
Cyclooxygenase Inhibition Safety and Efficacy in Inflammation-Based Psychiatric Disorders

 ,
,  , and
, and
Abstract
:
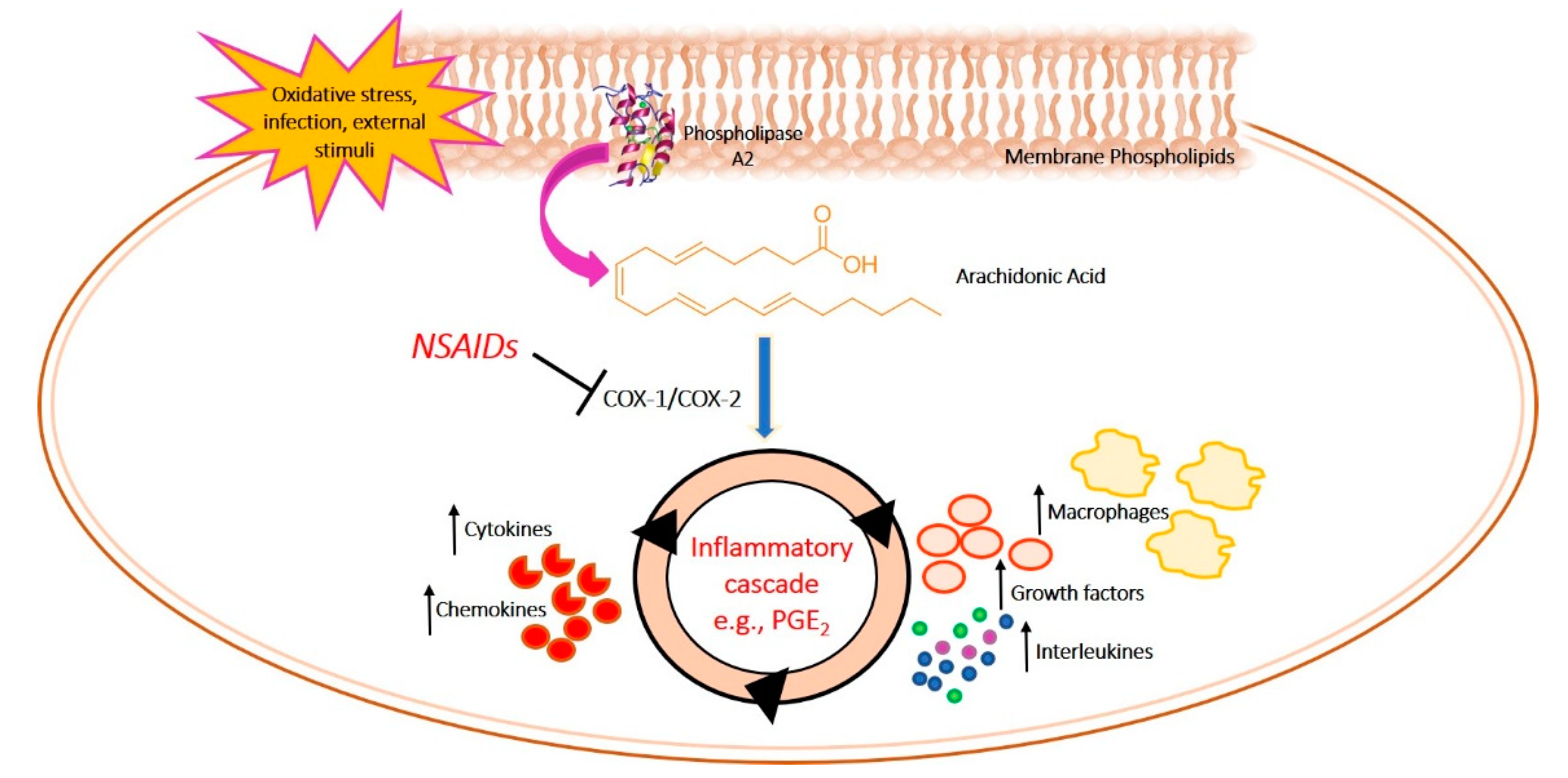
1. Introduction
2. Role of COX Inhibitors in Mental Disorders
- Aspirin, which at low-dose (≤100 mg daily) irreversibly inhibits platelet COX-1 with a COX-1 IC50 = 1.7 μM and >100 μM for COX-2 measured in a whole blood assay (HWBA) [43].
