
Study on the Synthesis, Antioxidant Properties, and Self-Assembly of Carotenoid–Flavonoid Conjugates

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
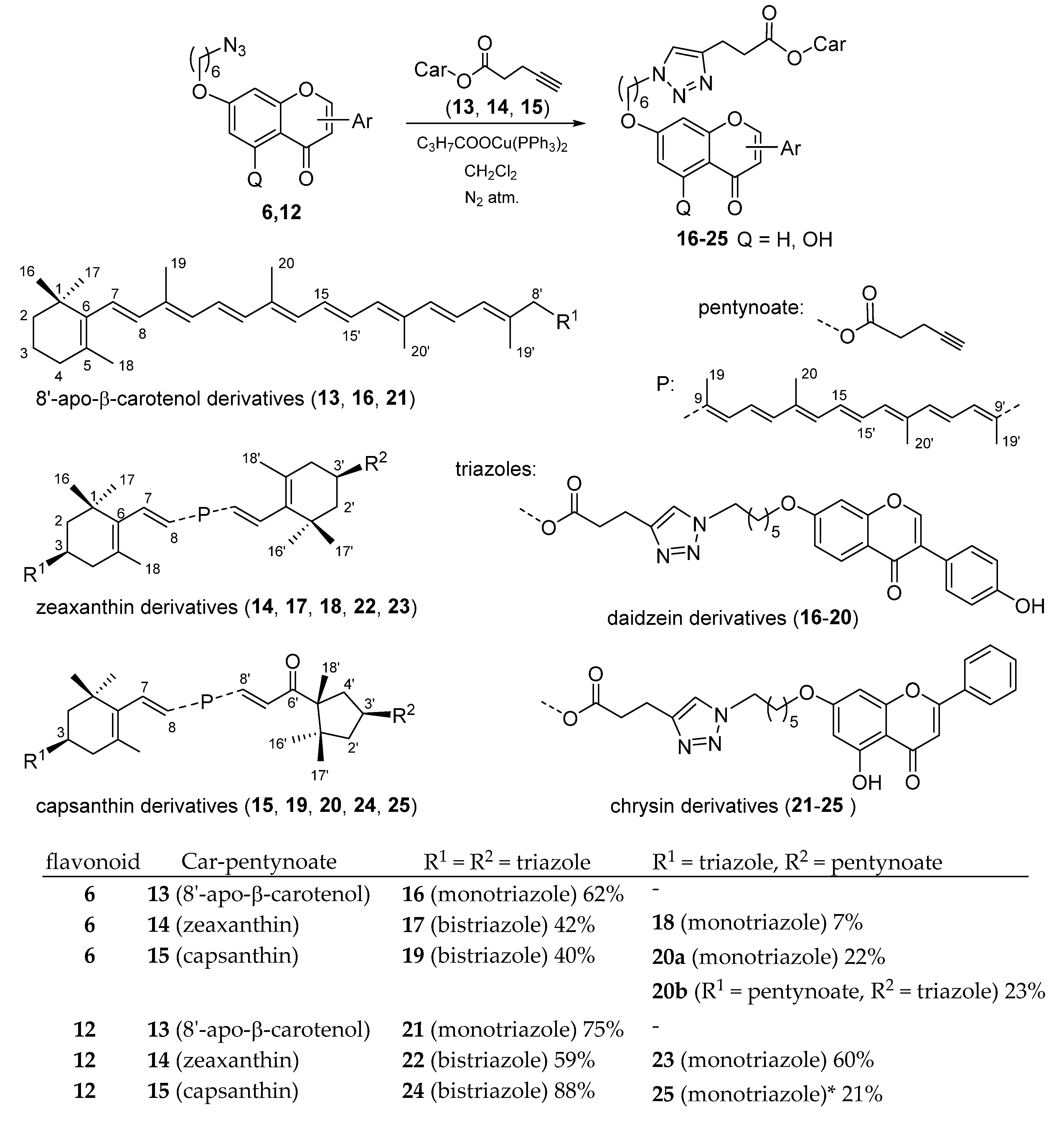
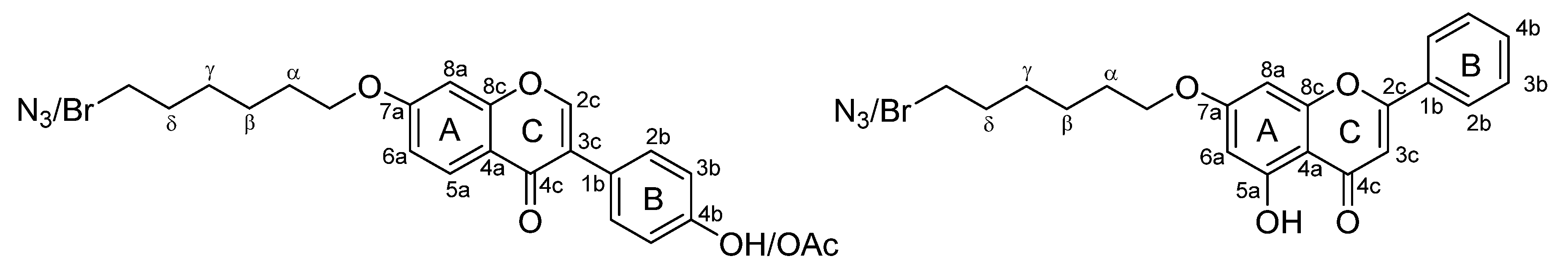
2.1. Synthesis
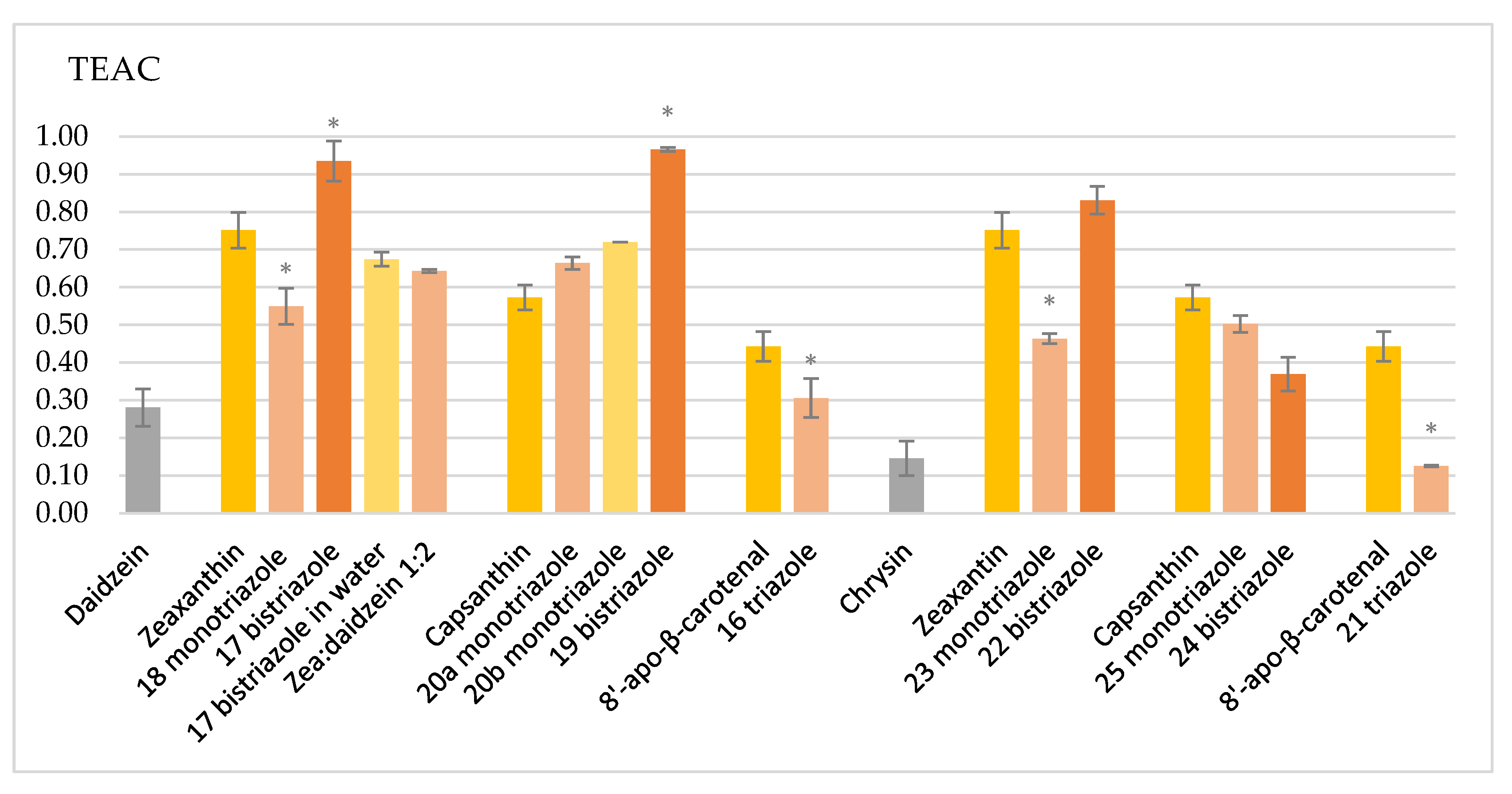
2.2. Measurement of Antioxidant Capacity
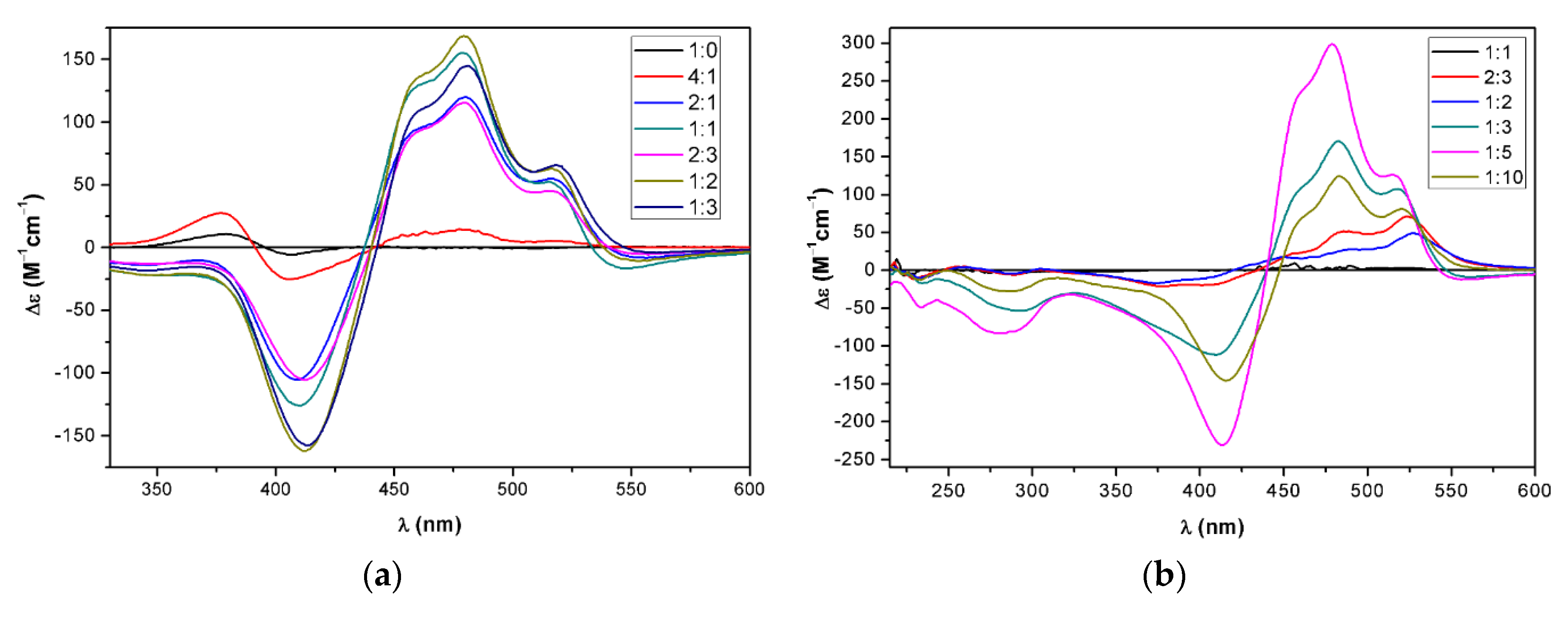
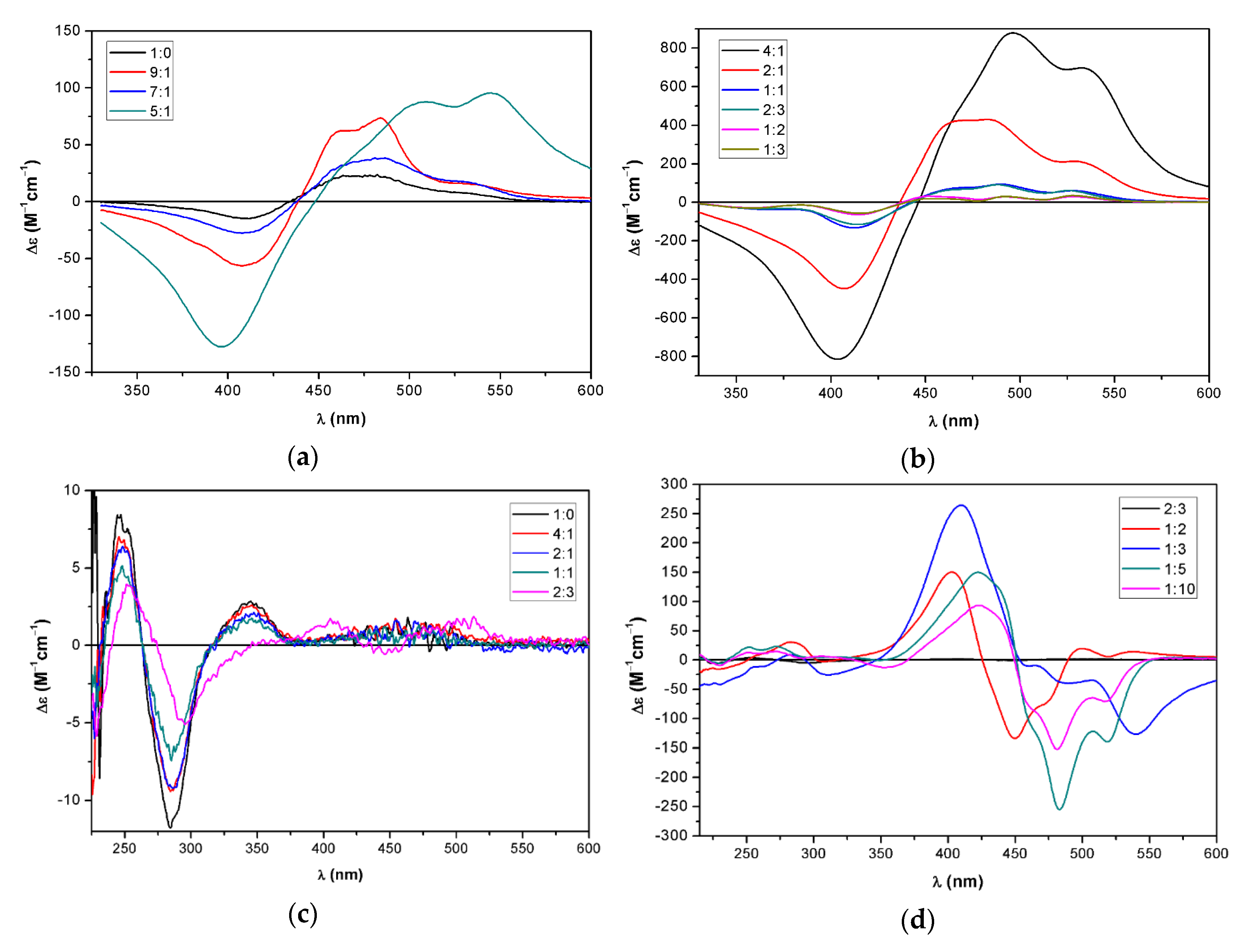
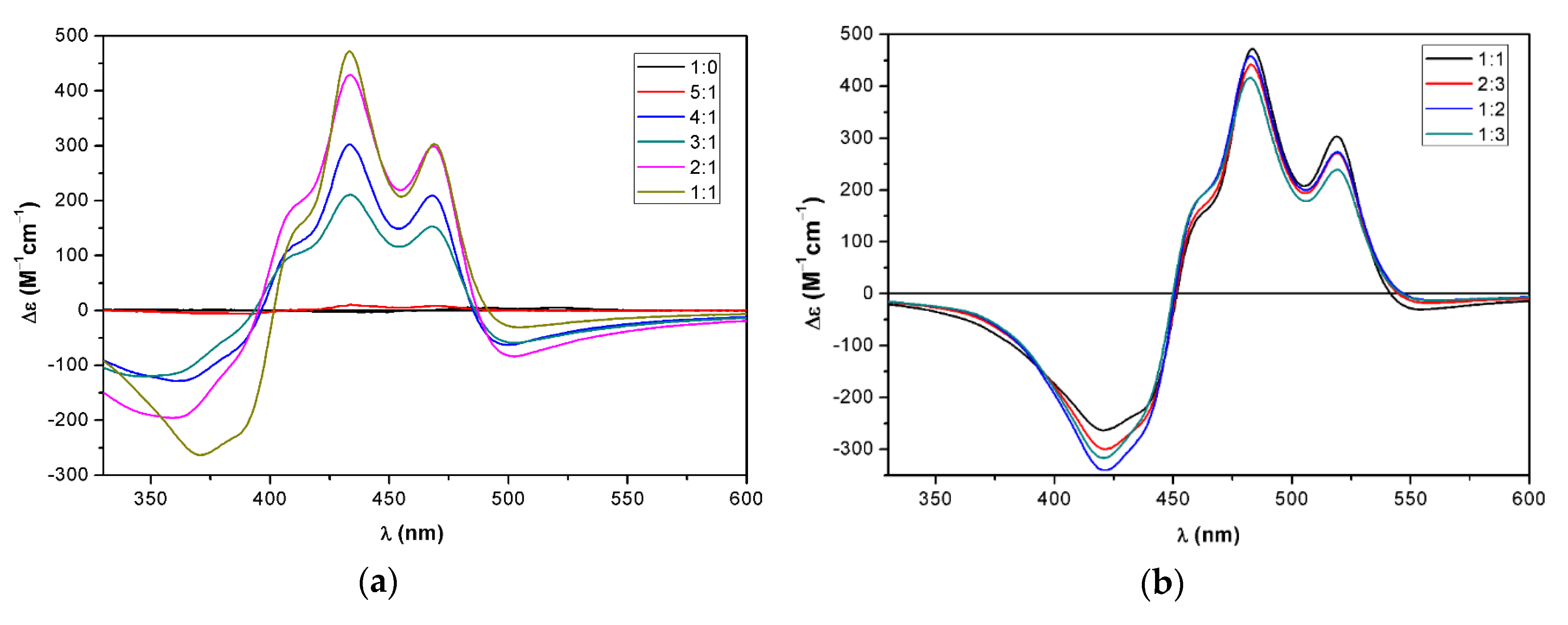
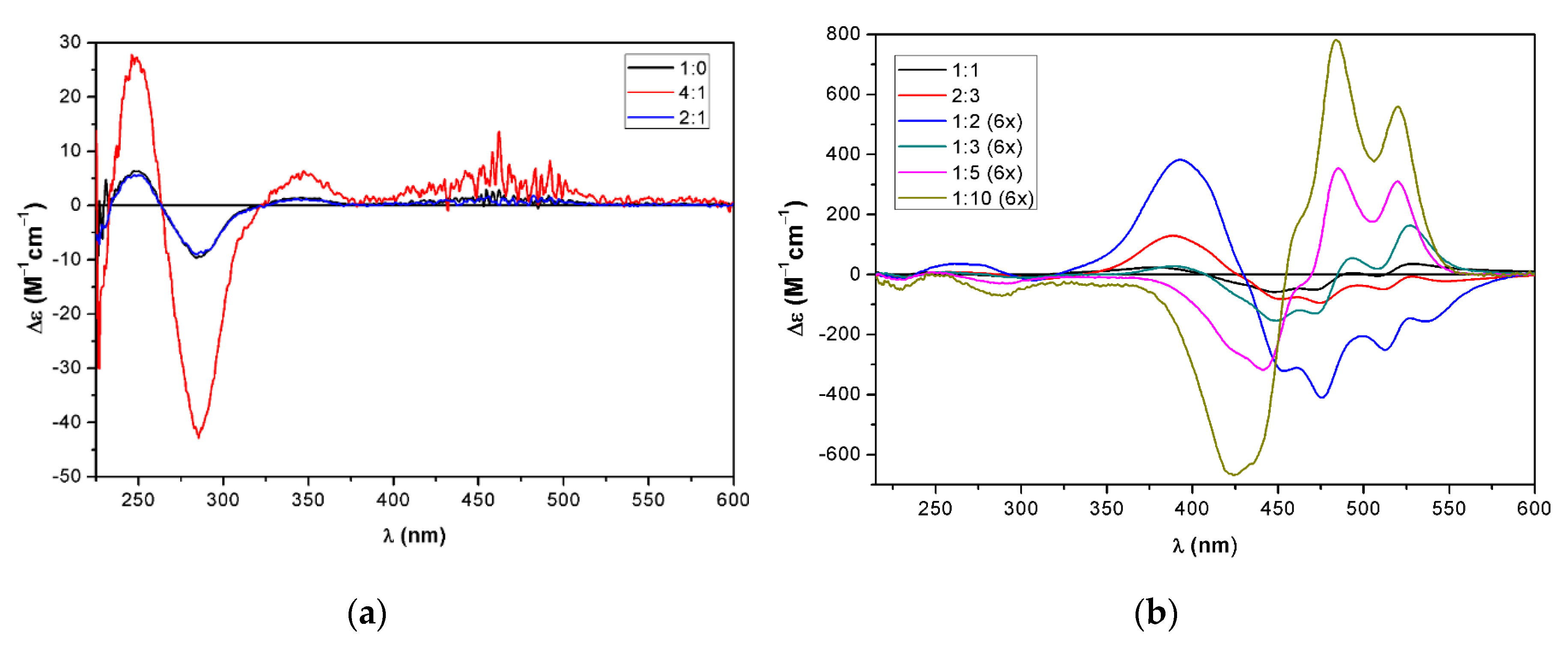
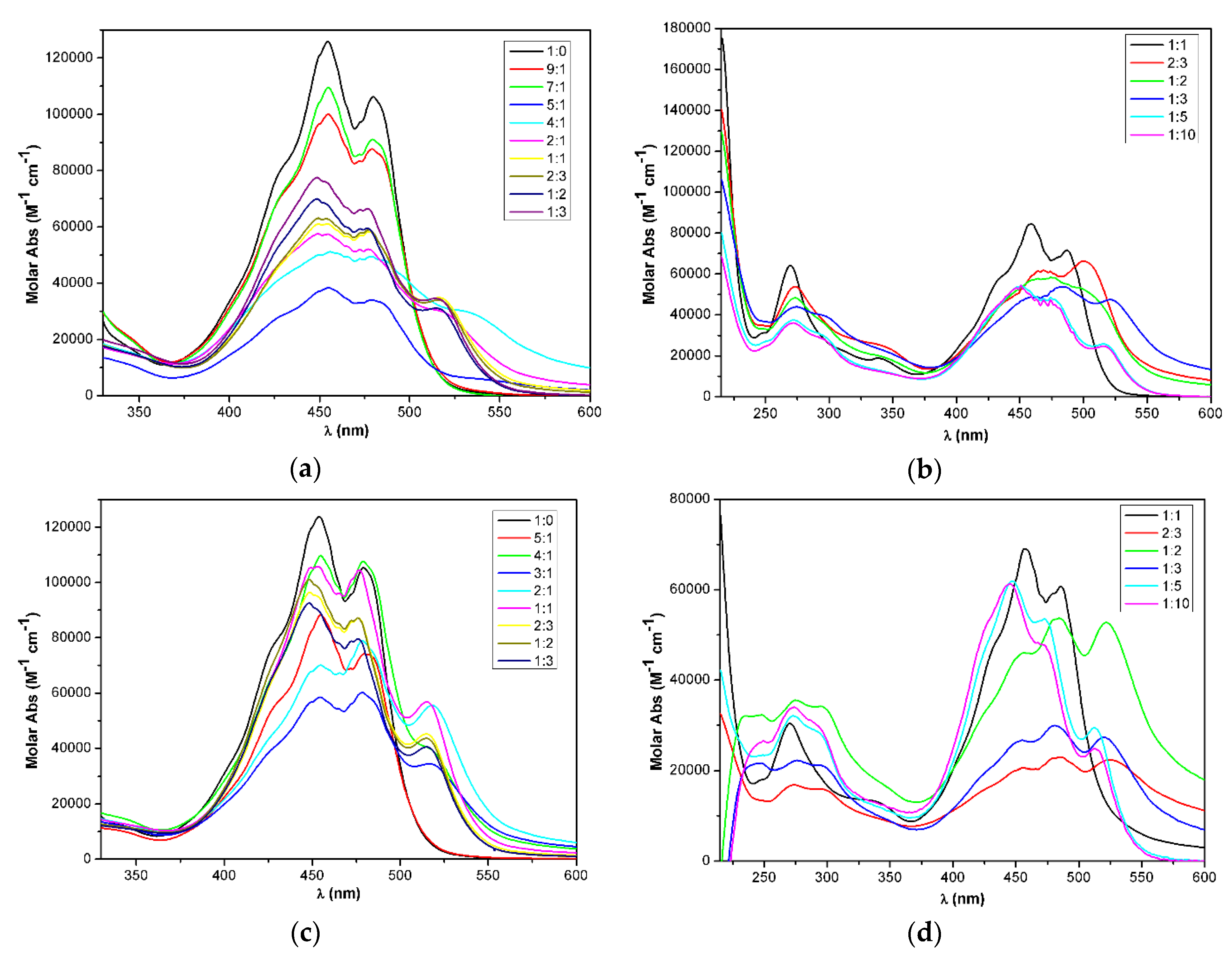
2.3. Supramolecular Assembly
3. Conclusions
4. Experimental Section
4.1. Materials and Methods
4.2. Assay for the Antioxidant Activity of the Carotenoids/TEAC Assay
4.3. Acetylation of Flavonoids
4.4. Selective Deacetylation of Flavonoid Diacetates
4.5. Mitsunobu Reactions
4.6. Deprotection
4.7. Azide Substitutions
4.8. Azide-Alkyne Click-Reactions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, R.H. Health benefits of fruit and vegetables are from additive and synergistic combinations of phytochemicals. Am. J. Clin. Nutr. 2003, 78, 517S–520S. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.H. Potential Synergy of Phytochemicals in Cancer Prevention: Mechanism of Action. J. Nutr. 2004, 134, 3479S–3485S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, N.E.; Mohn, E.S.; Hason, N.; Erdman, J.W., Jr.; Johnson, E.J. Intrinsic and Extrinsic Factors Impacting Absorption, Metabolism, and Health Effects of Dietary Carotenoids. Adv. Nutr. 2018, 9, 465–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.D.; Reddy, M.B. Effect of ascorbic acid intake on nonheme-iron absorption from a complete diet. Am. J. Clin. Nutr. 2001, 73, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Trombino, S.; Serini, S.; di Nicuolo, F.; Celleno, L.; Andò, S.; Picci, N.; Calviello, G.; Palozza, P. Antioxidant Effect of Ferulic Acid in Isolated Membranes and Intact Cells: Synergistic Interactions with α-Tocopherol, β-Carotene, and Ascorbic Acid. J. Agric. Food Chem. 2004, 52, 2411–2420. [Google Scholar] [CrossRef]
- Álvarez, R.; Vaz, B.; Gronemeyer, H.; de Lera, Á.R. Functions, Therapeutic Applications, and Synthesis of Retinoids and Carotenoids. Chem. Rev. 2014, 114, 1–125. [Google Scholar] [CrossRef] [PubMed]
- Lakey-Beitia, J.; Kumar, D.J.; Hegde, M.L.; Rao, K.S. Carotenoids as Novel Therapeutic Molecules Against Neurodegenerative Disorders: Chemistry and Molecular Docking Analysis. Int. J. Mol. Sci. 2019, 20, 5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-Y.; Yang, D.-P.; Tang, G.-Y. Multipotent antioxidants: From screening to design. Drug Discov. Today 2006, 11, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Jackman, K.A.; Woodman, O.L.; Sobey, C.G. Isoflavones, Equol and Cardiovascular Disease: Pharmacological and Therapeutic Insights. Curr. Med. Chem. 2007, 14, 2824–2830. [Google Scholar] [CrossRef] [PubMed]
- Tobe, H.; Komiyama, O.; Komiyama, Y.; Maruyama, H.B. Daidzein Stimulation of Bone Resorption in Pit Formation Assay. Biosci. Biotech. Biochem. 1997, 61, 370–371. [Google Scholar] [CrossRef]
- Rietjens, I.M.C.M.; Sotoca, A.M.; Vervoort, J.; Louisse, J. Mechanisms underlying the dualistic mode of action of major soy isoflavones in relation to cell proliferation and cancer risks. Mol. Nutr. Food Res. 2013, 57, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Salimi, A.; Pourahmad, J. Chapter 16—Role of Natural Compounds in Prevention and Treatment of Chronic Lymphocytic Leukemia. In Polyphenols: Prevention and Treatment of Human Disease, 2nd ed.; Watson, R., Preedy, V., Zibadi, S., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2018; Volume 2, pp. 195–203. [Google Scholar] [CrossRef]
