
Lights and Shadows of Gold Introduction into Beta Zeolite


Abstract
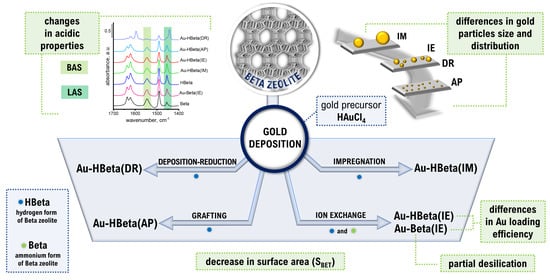
:
1. Introduction
2. Results and Discussion
2.1. Composition of Materials and Effectiveness of Gold Loading
2.2. Structure/Texture Characterization
2.3. Gold Particle Size
2.4. Surface and Electronic Properties of Zeolites—XPS Study
2.5. Acidity of Zeolites
3. Materials and Methods
3.1. Preparation of Au-HBeta(IE), Au-Beta(IE)_18h and Au-Beta(IE) by Ion Exchange Method
3.2. Preparation of Au-HBeta(IM) by Wet Impregnation Method
3.3. Preparation of Au-HBeta(DR) by Deposition-Reduction Method
3.4. Preparation of Au-HBeta(AP) by Anchoring of Gold Species on APTMS-Grafted HBeta
3.5. Characterization Techniques
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Riello, P.; Canton, P.; Benedetti, A. Au/C Catalyst: Experimental evidence of the coexistence of nanoclusters and larger Au particles. Langmuir 1998, 14, 6617–6619. [Google Scholar] [CrossRef]
- Pittayaporn, N.; Therdthianwong, A.; Therdthianwong, S. Au/C catalysts promoted with Ni for glycerol electrooxidation in alkaline media. J. Appl. Electrochem. 2018, 48, 251–262. [Google Scholar] [CrossRef]
- Sobczak, I.; Wolski, L. Au–Cu on Nb2O5 and Nb/MCF supports—Surface properties and catalytic activity in glycerol and methanol oxidation. Catal. Today 2015, 254, 72–82. [Google Scholar] [CrossRef]
- Kaminski, P.; Ziolek, M. Surface and catalytic properties of Ce-, Zr-, Au-, Cu-modified SBA-15. J. Catal. 2014, 312, 249–262. [Google Scholar] [CrossRef]
- Escamilla-Perea, L.; Nava, R.; Pawelec, B.; Rosmaninho, M.G.; Peza-Ledesma, C.L.; Fierro, J.L.G. SBA-15-supported gold nanoparticles decorated by CeO2: Structural characteristics and CO oxidation activity. Appl. Catal. A Gen. 2010, 381, 42–53. [Google Scholar] [CrossRef]
- Moreno, I.; Dummer, N.F.; Edwards, J.K.; Alhumaimess, M.; Sankar, M.; Sanz, R.; Pizarro, P.; Serrano, D.P.; Hutchings, G.J. Selective oxidation of benzyl alcohol using in situ generated H2O2 over hierarchical Au–Pd titanium silicalite catalysts. Catal. Sci. Technol. 2013, 3, 2425. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Edwards, J.; Carley, A.F.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using zeolite-supported Au catalysts. Catal. Today 2006, 114, 369–371. [Google Scholar] [CrossRef]
- Guillemot, D.; Borovkov, V.Y.; Kazansky, V.B.; Polisset-Thfoin, M.; Fraissard, J. Surface characterization of Au/HY by 129Xe NMR and diffuse reflectance IR spectroscopy of adsorbed CO. Formation of electron-deficient gold particles inside HY cavities. J. Chem. Soc. Faraday Trans. 1997, 93, 3587–3591. [Google Scholar] [CrossRef]
- Wojtaszek-Gurdak, A.; Sobczak, I.; Grzelak, K.; Ziolek, M.; Hartfelder, U.; van Bokhoven, J.A. The role of pillaring in MCM-22 on the dispersion of noble metals and catalytic activity. Mater. Res. Bull. 2016, 76, 169–178. [Google Scholar] [CrossRef]
- Højholt, K.T.; Laursen, A.B.; Kegnæs, S.; Christensen, C.H. Size-selective oxidation of aldehydes with zeolite encapsulated gold nanoparticles. Top. Catal. 2011, 54, 1026–1033. [Google Scholar] [CrossRef]
- Behravesh, E.; Kumar, N.; Balme, Q.; Roine, J.; Salonen, J.; Schukarev, A.; Mikkola, J.; Peurla, M.; Aho, A.; Eränen, K.; et al. Synthesis and characterization of Au nano particles supported catalysts for partial oxidation of ethanol: Influence of solution pH, Au nanoparticle size, support structure and acidity. J. Catal. 2017, 353, 223–238. [Google Scholar] [CrossRef]