- Ibuprofen, which is a non-preferential COX inhibitor with an IC50 of 7.6 μM and 7.2 µM (HWBA) for COX-1 and COX-2, respectively [43].
- Naproxen a preferential COX-1 inhibitor, inhibits COX-1 with an IC50 = 9.3 μM and COX-2 with IC50 = 28 µM (HWBA) [43].
2.1. Major Depressive Disorder (MDD)
2.2. Schizophrenia
2.3. Bipolar Disorder (BD)
2.4. Autism Spectrum Disorder (ASD)
3. Conclusions
Funding
Conflicts of Interest
References
- American Psychiatric Association. Available online: https://www.psychiatry.org/patients-families/what-is-mental-illness (accessed on 7 September 2020).
- Our World in Data. Available online: https://ourworldindata.org/mental-health (accessed on 15 September 2020).
- Kessler, R.C.; Amminger, G.P.; Aguilar-Gaxiola, S.; Alonso, J.; Lee, S.; Üstün, T.B. Age of onset of mental disorders: A review of recent literature. Curr. Opin. Psychiatry 2007, 20, 359–364. [Google Scholar] [CrossRef]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 2012, 37, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Raison, L. Are anti-inflammatory therapies viable treatments for psychiatric disorders? Where the rubber meets the road. JAMA Psychiatry 2015, 72, 527–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, B.E. Inflammation and depression: A causal or coincidental link to the pathophysiology? Acta Neuropsychiatr. 2018, 30, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-Z.; Wang, Y.-X.; Jiang, C.-L. Inflammation: The common pathway of stress-related disease. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.E.; Teixeira, A.L. Inflammation in psychiatric disorders: What comes first? Ann. N. Y. Acad. Sci. 2018, 1437, 57–67. [Google Scholar] [CrossRef]
- Tao, R.; Li, H. High serumuric acid level in adolescent depressive patients. J. Affect. Disord. 2015, 174, 464–466. [Google Scholar] [CrossRef]
- Bartoli, F.; Crocamo, C.; Mazza, M.G.; Clerici, M.; Carrà, G. Uric acid levels in subjects with bipolar disorder: A comparative meta-analysis. J. Psychiatr. Res. 2016, 81, 133–139. [Google Scholar] [CrossRef]
- Oral, E.; Ozcan, H.; Kirkan, T.S.; Askin, S.; Gulec, M.; Aydin, N. Luteal serum BDNF and HSP70 levels in women with premenstrual dysphoric disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 685–693. [Google Scholar] [CrossRef]
- Fleshner, M.; Frank, M.; Maier, S.F. Danger signals and inflammasomes: Stress-evoked sterile inflammation in mood disorders. Neuropsychopharmacology 2017, 42, 36–45. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.G.; Vitale, P.; Ferorelli, S.; Boccarelli, A.; Coluccia, M.; Pannunzio, A.; Campanella, F.; Di Mauro, G.; Bonaccorso, C.; Fortuna, C.G.; et al. Effect of mofezolac-galactose distance in conjugates targeting cyclooxygenase (COX)-1 and CNS GLUT-1 carrier. Eur. J Med. Chem. 2017, 141, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Calvello, R.; Panaro, M.A.; Carbone, M.L.; Cianciulli, A.; Perrone, M.G.; Vitale, P.; Malerba, P.; Scilimati, A. Novel selective COX-1 inhibitors suppress neuroinflammatory mediators in LPS-stimulated N13 microglial cells. Pharmacol. Res. 2012, 65, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Calvello, R.; Lofrumento, D.D.; Perrone, M.G.; Cianciulli, A.; Salvatore, R.; Vitale, P.; De Nuccio, F.; Giannotti, L.; Nicolardi, G.; Panaro, M.A.; et al. Highly selective COX-1 inhibitors P6 and mofezolac counteract inflammatory state both in vitro and in vivo models of neuroinflammation. Front. Neurol. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjalkens, R.B.; Popichak, K.A.; Kirkley, K.A. Inflammatory activation of microglia and astrocytes in manganese neurotoxicity. Adv. Neurobiol. 2017, 18, 159–181. [Google Scholar]
- DiSabato, D.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry. 2011, 70, 663–671. [Google Scholar] [CrossRef]
- Meyer, U.; Feldon, J.; Yee, B.K. A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr. Bull. 2009, 35, 959–972. [Google Scholar] [CrossRef] [Green Version]