- Asakura, H.; Tetsuji Kitahora, T. Chapter 23—Antioxidants and Polyphenols in Inflammatory Bowel Disease: Ulcerative Colitis and Crohn Disease. In Polyphenols: Prevention and Treatment of Human Disease, 2nd ed.; Watson, R., Preedy, V., Zibadi, S., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2018; Volume 2, pp. 279–292. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, B.; Sun, W.; Xing, Y. Chapter 36—Intervention of Prediabetes by Flavonoids From Oroxylum indicum. In Bioactive Food as Dietary Interventions for Diabetes, 2nd ed.; Watson, R., Preedy, V., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2019; pp. 559–575. [Google Scholar] [CrossRef]
- Focsan, A.L.; Polyakov, N.E.; Kispert, L.D. Supramolecular Carotenoid Complexes of Enhanced Solubility and Stability—The Way of Bioavailability Improvement. Molecules 2019, 24, 3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milde, J.; Elstner, E.F.; Graßmann, J. Synergistic effects of phenolics and carotenoids on human low-density lipoprotein oxidation. Mol. Nutr. Food Res. 2007, 51, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Han, R.-M.; Chen, C.-H.; Tian, Y.-X.; Zhang, J.-P.; Skibsted, L.H. Fast Regeneration of Carotenoids from Radical Cations by Isoflavonoid Dianions: Importance of the Carotenoid Keto Group for Electron Transfer. J. Phys. Chem. A 2010, 114, 126–132. [Google Scholar] [CrossRef]