- Tatibouët, J.M. Methanol oxidation as a catalytic surface probe. Appl. Catal. A Gen. 1997, 148, 213–252. [Google Scholar] [CrossRef]
- Kaskow, I.; Wojtaszek-Gurdak, A.; Sobczak, I. Methanol oxidation on AuAg-Zn/MCM-36—The effect of catalyst components and pretreatment. Catal. Today 2020, 354, 123–132. [Google Scholar] [CrossRef]
- Wolski, L.; Sobczak, I.; Ziolek, M. Variability of surface components in gold catalysts—The role of hydroxyls and state of gold on activity and selectivity of Au-Nb2O5 and Au-ZnNb2O6 in methanol oxidation. J. Catal. 2017, 354, 100–112. [Google Scholar] [CrossRef]
- Forzatti, P.; Tronconi, E.; Elmi, A.S.; Busca, G. Methanol oxidation over vanadia-based catalysts. Appl. Catal. A Gen. 1997, 157, 387–408. [Google Scholar] [CrossRef]
- Tranca, D.C.; Keil, F.J.; Tranca, I.; Calatayud, M.; Dzwigaj, S.; Trejda, M.; Tielens, F. Methanol Oxidation to Formaldehyde on VSiBEA Zeolite: A combined DFT/vdW/transition path sampling and experimental study. J. Phys. Chem. C 2015, 119, 13619–13631. [Google Scholar] [CrossRef]
- Rodrigues, C.S.D.; Silva, R.M.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Madeira, L.M. Wastewater treatment by catalytic wet peroxidation using nano gold-based catalysts: A review. Catalysts 2019, 9, 478. [Google Scholar] [CrossRef] [Green Version]
- Wolski, L. Factors affecting the activity and selectivity of niobia-based gold catalysts in liquid phase glycerol oxidation. Catal. Today 2020, 354, 36–43. [Google Scholar] [CrossRef]
- Liu, X.; Wang, A.; Yang, X.; Zhang, T.; Mou, C.; Su, D.-S.; Li, J. Synthesis of thermally stable and highly active bimetallic Au–Ag nanoparticles on inert supports. Chem. Mater. 2009, 21, 410–418. [Google Scholar] [CrossRef]
- Sobczak, I.; Dembowiak, E. The effect of AuAg–MCF and AuAg–NbMCF catalysts pretreatment on the gold–silver alloy formation and the catalytic behavior in selective methanol oxidation with oxygen. J. Mol. Catal. A Chem. 2015, 409, 137–148. [Google Scholar] [CrossRef]
- Li, H.; Pan, J.; Gao, C.; Ma, M.; Lu, L.; Xiong, Y.; Dong, F. Mercapto-functionalized porous organosilica monoliths loaded with gold nanoparticles for catalytic application. Molecules 2019, 24, 4366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsam, J.M.; Treacy, M.M.J.; Koetsier, W.T.; De Gruyter, C.B. Structural characterization of zeolite beta. Proc. R. Soc. Lond. A Math. Phys. Sci. 1988, 420, 375–405. [Google Scholar] [CrossRef]
- Higgins, J.B.; LaPierre, R.B.; Schlenker, J.L.; Rohrman, A.C.; Wood, J.D.; Kerr, G.T.; Rohrbaugh, W.J. The framework topology of zeolite beta. Zeolites 1988, 8, 446–452. [Google Scholar] [CrossRef]
- Lu, T.; Yan, W.; Xu, R. Chiral zeolite beta: Structure, synthesis, and application. Inorg. Chem. Front. 2019, 6, 1938–1951. [Google Scholar] [CrossRef]
- Bejblová, M.; Procházková, D.; Čejka, J. Acylation reactions over zeolites and mesoporous catalysts. ChemSusChem 2009, 2, 486–499. [Google Scholar] [CrossRef]
- Zeng, X.; Wang, Z.; Ding, J.; Wang, L.; Jiang, Y.; Stampfl, C.; Hunger, M. Catalytic arene alkylation over H-Beta zeolite: Influence of zeolite shape selectivity and reactant nucleophilicity. J. Catal. 2019, 380, 9–20. [Google Scholar] [CrossRef]
