
Permeation of β-Lactamase Inhibitors through the General Porins of Gram-Negative Bacteria

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
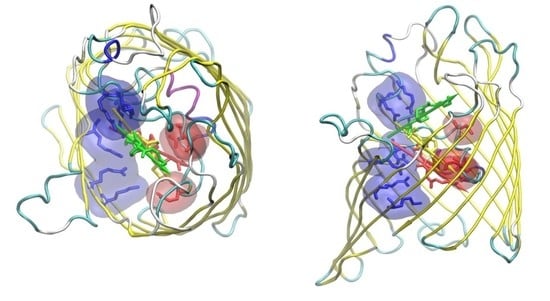
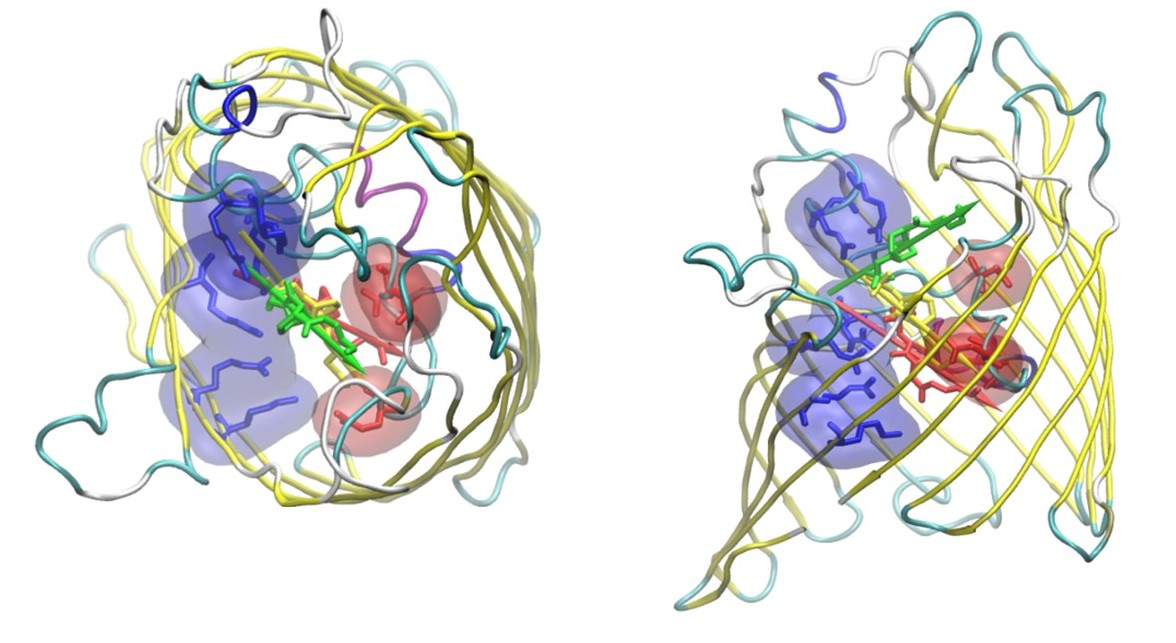