- Han, R.-M.; Zhang, J.-P.; Skibsted, L.H. Reaction Dynamics of Flavonoids and Carotenoids as Antioxidants. Molecules 2012, 17, 2140–2160. [Google Scholar] [CrossRef] [Green Version]
- Háda, M.; Nagy, V.; Deli, J.; Agócs, A. Hydrophilic Carotenoids: Recent Progress. Molecules 2012, 17, 5003–5012. [Google Scholar] [CrossRef] [Green Version]
- Gažák, R.; Purchartová, K.; Marhol, P.; Živná, L.; Sedmera, P.; Valentová, K.; Kato, N.; Matsumura, H.; Kaihatsu, K.; Křen, V. Antioxidant and antiviral activities of silybin fatty acid conjugates. Eur. J. Med. Chem. 2010, 45, 1059–1067. [Google Scholar] [CrossRef]
- Vavříková, E.; Křen, V.; Jezova-Kalachova, L.; Biler, M.; Chantemargue, B.; Pyszková, M.; Riva, S.; Kuzma, M.; Valentová, K.; Ulrichová, J.; et al. Novel flavonolignan hybrid antioxidants: From enzymatic preparation to molecular rationalization. Eur. J. Med. Chem. 2017, 127, 263–274. [Google Scholar] [CrossRef]
- Beutner, S.; Frixel, S.; Ernst, H.; Hoffmann, T.; Hernandez-Blanco, I.; Hundsdoerfer, C.; Kiesendahl, N.; Kock, S.; Martin, H.-D.; Mayer, B.; et al. Carotenylflavonoids, a novel group of potent, dual-functional antioxidants. Arkivoc 2007, 8, 279–295. [Google Scholar] [CrossRef] [Green Version]
- Hundsdörfer, C.; Stahl, W.; Müller, T.J.J.; De Spirt, S. UVA Photoprotective Properties of an Artificial Carotenylflavonoid Hybrid Molecule. Chem. Res. Toxicol. 2012, 25, 1692–1698. [Google Scholar] [CrossRef]
- An, C.-B.; Liang, R.; Ma, X.-H.; Fu, L.-M.; Zhang, J.-P.; Wang, P.; Han, R.-M.; Ai, X.-C.; Skibsted, L.H. Retinylisoflavonoid as a Novel Membrane Antioxidant. J. Phys. Chem. B 2010, 114, 13904–13910. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Bu, Y.Z.; Liang, R.; Duan, R.M.; Wang, S.; Han, R.M.; Wang, P.; Ai, X.C.; Zhang, J.P.; Skibsted, L.H. Quercetin and daidzein β-apo-14′-carotenoic acid esters as membrane antioxidants. Free Rad. Res. 2013, 47, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.S.; Biedermann, D.; Valentová, K.; Petrásková, L.; Viktorová, J.; Kuzma, M.; Křen, V. Preparation of Retinoyl-Flavonolignan Hybrids and Their Antioxidant Properties. Antioxidants 2019, 8, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Háda, M.; Nagy, V.; Takátsy, A.; Deli, J.; Hait, J.; Agócs, A. Introduction of click chemistry to carotenoids. Tetrahedron Lett. 2012, 53, 2480–2482. [Google Scholar] [CrossRef]
- Agócs, A.; Bokor, É.; Takátsy, A.; Lóránd, T.; Deli, J.; Somsák, L.; Nagy, V. Synthesis of carotenoid-monosaccharide conjugates via azide-alkyne click-reaction. Tetrahedron 2017, 73, 519–526. [Google Scholar] [CrossRef]
- Li, M.; Han, X.; Yu, B. Facile Synthesis of Flavonoid 7-O-Glycosides. J. Org. Chem. 2003, 68, 6842–6845. [Google Scholar] [CrossRef]
- Gonda, Z.; Novák, Z. Highly active copper-catalysts for azide-alkyne cycloaddition. Dalton Trans. 2010, 39, 726–729. [Google Scholar] [CrossRef]
- Nagane, M.; Yamashita, T.; Vörös, P.; Kálai, T.; Hideg, K.; Bognár, B. Synthesis and evaluation of paramagnetic caffeic acid phenethyl ester (CAPE) analogs. Monatshefte Für Chemie-Chemical Monthly 2019, 150, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Polyakov, N.E.; Magyar, A.; Kispert, L.D. Photochemical and Optical Properties of Water-Soluble Xanthophyll Antioxidants: Aggregation vs Complexation. J. Phys. Chem. B 2013, 117, 10173–10182. [Google Scholar] [CrossRef]
- Ruban, A.V.; Horton, P.; Young, A.J. Aggregation of higher plant xanthophylls: Differences in absorption spectra and in the dependency on solvent polarity. J. Photochem. Photobiol. B Biol. 1993, 21, 229–234. [Google Scholar] [CrossRef]