- Muller, N.; Myint, A.M.; Schwarz, M.J. Inflammation in schizophrenia. Adv. Protein Chem. Struct. Biol. 2012, 88, 49–68. [Google Scholar]
- Miller, B.J.; Culpepper, N.; Rapaport, M.H.; Buckley, P. Prenatal inflammation and neurodevelopment in schizophrenia: A review of human studies. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2013, 42, 92–100. [Google Scholar] [CrossRef]
- Potvin, S.; Stip, E.; Sepehry, A.A.; Gendron, A.; Bah, R.; Kouassi, E. Inflammatory cytokine alterations in schizophrenia: A systematic quantitative review. Biol. Psychiatry 2008, 63, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Drexhage, R.C.; Hoogenboezem, T.A.; Cohen, D.; Versnel, M.A.; Nolen, W.A.; van Beveren, N.J.M.; Drexhage, H.A. An activated set point of T-cell and monocyte inflammatory networks in recent-onset schizophrenia patients involves both pro- and anti-inflammatory forces. Int. J. Neuropsychopharmacol. 2011, 14, 746–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berk, M.; Kapczinski, F.; Andreazza, A.C.; Dean, O.M.; Giorlando, F.; Maes, M.; Yücel, M.; Gama, C.S.; Dodd, S.; Dean, B.; et al. Pathways underlying neuroprogression in bipolar disorder: Focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 2011, 35, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Drexhage, R.C.; Knijff, E.M.; Padmos, R.C.; van der Heul-Nieuwenhuijzen, L.; Beumer, W.; Versnel, M.A.; Drexhage, H.A. The mononuclear phagocyte system and its cytokine inflammatory networks in schizophrenia and bipolar disorder. Expert Rev. Neurother. 2010, 10, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.I.; Kemp, D.E.; Soczynska, J.K.; McIntyre, R.S. Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: A systematic review of the literature. J. Clin. Psychiatry 2009, 70, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Fond, G.; Hamdani, N.; Kapczinski, F.; Boukouaci, W.; Drancourt, N.; Dargel, A.; Oliveira, J.; Guen, E.L.; Marlinge, E.; Tamouza, R.; et al. Effectiveness and tolerance of anti-inflammatory drugs’ add-on therapy in major mental disorders: A systematic qualitative review. Acta Psychiatr. Scand. 2014, 129, 163–179. [Google Scholar] [CrossRef]
- Taams, L.S. Neuroimmune interactions: How the nervous and immune systems influence each other. Clin. Exp. Immunol. 2019, 197, 294–307. [Google Scholar] [CrossRef] [Green Version]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Hopkins, S.J. Central nervous system recognition of peripheral inflammation: A neural, hormonal collaboration. Acta Biomed 2007, 1, 231–247. [Google Scholar]
- Smith, W.L.; Urade, Y.; Jakobsson, P. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [Green Version]
- Ricciotti, E.; Garret, A.F. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Kam, P.C.A.; See, A.U.L. Cyclo-oxygenase isoenzymes: Physiological and pharmacological role. Anaesthesia 2000, 55, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.E.; Worley, P.F.; Pegg, J.; Bremer, M.; Isakson, P.C. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc. Natl. Acad. Sci. USA 1996, 93, 2317–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, W.E.; Andreasson, K.I.; Isakson, P.C.; Worley, P.F. Cyclooxygenases and the central nervous system. Prostaglandins 1997, 54, 601–624. [Google Scholar] [CrossRef]
- Maes, M. Targeting cyclooxygenase-2 in depression is not a viable therapeutic approach and may even aggravate the pathophysiology underpinning depression. Metab. Brain Dis. 2012, 27, 405–413. [Google Scholar] [CrossRef]
- Stefanovic, B.; Bosetti, F.; Silva, A.C. Modulatory role of cyclooxygenase-2 in cerebrovascular coupling. Neuroimage 2006, 32, 23–32. [Google Scholar] [CrossRef]