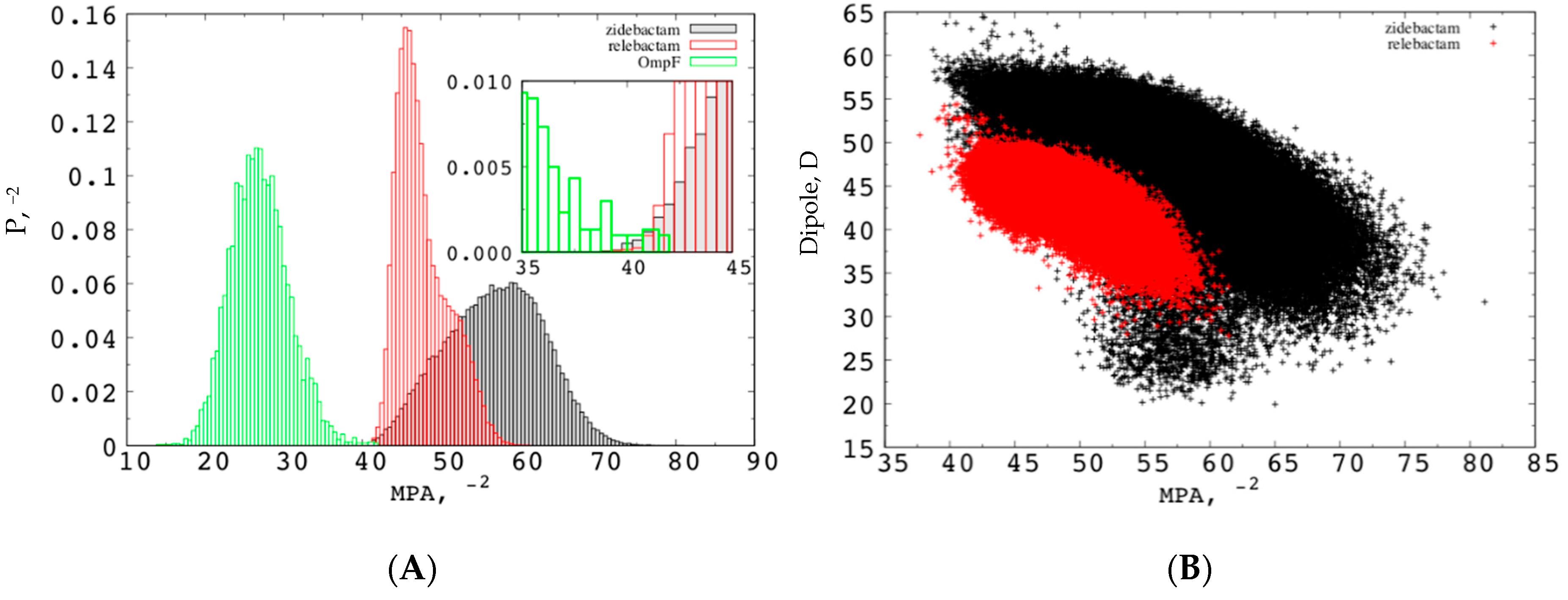
2.1. The Electrostatic Fingerprint of the OmpF and OmpC Orthologues
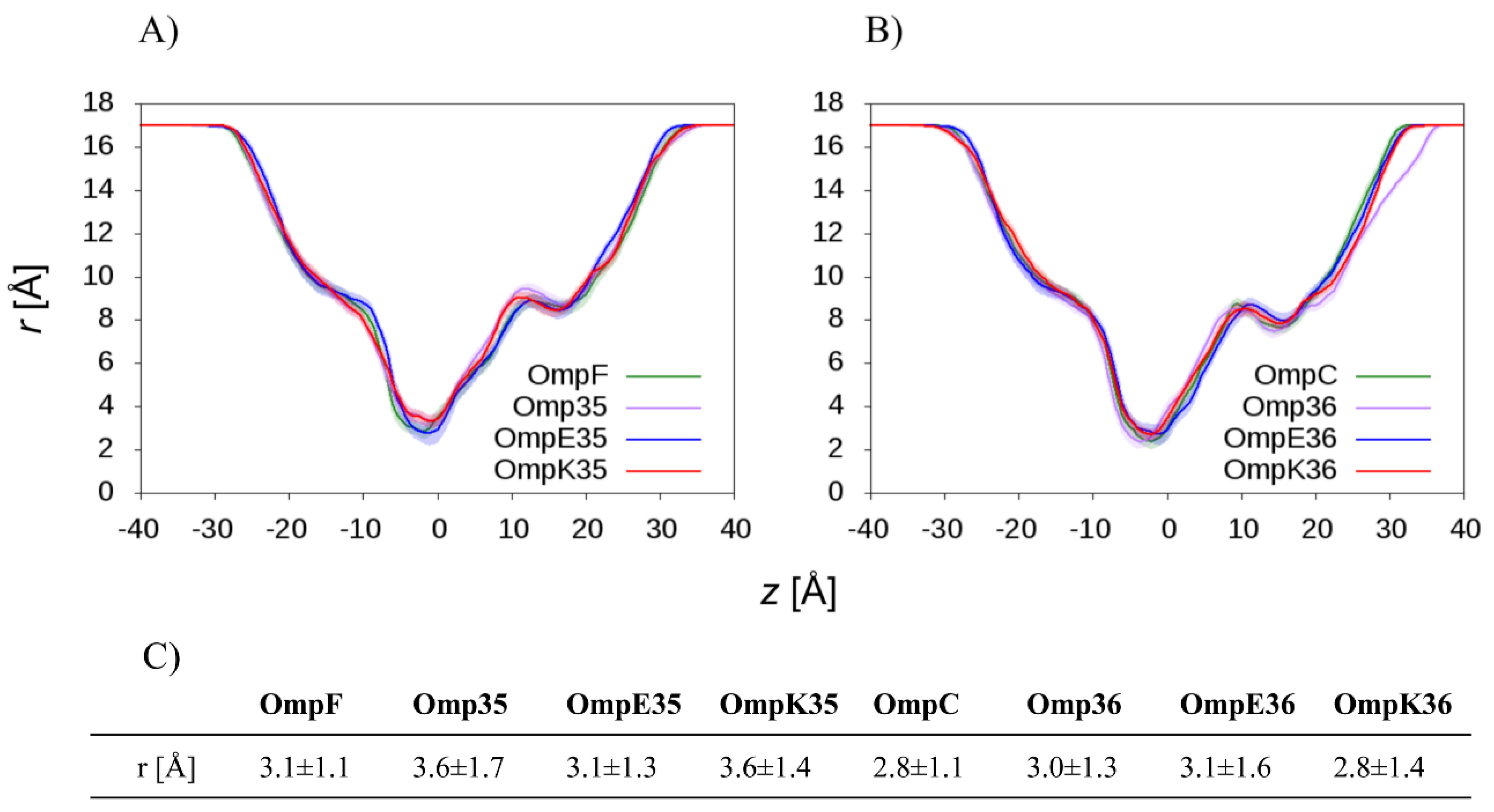
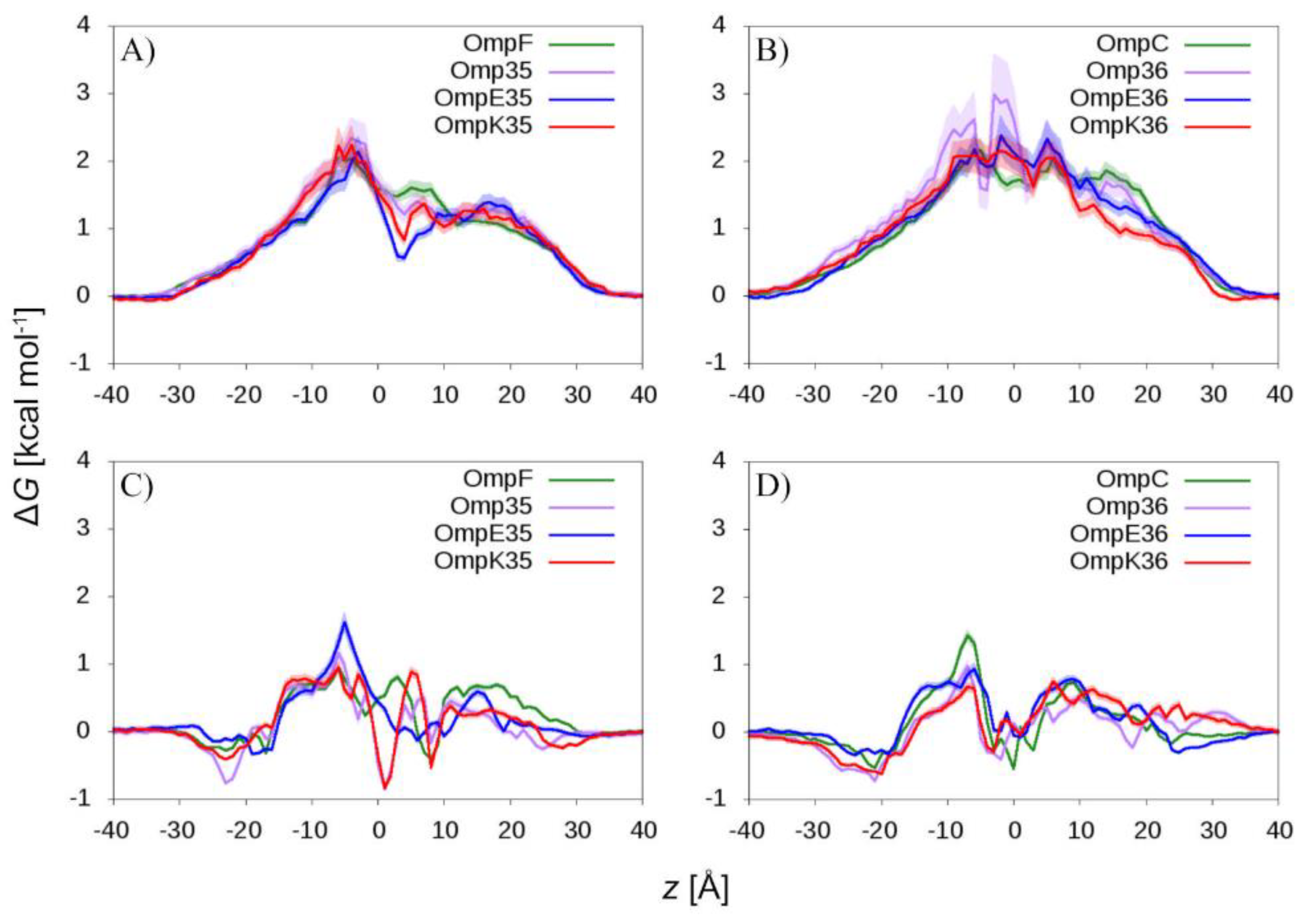