- Manrique, C.; Guzmán, A.; Pérez-Pariente, J.; Márquez-Álvarez, C.; Echavarría, A. Effect of synthesis conditions on zeolite Beta properties and its performance in vacuum gas oil hydrocracking activity. Microporous Mesoporous Mater. 2016, 234, 347–360. [Google Scholar] [CrossRef]
- LaPierre, R.B.; Partridge, R.D.; Chen, N.Y.; Wong, S.S. Catalytic Dewaxing Process with Zeolite Beta. U.S. Patent 45,019,26A, 26 February 1985. [Google Scholar]
- Sanada, T.; Murakami, C.; Góra-Marek, K.; Iida, K.; Katada, N.; Okumura, K. Fabrication and catalytic activity of thermally stable gold nanoparticles on ultrastable Y (USY) Zeolites. Catalysts 2013, 3, 599–613. [Google Scholar] [CrossRef]
- Chaves, T.F.; Pastore, H.O.; Hammer, P.; Cardoso, D. As-synthesized TEA-BEA zeolite: Effect of Si/Al ratio on the Knoevenagel condensation. Microporous Mesoporous Mater. 2015, 202, 198–207. [Google Scholar] [CrossRef]
- Murphy, P.J.; Stevens, G.; LaGrange, M.S. The effects of temperature and pressure on gold–chloride speciation in hydrothermal fluids: A Raman spectroscopic study. Geochim. Cosmochim. Acta 2000, 64, 479–494. [Google Scholar] [CrossRef]
- Ivanova, S.; Petit, C.; Pitchon, V. A new preparation method for the formation of gold nanoparticles on an oxide support. Appl. Catal. A Gen. 2004, 267, 191–201. [Google Scholar] [CrossRef]
- Mironov, I.V.; Makotchenko, E.V. The Hydrolysis of AuCl4− and the stability of aquachlorohydroxocomplexes of gold(III) in aqueous solution. J. Solution Chem. 2009, 38, 725–737. [Google Scholar] [CrossRef]
- Tossell, J.A. The speciation of gold in aqueous solution: A theoretical study. Geochim. Cosmochim. Acta 1996, 60, 17–29. [Google Scholar] [CrossRef]
- Ivanova, S.; Petit, C.; Pitchon, V. Application of heterogeneous gold catalysis with increased durability: Oxidation of CO and hydrocarbons at low temperature. Gold Bull. 2006, 39, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-M.; Wan, B.-Z. Preparation of gold in Y-type zeolite for carbon monoxide oxidation. Appl. Catal. A Gen. 1995, 128, 53–60. [Google Scholar] [CrossRef]
- Moreau, F.; Bond, G.; Taylor, A. Gold on titania catalysts for the oxidation of carbon monoxide: Control of pH during preparation with various gold contents. J. Catal. 2005, 231, 105–114. [Google Scholar] [CrossRef]
- Kang, Y.-M.; Wan, B.-Z. Gold and iron supported on Y-type zeolite for carbon monoxide oxidation. Catal. Today 1997, 35, 379–392. [Google Scholar] [CrossRef]
- Tong, M.; Zhang, D.; Fan, W.; Xu, J.; Zhu, L.; Guo, W.; Yan, W.; Yu, J.; Qiu, S.; Wang, J.; et al. Synthesis of chiral polymorph A-enriched zeolite Beta with an extremely concentrated fluoride route. Sci. Rep. 2015, 5, 11521. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, Z.S.; Sarawade, P.B.; Hussain, I.; Zhu, H.; Al-Johani, H.; Anjum, D.H.; Hedhili, M.N.; Maity, N.; D’Elia, V.; Basset, J.-M. Gold nanoparticles supported on fibrous silica nanospheres (KCC-1) as efficient heterogeneous catalysts for CO oxidation. ChemCatChem 2016, 8, 1671–1678. [Google Scholar] [CrossRef]
- Amendola, V.; Meneghetti, M. Size evaluation of gold nanoparticles by UV–vis spectroscopy. J. Phys. Chem. C 2009, 113, 4277–4285. [Google Scholar] [CrossRef]