- Hewett, S.J.; Bell, S.C.; Hewett, J.A. Contributions of cyclooxygenase-2 to neuroplasticity and neuropathology of the central nervous system. Pharmacol. Ther. 2006, 112, 335–357. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, X.; Zhong, S.; Zhang, X.; Liu, C.; Liu, F.; Zhao, C. Peripheral and central nervous system immune response crosstalk in amyotrophic lateral sclerosis. Front. Neurosci. 2020, 14, 575. [Google Scholar] [CrossRef]
- Burian, M.; Geisslinger, G. COX-dependent mechanisms involved in the antinociceptive actionof NSAIDs at central and peripheral sites. Pharmacol. Ther. 2005, 107, 139–154. [Google Scholar] [CrossRef]
- Rostevanov, I.S.; Boyko, M.; Ferorelli, S.; Scilimati, A.; Perrone, M.G.; Kaplanski, J.; Zlotnik, A.; Azab, A.N. Inhibition of cyclooxygenase-1 does not reduce mortality in post-ischemic stroke rats. Neurosci. Lett. 2020, 737, 135296. [Google Scholar] [CrossRef]
- Rao, P.P.N.; Knaus, E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008, 11, 81s–110s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, M.G.; Scilimati, A.; Simone, L.; Vitale, P. Selective COX-1 inhibition: A therapeutic target to be reconsidered. Curr. Med. Chem. 2010, 17, 3769–3805. [Google Scholar] [CrossRef] [PubMed]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-steroidal anti-inflammatory drugs and brain inflammation: Effects on microglial functions. Pharmaceuticals 2010, 3, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Novakova, I.; Subileau, E.-A.; Toegel, S.; Gruber, D.; Lachmann, B.; Urban, E.; Chesne, C.; Noe, C.R.; Neuhaus, W. Transport rankings of non-steroidal antiinflammatory drugs across blood-brain barrier in vitro models. PLoS ONE 2014, 9, e86806. [Google Scholar] [CrossRef]
- Berthold-Losleben, M.; Heitmann, S.; Himmerich, H. Anti-inflammatory drugs in psychiatry. Inflamm. Allergy Drug Targets 2009, 8, 266–276. [Google Scholar] [CrossRef]
- Riolo, S.A.; Nguyen, T.A.; Greden, J.F.; King, C.A. Prevalence of depression by race/ethnicity: Findings from the National Health and Nutrition Examination Survey III. Am. J. Public Health 2005, 95, 998–1000. [Google Scholar] [CrossRef]
- Kessler, R.C.; McGonagle, K.A.; Zhao, S.; Nelson, C.B.; Hughes, M.; Eshleman, S.; Wittchen, H.U.; Kendler, K.S. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch. Gen. Psychiatry 1994, 51, 8–19. [Google Scholar] [CrossRef]
- Trivedi, M.H.; Rush, A.J.; Wisniewski, S.R.; Nierenberg, A.A.; Warden, D.; Ritz, L.; Norquist, G.; Howland, R.H.; Lebowitz, B.; McGrath, P.J.; et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: Implications for clinical practice. Am. J. Psychiatry 2006, 163, 28–40. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, W.; Hashimoto, K. Brain-derived neurotrophic factor (BDNF)-TrkB signaling in inflammation-related depression and potential therapeutic targets. Curr. Neuropharmacol. 2016, 14, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.S. The macrophage theory of depression. Med. Hypotheses 1991, 35, 298–306. [Google Scholar] [CrossRef]
- Feltes, P.K.; Doorduin, J.; Klein, H.C.; Juárez-Orozco, L.E.; Dierckx, R.A.; Moriguchi-Jeckel, C.M.; de Vries, E.F.J. Anti-inflammatory treatment for major depressive disorder: Implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J. Psychopharmacol. 2017, 31, 1149–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzer, R.; O’Connor, J.C.; Lawson, M.A.; Kelley, K.W. Inflammation-associated depression: From serotonin to kynurenine. Psychoneuroendocrinology 2011, 36, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, O.; Krogh, J.; Mors, O.; Benros, M.E. Inflammation in depression and the potential for anti-inflammatory treatment. Curr. Neuropharmacol. 