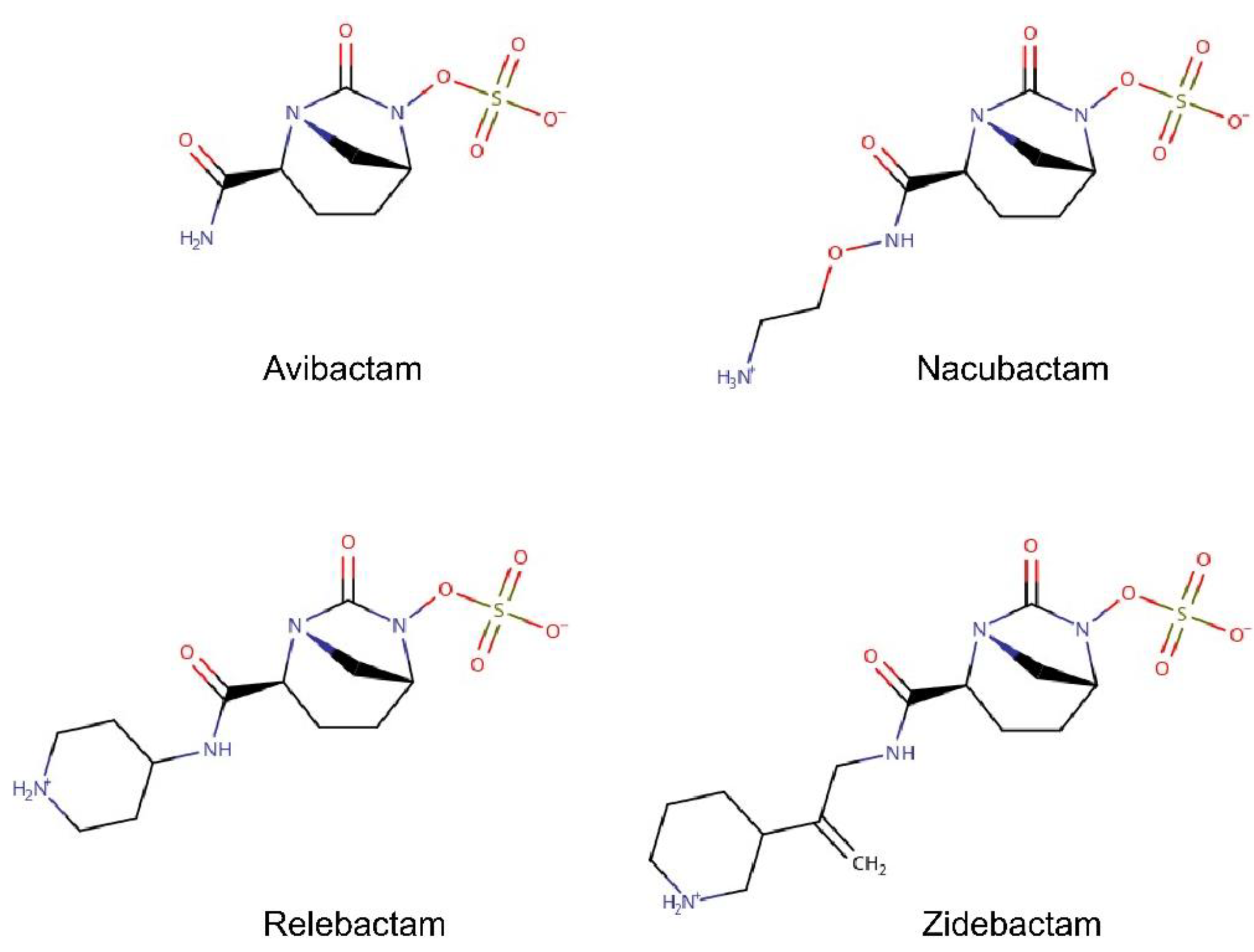
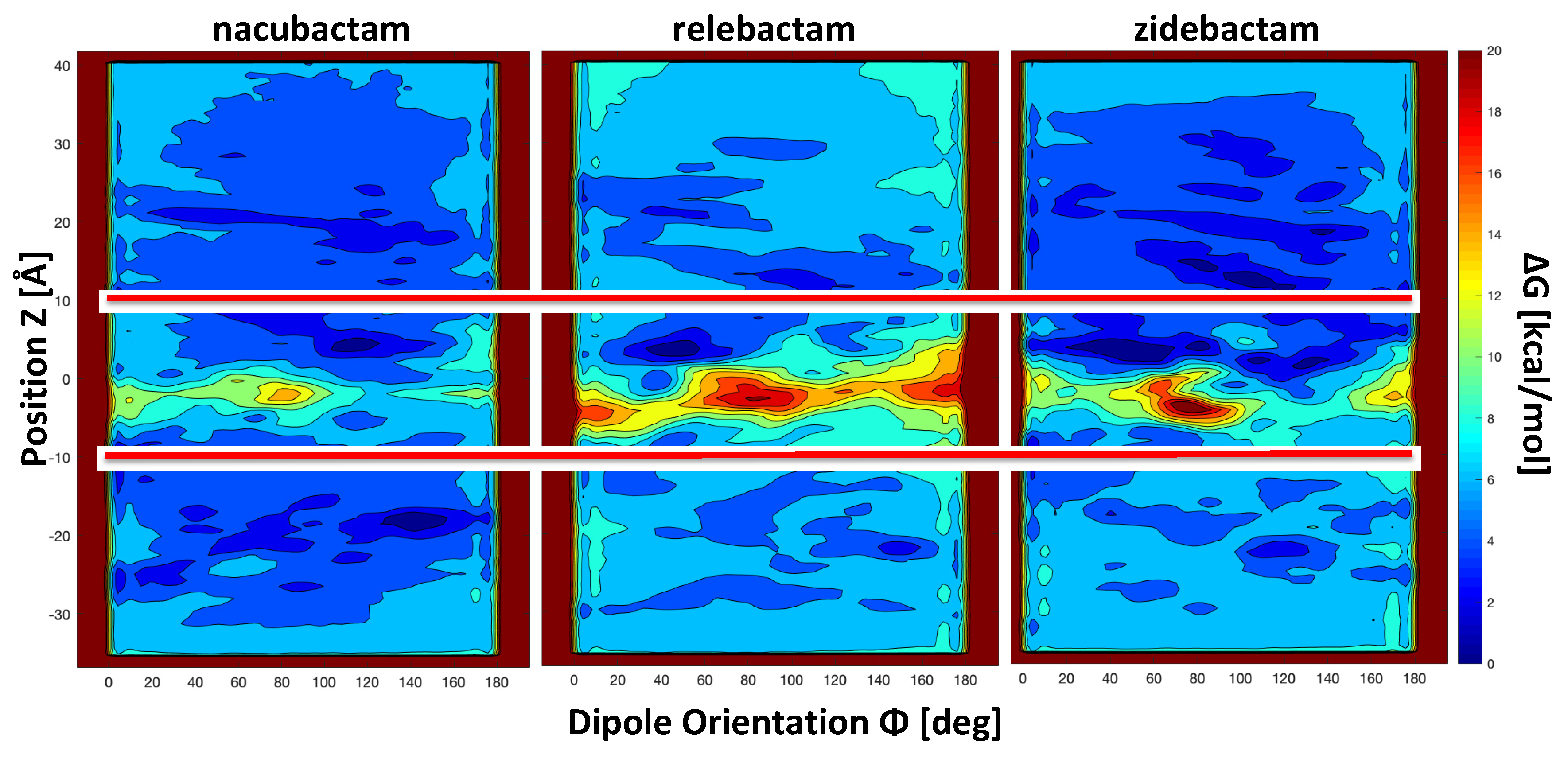
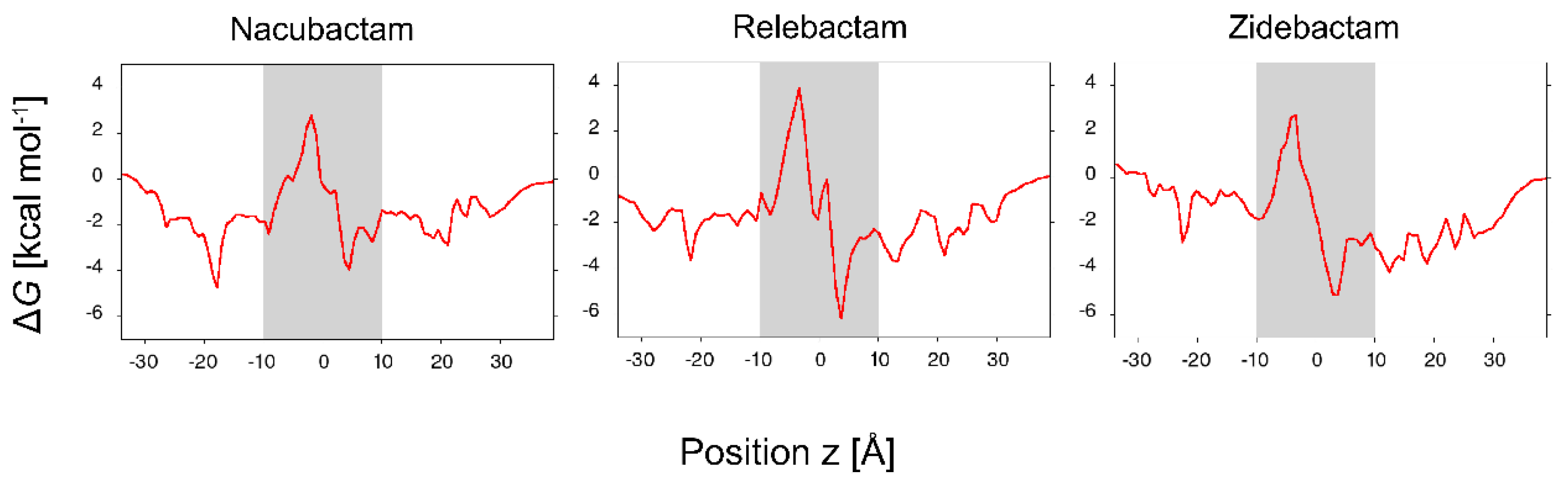
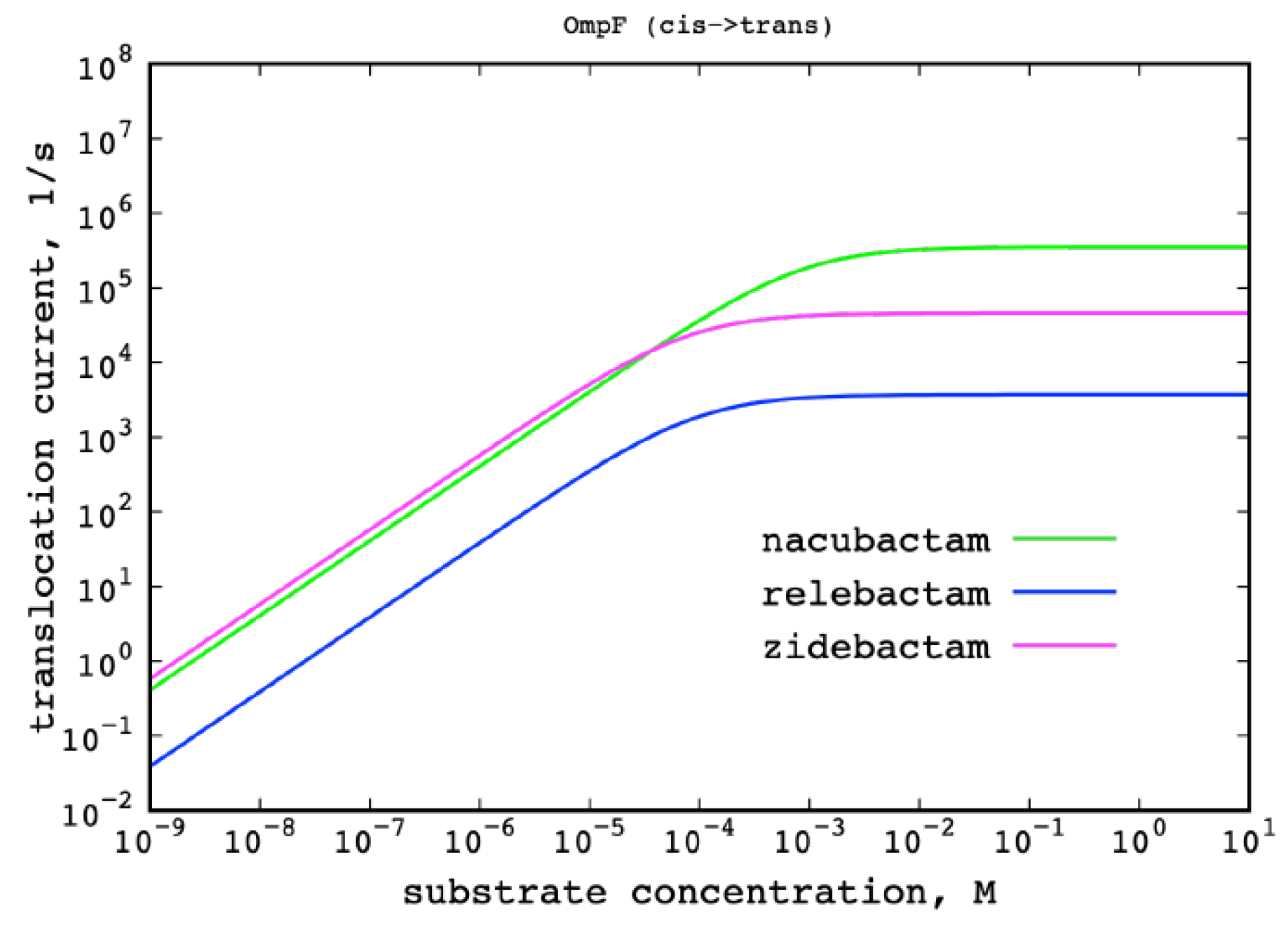
2.2. The Permeation Free-Energy of Diazobicyclooctans β-Lactamase Inhibitors through OmpF
3. Methods
3.1. Molecular Dynamics
3.2. Substrate Parameterization
3.3. Enhancing Sampling: Metadynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Lack of New Antibiotics Threatens Global Efforts to Contain Drug-Resistant Infections. Available online: https://www.who.int/news-room/detail/17-01-2020-lack-of-new-antibiotics-threatens-global-efforts-to-contain-drug-resistant-infections, (accessed on 3 April 2020).