- Tay-Agbozo, S.; Street, S.; Kispert, L.D. The carotenoid bixin: Optical studies of aggregation in polar/water solvents. J. Photochem. Photobiol. B Chem. 2018, 362, 31–39. [Google Scholar] [CrossRef]
- Horn, D.; Rieger, J. Organic Nanoparticles in the Aqueous Phase–Theory, Experiment, and Use. Angew. Chem. Int. Ed. 2001, 40, 4330–4361. [Google Scholar] [CrossRef]
- Simonyi, M.; Bikádi, Z.; Zila, F.; Deli, J. Supramolecular Exciton Chirality of Carotenoid Aggregates. Chirality 2003, 15, 680–698. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.A.; Machalska, E.; Oleszkiewicz, T.; Grzebelus, E.; Baranski, R.; Szcześniak, P.; Mlynarski, J.; Zajac, G.; Kaczor, A.; Baranska, M. Chiral Amplification in Nature: Cell-extracted Chiral Carotenoid Microcrystals Studied Via RROA of Model Systems. Angew. Chem. Int. Ed. 2019, 58, 8383–8388. [Google Scholar] [CrossRef]
- Sliwka, H.-R.; Partali, V.; Lockwood, S.F. Hydrophilic Carotenoids: Carotenoid Aggregates. In Carotenoids: Physical, Chemical, and Biological Functions and Properties; Landrum, J.T., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2010; Chapter 3; pp. 31–58. [Google Scholar]
- Billsten, H.H.; Sundström, V.; Polívka, T. Self-Assembled Aggregates of the Carotenoid Zeaxanthin: Time-Resolved Study of Excited States. J. Phys. Chem. A 2005, 109, 1521–1529. [Google Scholar] [CrossRef]
- Wang, C.; Berg, C.J.; Hsu, C.-C.; Merrill, B.A.; Tauber, M.J. Characterization of Carotenoid Aggregates by Steady-State Optical Spectroscopy. J. Phys. Chem. B 2012, 116, 10617–10630. [Google Scholar] [CrossRef]
Sample Availability: Samples of all the compounds (1–25) are available from the authors. |