2016, 14, 732–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Benros, M.E.; Waltoft, B.L.; Nordentoft, M.; Ostergaard, S.D.; Eaton, W.W.; Krogh, J.; Mortensen, P.B. Autoimmune diseases and severe infections as risk factors for mood disorders: A nationwide study. JAMA Psychiatry 2013, 70, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, R.; Capuron, L.; Irwin, M.R.; Miller, A.H.; Ollat, H.; Perry, V.H.; Rousey, S.; Yirmiya, R. Identification and treatment of symptoms associated with inflammation in medically ill patients. Psychoneuroendocrinology 2008, 33, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Pucak, M.L.; Kaplin, A.I. Unkind cytokines: Current evidence for the potential role of cytokines in immune-mediated depression. Int. Rev. Psychiatry 2005, 17, 477–483. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; Suy, E.; Vandervorst, C.; DeJonckheere, C.; Raus, J. Depression-related disturbances in mitogen-induced lymphocyte responses and interleukin-1 beta and soluble interleukin-2 receptor production. Acta Psychiatr. Scand. 1991, 84, 379–386. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Dahl, J.; Ormstad, H.; Aass, H.C.; Malt, U.F.; Bendz, L.T.; Sandvik, L.; Brundin, L.; Andreassen, O.A. The plasma levels of various cytokines are increased during ongoing depression and are reduced to normal levels after recovery. Psychoneuroendocrinology 2014, 45, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ho, R.C.; Mak, A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: A metaanalysis and meta-regression. J. Affect. Disord. 2012, 139, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Mikova, O.; Yakimova, R.; Bosmans, E.; Kenis, G.; Maes, M. Increased serum tumor necrosis factor alpha concentrations in major depression and multiple sclerosis. Eur. Neuropsychopharmacol. 2001, 11, 203–208. [Google Scholar] [CrossRef]
- Berk, M.; Wadee, A.A.; Kuschke, R.H.; O’Neill-Kerr, A. Acute phase proteins in major depression. J. Psychosom. Res. 1997, 43, 529–534. [Google Scholar] [CrossRef]
- Wium-Andersen, M.K.; Ørsted, D.D.; Nielsen, S.F.; Nordestgaard, B.G. Elevated C-reactive protein levels, psychological distress, and depression in 73, 131 individuals. JAMA Psychiatry 2013, 70, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Scharpé, S.; Van Grootel, L.; Uyttenbroeck, W.; Cooreman, W.; Cosyns, P.; Suy, E. Higher alpha 1-antitrypsin, haptoglobin, ceruloplasmin and lower retinol binding protein plasma levels during depression: Further evidence for the existence of an inflammatory response during that illness. J Affect. Disord. 1992, 24, 183–192. [Google Scholar] [CrossRef]
- Kim, Y.K.; Na, K.S.; Shin, K.H.; Jung, H.Y.; Choi, S.H.; Kim, J.B. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2007, 31, 1044–1053. [Google Scholar] [CrossRef]
- Krishnadas, R.; Cavanagh, J. Depression: An inflammatory illness? J. Neurol. Neurosurg. Psychiatry 2012, 83, 495–502. [Google Scholar] [CrossRef] [Green Version]
- El-Sisi, A.E.-S.; Sokkar, S.S.; El-Sayad, M.E.-S.; Ramadan, E.S.; Osman, E.Y. Celecoxib and omega-3 fatty acids alone and in combination with risperidone affect the behavior and brain biochemistry in amphetamine-induced model of schizophrenia. Biomed. Pharmacother. 2016, 82, 425–431. [Google Scholar] [CrossRef]
- Liu, H.; Luiten, P.G.M.; Eisel, U.L.M.; Dejongste, M.J.L.; Schoemaker, R.G. Depression after myocardial infarction: TNF-α-induced alterations of the blood-brain barrier and its putative therapeutic implications. Neurosci. Biobehav. Rev. 2013, 37, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Baune, B.T. Are non-steroidal anti-inflammatory drugs clinically suitable for the treatment of symptoms in depression-associated inflammation? Curr. Top. Behav. Neurosci. 2017, 31, 303–319. [Google Scholar] [PubMed]
- Ruhe, H.G.; Mason, N.S.; Schene, A.H. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: A meta-analysis of monoamine depletion studies. Mol. Psychiatry 2007, 12, 331–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satyanarayana, U.; Narasinga Rao, B.S. Dietary tryptophan level and the enzymes of tryptophan NAD pathway. Br. J. Nutr. 1980, 43, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlin, J.M.; Borden, E.C.; Sondel, P.M.; Byrne, G.I. Interferon-induced indoleamine 2,3-dioxygenase activity in human mononuclear phagocytes. J. Leukoc. Biol. 1989, 45, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.W.; Feng, G. Relationship between interferon-γ, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991, 5, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Gałecki, P.; Talarowska, M. Inflammatory theory of depression. Psychiatr. Pol. 2018, 52, 437–447. [Google Scholar] [CrossRef]