- WHO. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Silver, L.L. Challenges of Antibacterial Discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tommasi, R.; Brown, D.G.; Walkup, G.K.; Manchester, J.I.; Miller, A.A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov. 2015, 14, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nat. Cell Biol. 2017, 545, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta-Gutiérrez, S.; Ferrara, L.; Pathania, M.; Masi, M.; Wang, J.; Bodrenko, I.; Zahn, M.; Winterhalter, M.; Stavenger, R.A.; Pagés, J.-M.; et al. Getting Drugs into Gram-Negative Bacteria: Rational Rules for Permeation through General Porins. ACS Infect. Dis. 2018, 4, 1487–1498. [Google Scholar] [CrossRef] [Green Version]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp-Wallace, K.M. The latest advances in β-lactam/β-lactamase inhibitor combinations for the treatment of Gram-negative bacterial infections. Expert Opin. Pharmacother. 2019, 20, 2169–2184. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- The PEW Charitable Trusts, Antibiotics Currently in Clinical Development. Available online: http://www.pewtrusts.org/en/multimedia/data-visualizations/2014/antibiotics-currently-in-clinical-development (accessed on 30 September 2020).
- O’Neill, J.; Audi, H.; Knox, J.; Hall, W.; McDonnell, A.; Seshadri, A.; Mudd, J.; Truscott-Reid, N.; Toxvaerd, F.; Woodford, N.; et al. Review on Antimicrobial Resistance; Wellcome Trust: London, UK, 2016. [Google Scholar]
- Venter, H. Reversing resistance to counter antimicrobial resistance in the World Health Organisation’s critical priority of most dangerous pathogens. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.; Hancock, R.E.W. Adaptive and Mutational Resistance: Role of Porins and Efflux Pumps in Drug Resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, T.S.; Parkin, J.; Khalid, S. The Free Energy of Small Solute Permeation through the Escherichia coli Outer Membrane Has a Distinctly Asymmetric Profile. J. Phys. Chem. Lett. 2016, 7, 3446–3451. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H.; Vaara, M. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 1985, 49, 1–32. [Google Scholar] [CrossRef]
- Bartsch, A.; Llabrés, S.; Pein, F.; Kattner, C.; Schön, M.; Diehn, M.; Tanabe, M.; Munk, A.; Zachariae, U.; Steinem, C. High-resolution experimental and computational electrophysiology reveals weak β-lactam binding events in the porin PorB. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagés, J.-M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Genet. 2008, 6, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Deng, Z.; Yan, A. Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochem. Biophys. Res. Commun. 2014, 453, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galdadas, I.; Lovera, S.; Pérez-Hernández, G.; Barnes, M.D.; Healy, J.; Afsharikho, H.; Woodford, N.; Bonomo, R.A.; Gervasio, F.L.; Haider, S. Defining the architecture of KPC-2 Carbapenemase: Identifying allosteric networks to fight antibiotics resistance. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Vergalli, J.; Bodrenko, I.V.; Masi, M.; Moynié, L.; Acosta-Gutiérrez, S.; Naismith, J.H.; Davin-Regli, A.; Ceccarelli, M.; Berg, B.V.D.; Winterhalter, M.; et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat. Rev. Genet. 2019, 18, 164–176. [Google Scholar] [CrossRef]
- Hancock, R.E.; Brinkman, F.S.L. Function ofPseudomonasPorins in Uptake and Efflux. Annu. Rev. Microbiol. 2002, 56, 17–38. [Google Scholar] [CrossRef] [Green Version]
- Eren, E.; Vijayaraghavan, J.; Liu, J.; Cheneke, B.R.; Touw, D.S.; Lepore, B.W.; Indic, M.; Movileanu, L.; Berg, B.V.D. Substrate Specificity within a Family of Outer Membrane Carboxylate Channels. PLoS Biol. 2012, 10, e1001242. [Google Scholar] [CrossRef] [Green Version]
- Benkerrou, D.; Ceccarelli, M. Free energy calculations and molecular properties of substrate translocation through OccAB porins. Phys. Chem. Chem. Phys. 2018, 20, 8533–8546. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Porins and specific diffusion channels in bacterial outer membranes. J. Biol. Chem. 1994, 269, 3905–3908. [Google Scholar] [PubMed]