- Huang, X.; El-Sayed, M.A. Gold nanoparticles: Optical properties and implementations in cancer diagnosis and photothermal therapy. J. Adv. Res. 2010, 1, 13–28. [Google Scholar] [CrossRef] [Green Version]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.M.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the characterization of porous solids (Technical Report). Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Bordoloi, A.; Devassy, B.M.; Niphadkar, P.S.; Joshi, P.N.; Halligudi, S.B. Shape selective synthesis of long-chain linear alkyl benzene (LAB) with AlMCM-41/Beta zeolite composite catalyst. J. Mol. Catal. A Chem. 2006, 253, 239–244. [Google Scholar] [CrossRef]
- Khoramzadeh, E.; Mofarahi, M.; Lee, C. Equilibrium adsorption study of CO2 and N2 on synthesized zeolites 13X, 4A, 5A, and Beta. J. Chem. Eng. Data 2019, 64, 5648–5664. [Google Scholar] [CrossRef]
- Ishida, T.; Murayama, T.; Taketoshi, A.; Haruta, M. Importance of size and contact structure of gold nanoparticles for the genesis of unique catalytic processes. Chem. Rev. 2020, 120, 464–525. [Google Scholar] [CrossRef] [Green Version]
- Patterson, A.L. The Scherrer formula for X-ray particle size determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent Developments in the synthesis of supported catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wang, A.; Zhang, T.; Mou, C.-Y. Catalysis by gold: New insights into the support effect. Nano Today 2013, 8, 403–416. [Google Scholar] [CrossRef]
- Xie, X.; Long, J.; Xu, J.; Chen, L.; Wang, Y.; Wang, X. Nitrogen-doped graphene stabilized gold nanoparticles for aerobic selective oxidation of benzylic alcohols. RSC Adv. 2012, 12438–12446. [Google Scholar] [CrossRef]
- Hegde, S.G.; Kumar, R.; Bhat, R.N.; Ratnasamy, P. Characterization of the acidity of zeolite Beta by FT i.r. spectroscopy and t.p.d. of NH3. Zeolites 1989, 9, 231–237. [Google Scholar] [CrossRef]
- Jia, C.; Massiani, P.; Barthomeuf, D. Characterization by infrared and nuclear magnetic resonance spectroscopies of calcined beta zeolite. J. Chem. Soc. Faraday Trans. 1993, 89, 3659. [Google Scholar] [CrossRef]
- Kiricsi, I.; Flego, C.; Pazzuconi, G.; Parker, W.O.J.; Millini, R.; Perego, C.; Bellussi, G. Progress toward understanding zeolite Beta acidity: An IR and 27Al NMR spectroscopic study. J. Phys. Chem. 1994, 98, 4627–4634. [Google Scholar] [CrossRef]
- Guisnet, M.; Ayrault, P.; Coutanceau, C.; Fernanda Alvarez, M.; Datka, J. Acid properties of dealuminated beta zeolites studied by IR spectroscopy. J. Chem. Soc. Faraday Trans. 1997, 93, 1661–1665. [Google Scholar] [CrossRef]
- Busca, G. Spectroscopic characterization of the acid properties of metal oxide catalysts. Catal. Today 1998, 41, 191–206. [Google Scholar] [CrossRef]
- Busch, O.M.; Brijoux, W.; Thomson, S.; Schüth, F. Spatially resolving infrared spectroscopy for parallelized characterization of acid sites of catalysts via pyridine sorption: Possibilities and limitations. J. Catal. 2004, 222, 174–179. [Google Scholar] [CrossRef]
- Busca, G. The surface acidity of solid oxides and its characterization by IR spectroscopic methods. An attempt at systematization. Phys. Chem. Chem. Phys. 1999, 1, 723–736. [Google Scholar] [CrossRef]
- Gil, B.; Marszałek, B.; Micek-Ilnicka, A.; Olejniczak, Z. The Influence of Si/Al Ratio on the Distribution of OH Groups in Zeolites with MWW Topology. Top. Catal. 2010, 53, 1340–1348. [Google Scholar] [CrossRef] [Green Version]
- Suárez, N.; Pérez-Pariente, J.; Márquez-Álvarez, C.; Grande Casas, M.; Mayoral, A.; Moreno, A. Preparation of mesoporous Beta zeolite by fluoride treatment in liquid phase. Textural, acid and catalytic properties. Microporous Mesoporous Mater. 2019, 284, 296–303. [Google Scholar] [CrossRef]