- Anderson, G. Editorial: The kynurenine and melatonergic pathways in psychiatric and CNS disorders. Curr. Pharm. Des. 2016, 22, 947–948. [Google Scholar] [CrossRef]
- Wirleitner, B.; Neurauter, G.; Schröcksnadel, K.; Frick, B.; Fuchs, D. Interferon-gamma-induced conversion of tryptophan: Immunologic and neuropsychiatric aspects. Curr. Med. Chem. 2003, 10, 1581–1591. [Google Scholar] [CrossRef]
- Myint, A.M.; Kim, Y.K. Cytokine-serotonin interaction through IDO: A neurodegeneration hypothesis of depression. Med. Hypotheses 2003, 61, 519–525. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Wang, L.; Brew, B.J. Quinolinic acid selectively induces apoptosis of human astocytes: Potential role in AIDS dementia complex. J. Neuroinflammation 2005, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005. [Google Scholar] [CrossRef] [PubMed]
- Spalletta, G.; Bossù, P.; Ciaramella, A.; Bria, P.; Caltagirone, C.; Robinson, R.G. The etiology of poststroke depression: A review of the literature and a new hypothesis involving inflammatory cytokines. Mol. Psychiatry 2006, 11, 984–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehrmann, E.; Guidetti, P.; Löve, A.; Williamson, J.; Bertram, E.H.; Schwarcz, R. Glial activation precedes seizures and hippocampal neurodegeneration in measles virus-infected mice. Epilepsia 2008, 49, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Schwarz, M.J. The immune-mediated alteration of serotonin and glutamate: Towards an integrated view of depression. Mol. Psychiatry 2007, 12, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.J.; Swiergiel, A.H.; De Beaurepaire, R. Cytokines as mediators of depression: What can we learn from animal studies? Neurosci. Biobehav. Rev. 2005, 29, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.-B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Malynn, S.; Campos-Torres, A.; Moynagh, P.; Haase, J. The pro-inflammatory cytokine TNF-α regulates the activity and expression of the serotonin transporter (SERT) in astrocytes. Neurochem. Res. 2013, 38, 694–704. [Google Scholar] [CrossRef]
- Cai, W.; Khaoustov, V.I.; Xie, Q.; Pan, T.; Le, W.; Yoffe, B. Interferon-α-induced modulation of glucocorticoid and serotonin receptors as a mechanism of depression. J. Hepatol. 2005, 42, 880–887. [Google Scholar] [CrossRef]
- Pariante, C.M. Depression, stress and the adrenal axis. J. Neuroendocrinol. 2003, 15, 811–812. [Google Scholar]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [Green Version]
- Raison, C.L.; Miller, A.H. When not enough is too much: The role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am. J. Psychiatry 2003, 160, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Schuld, A.; Schmid, D.A.; Haack, M.; Holsboer, F.; Friess, E.; Pollmächer, T. Hypothalamo-pituitary-adrenal function in patients with depressive disorders is correlated with baseline cytokine levels, but not with cytokine responses to hydrocortisone. J. Psychiatr. Res. 2003, 37, 463–470. [Google Scholar] [CrossRef]
- Anderson, G.; Berk, M.; Dean, O.; Moylan, S.; Maes, M. Role of immune-inflammatory and oxidative and nitrosative stress pathways in the etiology of depression: Therapeutic implications. CNS Drugs 2014, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Casolini, P.; Catalani, A.; Zuena, A.R.; Angelucci, L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J. Neurosci. Res. 2002, 68, 337–343. [Google Scholar] [CrossRef]