- Masi, M.; Pagès, J.-M. Structure, Function and Regulation of Outer Membrane Proteins Involved in Drug Transport in Enterobactericeae: The OmpF/C—TolC Case. Open Microbiol. J. 2013, 7, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, S.; Nikaido, H. High Salt Concentrations Increase Permeability through OmpC Channels of Escherichia coli. J. Biol. Chem. 2014, 289, 26464–26473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.R.; Ceccarelli, M.; Lovelle, M.; Winterhalter, M.; Mahendran, K.R. Antibiotic Permeation across the OmpF Channel: Modulation of the Affinity Site in the Presence of Magnesium. J. Phys. Chem. B 2012, 116, 4433–4438. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Nikaido, H. Permeation rates of penicillins indicate that Escherichia coli porins function principally as nonspecific channels. Proc. Natl. Acad. Sci. 2013, 110, E2629–E2634. [Google Scholar] [CrossRef] [Green Version]
- Mahendran, R.; Kreir, M.; Weingart, H.; Fertig, N.; Winterhalter, M. Permeation of antibiotics through escherichia coli OmpF and OmpC porins: Screening for influx on a single-molecule level. J. Biomol. Screen. 2010, 15, 302–307. [Google Scholar] [CrossRef]
- Mahendran, K.R.; Hajjar, E.; Mach, T.; Lovelle, M.; Kumar, A.; Sousa, I.; Spiga, E.; Weingart, H.; Gameiro, P.; Winterhalter, M.; et al. Molecular Basis of Enrofloxacin Translocation through OmpF, an Outer Membrane Channel of Escherichia coli—When Binding Does Not Imply Translocation. J. Phys. Chem. B 2010, 114, 5170–5179. [Google Scholar] [CrossRef]
- Lou, H.; Chen, M.; Black, S.S.; Bushell, S.R.; Ceccarelli, M.; Mach, T.; Beis, K.; Low, A.S.; Bamford, V.A.; Booth, I.R.; et al. Altered Antibiotic Transport in OmpC Mutants Isolated from a Series of Clinical Strains of Multi-Drug Resistant E. coli. PLoS ONE 2011, 6, e25825. [Google Scholar] [CrossRef] [Green Version]
- Tran, Q.-T.; Williams, S.; Farid, R.; Erdemli, G.; Pearlstein, R. The translocation kinetics of antibiotics through porin OmpC: Insights from structure-based solvation mapping using WaterMap. Proteins Struct. Funct. Bioinform. 2013, 81, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; May-Dracka, T.L.; Gagnon, M.M.; Tommasi, R. Trends and Exceptions of Physical Properties on Antibacterial Activity for Gram-Positive and Gram-Negative Pathogens. J. Med. Chem. 2014, 57, 10144–10161. [Google Scholar] [CrossRef] [PubMed]
- Ziervogel, B.K.; Roux, B. The Binding of Antibiotics in OmpF Porin. Structure 2013, 21, 76–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, R.; Moser, H.E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Danelon, C.; Nestorovich, E.M.; Winterhalter, M.; Ceccarelli, M.; Bezrukov, S.M. Interaction of Zwitterionic Penicillins with the OmpF Channel Facilitates Their Translocation. Biophys. J. 2006, 90, 1617–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiu, F.; Yang, S.; Simmons, R.L.; Casarez, A.; Jones, A.K.; Li, C.; Jansen, J.M.; Moser, H.E.; Dean, C.R.; Reck, F.; et al. Size Matters and How You Measure It: A Gram-Negative Antibacterial Example Exceeding Typical Molecular Weight Limits. ACS Infect. Dis. 2019, 5, 1688–1692. [Google Scholar] [CrossRef]
- Levadny, V.; Aguilella, V.M.; Aguilella-Arzo, M.; Belaya, M. Interaction of a polar molecule with an ion channel. Phys. Rev. E 2004, 70, 041912. [Google Scholar] [CrossRef]
- Kumar, A.; Hajjar, E.; Ruggerone, P.; Ceccarelli, M. Structural and dynamical properties of the porins OmpF and OmpC: Insights from molecular simulations. J. Phys. Condens. Matter 2010, 22, 454125. [Google Scholar] [CrossRef]
- Scorciapino, M.A.; Acosta-Gutiérrez, S.; Benkerrou, D.; D’Agostino, T.; Malloci, G.; Samanta, S.; Bodrenko, I.; Ceccarelli, M. Rationalizing the permeation of polar antibiotics into Gram-negative bacteria. J. Phys. Condens. Matter 2017, 29, 113001. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Gutiérrez, S.; Scorciapino, M.A.; Bodrenko, I.V.; Ceccarelli, M. Filtering with Electric Field: The Case of E. coli Porins. J. Phys. Chem. Lett. 2015, 6, 1807–1812. [Google Scholar] [CrossRef]
- Low, A.S.; MacKenzie, F.M.; Gould, I.M.; Booth, I.R. Protected environments allow parallel evolution of a bacterial pathogen in a patient subjected to long-term antibiotic therapy. Mol. Microbiol. 2008, 42, 619–630. [Google Scholar] [CrossRef]
- Acosta-Gutiérrez, S.; Bodrenko, I.; Scorciapino, M.A.; Ceccarelli, M. Macroscopic electric field inside water-filled biological nanopores. Phys. Chem. Chem. Phys. 2016, 18, 8855–8864. [Google Scholar] [CrossRef]