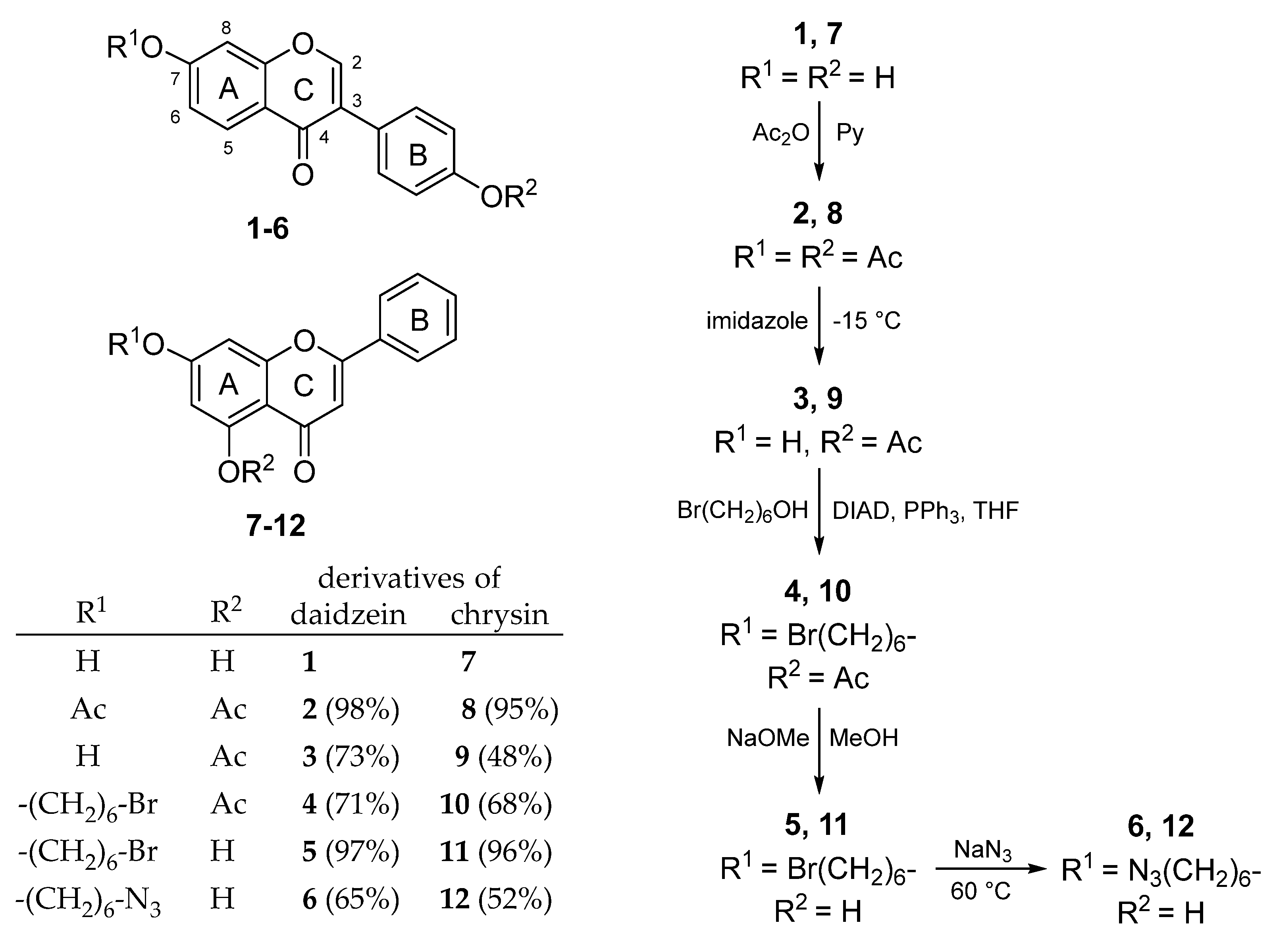{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | δ H-3 m (ppm) | δ H-3′ m (ppm) |
|---|---|---|
| capsanthin dipentynoate 15 | 5.16–5.10 | 5.33–5.29 |
| bistriazole 19 | 5.09–5.03 | 5.26–5.22 |
| monotriazole 20a | 5.13–5.03 | 5.31–5.25 |
| monotriazole 20b | 5.14–5.07 | 5.27–5.22 |
| Compound | TEAC | SD% | Variance | Compound | TEAC | SD% | Variance |
|---|---|---|---|---|---|---|---|
| Daidzein | 0.2804 | 4.9981 | 0.0070 | Chrysin | 0.1458 | 4.5581 | 0.0132 |
| Zeaxanthin | 0.7514 | 4.7639 | 0.0208 | Zeaxanthin | 0.7514 | 4.7639 | 0.0208 |
| 18 monotriazole | 0.5496 | 4.8011 | 0.0035 | 23 monotriazole | 0.4636 | 1.3732 | 0.0066 |
| 17 bistriazole | 0.9353 | 5.3524 | 0.1123 | 22 bistriazole | 0.8313 | 3.7065 | 0.0064 |
| 17 bistriazole in water | 0.6744 | 1.8747 | 0.0516 | ||||
| Zea:daidzein 1:2 mixt. | 0.6434 | 0.4262 | 0.0316 | ||||
| Capsanthin | 0.5728 | 3.2883 | 0.0095 | Capsanthin | 0.5728 | 3.2883 | 0.0095 |
| 20a monotriazole | 0.6643 | 1.6416 | 0.0095 | 25 monotriazole | 0.5026 | 2.1947 | 0.0091 |
| 20b monotriazole | 0.7199 | 0.0446 | 0.0033 | 24 bistriazole | 0.3698 | 4.4718 | 0.0242 |
| 19 bistriazole | 0.9663 | 0.5469 | 0.0463 | ||||
| 8′-apo-β-carotenal | 0.4433 | 3.9429 | 0.0068 | 8′-apo-β-car. | 0.4433 | 3.9429 | 0.0068 |
| 16 triazole | 0.3057 | 5.1589 | 0.0157 | 21 triazole | 0.1256 | 0.2122 | 0.0284 |
| Comparison | F Test | T Test | Comparison | F Test | T Test |
|---|---|---|---|---|---|
| Daidzein:18 | 74.28% | 0.04% | Daidzein:16 | 27.71% | 94.39% |
| Daidzein:17 | 0.32% | 4.18% | 8′-apo-β-car:16 | 67.89% | 4.93% |
| Daidzein:17 in water | 45.15% | 0.00% | |||
| Daidzein:Zea-daid. mixt. | 9.80% | 0.00% | Chrysin:23 | 23.74% | 3.63% |