- Sandrini, M.; Vitale, G.; Pini, L.A. Effect of rofecoxib on nociception and the serotonin system in the rat brain. Inflamm. Res. 2002, 51, 154–159. [Google Scholar] [CrossRef]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; de Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef]
- Na, K.S.; Lee, K.J.; Lee, J.S.; Cho, Y.S.; Jung, H.Y. Efficacy of adjunctive celecoxib treatment for patients with major depressive disorder: A meta-analysis. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2014, 48, 79–85. [Google Scholar] [CrossRef]
- Sethi, R.; Gómez-Coronado, N.; Walker, A.J.; Robertson, O.D.; Agustini, B.; Berk, M.; Dodd, S. Neurobiology and therapeutic potential of cyclooxygenase-2 (COX-2) inhibitors for inflammation in neuropsychiatric disorders. Front. Psychiatry 2019, 10, 1–21. [Google Scholar] [CrossRef]
- Eyre, H.A.; Air, T.; Pradhan, A.; Johnston, J.; Lavretsky, H.; Stuart, M.J.; Baune, B.T. A meta-analysis of chemokines in major depression. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2016, 68, 1–8. [Google Scholar] [CrossRef]
- Yui, K.; Imataka, G.; Nakamura, H.; Ohara, N.; Naito, Y. Eicosanoids derived from arachidonic acid and their family prostaglandins and cyclooxygenase in psychiatric disorders. Curr. Neuropharmacol. 2015, 13, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendlewicz, J.; Kriwin, P.; Oswald, P.; Souery, D.; Alboni, S.; Brunello, N. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: A pilot open-label study. Int. Clin. Psychopharmacol. 2006, 21, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Müller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: Results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Akhondzadeh, S.; Jafari, S.; Raisi, F.; Nasehi, A.A.; Ghoreishi, A.; Salehi, B.; Mohebbi-Rasa, S.; Raznahan, M.; Kamalipour, A. Clinical trial of adjunctive celecoxib treatment in patients with major depression: A double blind and placebo controlled trial. Depress. Anxiety 2009, 26, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.H.; Hosseini, F.; Modabbernia, A.; Ashrafi, M.; Akhondzadeh, S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: Randomized double-blind placebo-controlled study. J. Affect. Disord. 2012, 141, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, R.L.; Gandhi, S.; Aneja, A.; Thorpe, K.; Razzouk, L.; Greenberg, J.; Mosovich, S.; Farkouh, M.E. NSAIDs are associated with lower depression scores in patients with osteoarthritis. Am. J. Med. 2013, 126, e11–e1017. [Google Scholar] [CrossRef]
- Majd, M.; Hashemian, F.; Hosseinib, S.M.; Shariatpanahi, M.V.; Sharifid, A. A randomized, double-blind, placebo-controlled trial of celecoxib augmentation of sertraline in treatment of drug-naive depressed women: A pilot study. Iran. J. Pharm. Res. 2015, 14, 891–899. [Google Scholar]
- Fields, C.; Drye, L.; Vaidya, V.; Lyketsos, C. Celecoxib or naproxen treatment does not benefit depressive symptoms in persons age 70 and older: Findings from a randomized controlled trial. Am. J. Geriatr. Psychiatry 2012, 20, 505–513. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Schizophrenia [Fact Sheet]. Available online: www.who.int/en/news-room/fact-sheets/detail/schizophrenia9April2018 (accessed on 23 September 2020).
- Schmidt, L.; Phelps, E.; Friedel, J.; Shokraneh, F. Acetylsalicylic acid (Aspirin) for schizophrenia. Cochrane Database Syst. Rev. 2019, 8, CD012116. [Google Scholar] [CrossRef]
- Mansur, R.B.; Zugman, A.; Asevedo, E.D.M.; Da Cunha, G.R.; Bressan, R.A.; Brietzke, E. Cytokines in schizophrenia: Possible role of anti-inflammatory medications in clinical and preclinical stages. Psychiatry Clin. Neurosci. 2012, 66, 247–260. [Google Scholar] [CrossRef]
- Tandon, R.; Gaebel, W.; Barch, D.M.; Bustillo, J.; Gur, R.E.; Heckers, S.; Malaspina, D.; Owen, M.J.; Schultz, S.; Tsuang, M.; et al. Definition and description of schizophrenia in the DSM-5. Schizophr. Res. 2013, 150, 3–10. [Google Scholar] [CrossRef]