- Bajaj, H.; Scorciapino, M.A.; Moynié, L.; Page, M.G.P.; Naismith, J.H.; Ceccarelli, M.; Winterhalter, M. Molecular Basis of Filtering Carbapenems by Porins from β-Lactam-resistant Clinical Strains of Escherichia coli. J. Biol. Chem. 2015, 291, 2837–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorciapino, M.A.; D’Agostino, T.; Acosta-Gutiérrez, S.; Malloci, G.; Bodrenko, I.; Ceccarelli, M. Exploiting the porin pathway for polar compound delivery into Gram-negative bacteria. Futur. Med. Chem. 2016, 8, 1047–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, H.; Acosta-Gutiérrez, S.; Bodrenko, I.; Malloci, G.; Scorciapino, M.A.; Winterhalter, M.; Ceccarelli, M. Bacterial Outer Membrane Porins as Electrostatic Nanosieves: Exploring Transport Rules of Small Polar Molecules. ACS Nano 2017, 11, 5465–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bafna, J.A.; Sans-Serramitjana, E.; Acosta-Gutiérrez, S.; Bodrenko, I.; Hörömpöli, D.; Berscheid, A.; Brötz-Oesterhelt, H.; Winterhalter, M.; Ceccarelli, M. Kanamycin uptake into E. coli is facilitated by OmpF and OmpC porin channels located in the outer membrane. ACS Infect. Dis. 2020, 6, 1855–1865. [Google Scholar] [CrossRef]
- Acosta-Gutiérrez, S.; Bodrenko, I.; Ceccarelli, M. permeability through bacterial porins dictates whole cell compound accumulation. arXiv 2020. [Google Scholar] [CrossRef]
- Cowan, S.; Schirmer, T.; Rummel, G.; Steiert, M.; Ghosh, R.; Pauptit, R.; Jansonius, J.; Rosenbusch, J. Crystal-structures explain functional-properties of 2 escherichia-coli porins. Nature 1992, 358, 727–733. [Google Scholar] [CrossRef]
- Baslé, A.; Rummel, G.; Storici, P.; Rosenbusch, J.P.; Schirmer, T. Crystal Structure of Osmoporin OmpC from E. coli at 2.0 Å. J. Mol. Biol. 2006, 362, 933–942. [Google Scholar] [CrossRef]
- Ghai, I.; Pira, A.; Scorciapino, M.A.; Bodrenko, I.V.; Benier, L.; Ceccarelli, M.; Winterhalter, M.; Wagner, R. General Method to Determine the Flux of Charged Molecules through Nanopores Applied to β-Lactamase Inhibitors and OmpF. J. Phys. Chem. Lett. 2017, 8, 1295–1301. [Google Scholar] [CrossRef]
- ChemAxon. MarvinSketch; ChemAxon: Budapest, Hungary, 2015. [Google Scholar]
- Bodrenko, I.V.; Salis, S.; Acosta-Gutiérrez, S.; Ceccarelli, M. Diffusion of large particles through small pores: From entropic to enthalpic transport. J. Chem. Phys. 2019, 150, 211102. [Google Scholar] [CrossRef]
- Coines, J.; Acosta-Gutiérrez, S.; Bodrenko, I.V.; Rovira, C.; Ceccarelli, M.; Lopez-Nieto, J.C. Glucose transport via the pseudomonad porin OprB: Implications for the design of Trojan Horse anti-infectives. Phys. Chem. Chem. Phys. 2019, 21, 8457–8463. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.; Chiu, S.-W.; Jakobsson, E. The Influence of Amino Acid Protonation States on Molecular Dynamics Simulations of the Bacterial Porin OmpF. Biophys. J. 2006, 90, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, M.J.; Giupponi, G.; De Fabritiis, G. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feenstra, A.; Hess, B.; Berendsen, H.J.C. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 1999, 20, 786–798. [Google Scholar] [CrossRef]
- Malloci, G.; Vargiu, A.V.; Serra, G.; Bosin, A.; Ruggerone, P.; Ceccarelli, M. A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds. Molecules 2015, 20, 13997–14021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Chemin. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [Green Version]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Raiteri, P.; Laio, A.; Gervasio, F.L.; Micheletti, C.; Parrinello, M. Efficient Reconstruction of Complex Free Energy Landscapes by Multiple Walkers Metadynamics. J. Phys. Chem. B 2006, 110, 3533–3539. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OmpF | Omp35 | OmpE35 | OmpK35 | OmpC | Omp36 | OmpE36 | OmpK36 | |
|---|---|---|---|---|---|---|---|---|
| 1.3 | 1.7 | 1.4 | 1.4 | 2.2 | 2.2 | 2.1 | 1.7 |
| Net Charge at Neutral pH (/e C) | Electric Dipole Moment (D) | Minimal Projection Area (Å2) | Radius of MPA (Å) | Van der Waals Volume (Å3) | |
|---|---|---|---|---|---|
| avibactam | −1 | 18 ± 1 | 43 ± 2 | 3.7 ± 0.1 | 199 |
| nacubactam | 0 (zwitterion) | 46 ± 4 | 46 ± 3 | 3.8 ± 0.1 | 257 |
| relebactam | 0 (zwitterion) | 44 ± 3 | 48 ± 3 | 3.9 ± 0.1 | 286 |
| zidebactam | 0 (zwitterion) | 47 ± 6 | 57 ± 6 | 4.2 ± 0.2 | 331 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pira, A.; Scorciapino, M.A.; Bodrenko, I.V.; Bosin, A.; Acosta-Gutiérrez, S.; Ceccarelli, M. Permeation of β-Lactamase Inhibitors through the General Porins of Gram-Negative Bacteria. Molecules 2020, 25, 5747. https://doi.org/10.3390/molecules25235747
Pira A, Scorciapino MA, Bodrenko IV, Bosin A, Acosta-Gutiérrez S, Ceccarelli M. Permeation of β-Lactamase Inhibitors through the General Porins of Gram-Negative Bacteria. Molecules. 2020; 25(23):5747. https://doi.org/10.3390/molecules25235747
Chicago/Turabian StylePira, Alessandro, Mariano Andrea Scorciapino, Igor V. Bodrenko, Andrea Bosin, Silvia Acosta-Gutiérrez, and Matteo Ceccarelli. 2020. "Permeation of β-Lactamase Inhibitors through the General Porins of Gram-Negative Bacteria" Molecules 25, no. 23: 5747. https://doi.org/10.3390/molecules25235747