| Zeaxanthin:18 | 30.03% | 4.99% | Chrysin:22 | 65.04% | 0.86% |
| Zeaxanthin:17 | 6.57% | 1.05% | Zeaxanthin:23 | 52.52% | 2.10% |
| Zeaxanthin:17 in water | 29.05% | 15.51% | Zeaxanthin:22 | 50.99% | 27.87% |
| Zeaxanthin: Zea-daid. mixt. | 55.19% | 74.42% | |||
| Chrysin:25 | 81.78% | 0.67% | |||
| Daidzein:20a | 62.55% | 0.51% | Chrysin:24 | 80.59% | 3.33% |
| Daidzein:20b | 41.90% | 0.00% | Capsanthin:25 | 88.47% | 7.40% |
| Daidzein:19 | 4.06% | 0.00% | Capsanthin:24 | 34.33% | 22.00% |
| Capsanthin:20a | 86.17% | 30.11% | |||
| Capsanthin:20b | 27.09% | 22.39% | Chrysin:21 | 69.35% | 32.28% |
| Capsanthin:19 | 13.29% | 4.94% | 8′-apo-β-car:21 | 41.10% | 3.86% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Línzembold, I.; Czett, D.; Böddi, K.; Kurtán, T.; Király, S.B.; Gulyás-Fekete, G.; Takátsy, A.; Lóránd, T.; Deli, J.; Agócs, A.; et al. Study on the Synthesis, Antioxidant Properties, and Self-Assembly of Carotenoid–Flavonoid Conjugates. Molecules 2020, 25, 636. https://doi.org/10.3390/molecules25030636
Línzembold I, Czett D, Böddi K, Kurtán T, Király SB, Gulyás-Fekete G, Takátsy A, Lóránd T, Deli J, Agócs A, et al. Study on the Synthesis, Antioxidant Properties, and Self-Assembly of Carotenoid–Flavonoid Conjugates. Molecules. 2020; 25(3):636. https://doi.org/10.3390/molecules25030636
Chicago/Turabian StyleLínzembold, Ildikó, Dalma Czett, Katalin Böddi, Tibor Kurtán, Sándor Balázs Király, Gergely Gulyás-Fekete, Anikó Takátsy, Tamás Lóránd, József Deli, Attila Agócs, and et al. 2020. "Study on the Synthesis, Antioxidant Properties, and Self-Assembly of Carotenoid–Flavonoid Conjugates" Molecules 25, no. 3: 636. https://doi.org/10.3390/molecules25030636