2,3,4-de]acridin-8-yl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (6): To the solution of 4 (100 mg, 0.40 mmol) in chloroform (6 mL), anhydrous MgSO4 (150 mg, 1.25 mmol) and 1,3-dimethylbarbituric acid (370 mg, 2.42 mmol) were added and the mixture was stirred for 10 days. The MgSO4 was filtered off and chloroform was removed under reduced pressure. The residue was taken up in chloroform (30 mL) and extracted with concentrated NaHCO3 solution (3 × 10 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The resultant product was isolated as purple solid (100 mg, 64%). 1H NMR (400 MHz, CDCl3): δ = 2.26 (m, 1 H, 3-Ha), 2.36 (m, 1 H, 3-Hb), 3.04 (s, 3 H, NMe), 3.10 (s, 3 H, NMe), 3.61 (d, J = 4.4 Hz, 1 H, 10-H), 3.93 (m, 1 H, 2-Ha), 4.10 (m, 2 H, 4-H), 4.38 (m, 1 H, 2-Hb), 4.90 (d, J = 4.4 Hz, 1 H, 10-H), 6.90–6.96 (m, 6-H, 7-H, 8-H, 11-H, 13-H, 5H), 7.03 (d, J = 6.8 Hz, 1 H, 9-H), 7.29 (m, 1 H, 12-H). 13C NMR (100 MHz, CDCl3): δ = 27.7 (C-3), 28.0 (N-CH3), 28.2 (N-CH3), 47.4 (C-4), 49.5 (C-10), 59.0 (C-1′), 70.4 (C-2), 112.3 (C-13), 120.4 (C-9a), 120.6 (C-6), 120.9 (C-8), 122.4 (C-11), 122.6 (C-12), 125.1 (C-10b), 128.1 (C-7), 129.2 (C-9), 134.5 (C-10a), 143.8 (C-13a), 149.2 (C-5a), 151.2 (C-4′), 166.4 (C-6′), 167.1 (C-2′).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