- Auroux, A.; Gervasini, A.; Guimon, C. Acidic Character of Metal-Loaded Amorphous and Crystalline Silica–Aluminas Determined by XPS and Adsorption Calorimetry. J. Phys. Chem. B 1999, 103, 7195–7205. [Google Scholar] [CrossRef]
- Wisniewska, J.; Sobczak, I.; Ziolek, M. Gold based on SBA-15 supports—Promising catalysts in base-free glucose oxidation. Chem. Eng. J. 2020, 127548. [Google Scholar] [CrossRef]
- Sobczak, I.; Pawlowski, H.; Chmielewski, J.; Ziolek, M. Gold and gold–iron modified zeolites—Towards the adsorptive deodourisation. J. Hazard. Mater. 2010, 179, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, I. The role of niobium in MCM-41 supported with Pt and Au—A comparative study of physicochemical and catalytic properties. Catal. Today 2009, 142, 258–266. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Au Content a [wt.% Au] | Si/Al Molar Ratio (ICP-OES) | Si/Al Molar Ratio b (XPS) | dAu (XRD) c [nm] | dAu (TEM) [nm] |
|---|---|---|---|---|---|
| Beta | - | 17.8 | - | - | - |
| Au-Beta(IE) | 1.5 | 12.7 | 11.7 | 32 | 24 |
| Au-Beta(IE)_18h | 1.2 | 16.9 | - | - | - |
| HBeta | - | 18.4 | 17.3 | - | - |
| Au-HBeta(IM) | 1.5 | 18.5 | 18.2 | 88 | - d |
| Au-HBeta(IE) | 0.2 | 17.5 | 14.7 | - e | - f |
| Au-HBeta(AP) | 1.4 | 18.6 | 17.9 | 8 | 6 |
| Au-HBeta(DR) | 0.7 | 18.4 | 19.4 | 52 | - f |
| Sample | BET Surface Area [m2·g−1] | t-Plot Micropore Area [m2·g−1] | t-Plot External Surface Area [m2·g−1] | BJH Culminative Volume of Pores (Adsorption Branch) [cm3·g−1] |
|---|---|---|---|---|
| Beta | 521 | 379 | 141 | 0.14 |
| Au-Beta(IE) | 516 | 349 | 167 | 0.16 |
| HBeta | 527 | 377 | 149 | 0.13 |
| Au-HBeta(IM) | 492 | 342 | 150 | 0.14 |
| Au-HBeta(IE) | 520 | 344 | 176 | 0.16 |
| Au-HBeta(AP) | 360 | 260 | 99 | 0.11 |
| Au-HBeta(DR) | 466 | 328 | 138 | 0.13 |
| Sample | Evacuation Temp. [°C] | No. of BAS Occupied by Pyridine [μmol·g−1] | Pyridine Desorbed at 300 °C from BAS [%] a | No. of LAS Occupied by Pyridine after Evacuation at 250 °C [μmol·g−1] | BAS/LAS Ratio after Evacuation at 250 °C |
|---|---|---|---|---|---|
| Beta | 150 | 594 | 26 | 87 | 7.03 |
| 200 | 694 | ||||
| 250 | 612 | ||||
| 300 | 515 | ||||
| Au-Beta(IE) | 150 | 475 | 26 | 166 | 2.90 |
| 200 | 522 | ||||
| 250 | 481 | ||||
| 300 | 389 | ||||
| HBeta | 150 | 434 | 34 | 196 | 1.84 |
| 200 | 452 | ||||
| 250 | 360 | ||||
| 300 | 297 | ||||
| Au-HBeta(IM) | 150 | 431 | 24 | 223 | 1.80 |
| 200 | 455 | ||||
| 250 | 401 | ||||
| 300 | 345 | ||||
| Au-HBeta(IE) | 150 | 383 | 27 | 160 | 2.46 |
| 200 | 429 | ||||
| 250 | 394 | ||||
| 300 | 313 | ||||
| Au-HBeta(AP) | 150 | 339 | 38 | 137 | 2.01 |
| 200 | 345 | ||||
| 250 | 275 | ||||
| 300 | 215 | ||||
| Au-HBeta(DR) | 150 | 64 | 12 | 119 | 0.76 |
| 200 | 87 | ||||
| 250 | 91 | ||||
| 300 | 77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walkowiak, A.; Wolski, L.; Ziolek, M. Lights and Shadows of Gold Introduction into Beta Zeolite. Molecules 2020, 25, 5781. https://doi.org/10.3390/molecules25245781
Walkowiak A, Wolski L, Ziolek M. Lights and Shadows of Gold Introduction into Beta Zeolite. Molecules. 2020; 25(24):5781. https://doi.org/10.3390/molecules25245781
Chicago/Turabian StyleWalkowiak, Adrian, Lukasz Wolski, and Maria Ziolek. 2020. "Lights and Shadows of Gold Introduction into Beta Zeolite" Molecules 25, no. 24: 5781. https://doi.org/10.3390/molecules25245781