- Simpson, E.H.; Kellendonk, C.; Kandel, E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron 2010, 65, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Levinson, D.F.; Duan, J.; Sanders, A.R.; Zheng, Y.; Péer, I.; Dudbridge, F.; Holmans, P.A.; Whittemore, A.S.; Mowry, B.J.; et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009, 460, 753–757. [Google Scholar] [CrossRef]
- Keller, W.R.; Kum, L.M.; Wehring, H.J.; Koola, M.M.; Buchanan, R.W.; Kelly, D.L. A review of anti-inflammatory agents for symptoms of schizophrenia. J. Psychopharmacol. 2013, 27, 337–342. [Google Scholar] [CrossRef] [Green Version]
- WHO. Burden of mental and behavioral disorders. In Mental Health: New Understanding, New Hope; World Health Organization: Geneva, Switzerland, 2001; pp. 19–45. [Google Scholar]
- Lieberman, J.A.; Buckley, P.F.; Perkins, D.O. Neuroprotection: A new strategy in the treatment of schizophrenia. CNS Spectr. 2007, 12, 4–6. [Google Scholar] [CrossRef]
- Ono, T.; Hashimoto, E.; Ukai, W.; Ishii, T.; Saito, T. The role of neural stem cells for in vitro models of schizophrenia: Neuroprotection via Akt/ERK signal regulation. Schizophr. Res. 2010, 122, 239–247. [Google Scholar] [CrossRef]
- Torrey, E.F.; Peterson, M.R. Slow and latent viruses in schizophrenia. Lancet 1973, 2, 22–24. [Google Scholar] [CrossRef]
- Sommer, I.E.; Van Westrhenen, R.; Begemann, M.J.H.; De Witte, L.D.; Leucht, S.; Kahn, R.S. Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: An update. Schizophr. Bull. 2014, 40, 181–191. [Google Scholar] [CrossRef]
- Fineberg, A.M.; Ellman, L.M. Inflammatory cytokines and neurological and neurocognitive alterations in the course of schizophrenia. Biol. Psychiatry 2013, 73, 951–966. [Google Scholar] [CrossRef] [Green Version]
- Müller, N. COX-2 inhibitors, aspirin, and other potential anti-inflammatory treatments for psychiatric disorders. Front. Psychiatry 2019, 375, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sekar, A.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Zubin, J.; Spring, B. Vulnerability: A new view of schizophrenia. J. Abnorm. Psychol. 1977, 6, 103–126. [Google Scholar] [CrossRef]
- Müller, N. What role does inflammation play in schizophrenia? Expert Rev. Neurother. 2016, 16, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Monji, A.; Kato, T.; Kanba, S. Cytokines and schizophrenia: Microglia hypothesis of schizophrenia. Psychiatry Clin. Neurosci. 2009, 63, 257–265. [Google Scholar] [CrossRef]
- Drexhage, R.C.; Weigelt, K.; van Beveren, N.; Cohen, D.; Versnel, M.A.; Nolen, W.A.; Drexhage, H.A. Immune and neuroimmune alterations in mood disorders and schizophrenia. Int. Rev. Neurobiol. 2011, 101, 169–201. [Google Scholar]
- Chew, L.J.; Fusar-Poli, P.; Schmitz, T. Oligodendroglial alterations and the role of microglia in white matter injury: Relevance to schizophrenia. Int. J. Dev. Neurosci. 2013, 27, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Schwarz, S.M. Schizophrenia as an inflammation-mediated dysbalance of glutamatergic neurotransmission. Neurotox. Res. 2006, 10, 131–148. [Google Scholar] [CrossRef]
- Muller, N.; Dursun, S.M. Schizophrenia genes, epigenetics and psychoneuroimmunology therapeutics: All make sense now? J. Psychopharmacol. 2011, 25, 713–714. [Google Scholar] [CrossRef]
- Chiang, S.S.W.; Riedel, M.; Schwarz, M.; Mueller, N. Is T-helper type 2 shift schizophrenia-specific? Primary results from a comparison of related psychiatric disorders and healthy controls. Psychiatry Clin. Neurosci. 2013, 67, 228–236. [Google Scholar] [CrossRef]
- Spellberg, B.; Edwards, J.E. Type 1/type 2 immunity in infectious diseases. Clin. Infect. Dis. 2001, 32, 76–102. [Google Scholar] [CrossRef]
- Müller, N.; Schwarz, M.J. Immune system and schizophrenia. Curr. Immunol. Rev. 2010, 6, 213–220. [Google Scholar] [CrossRef]
- Wilke, I.; Arolt, V.; Rothermundt, M.; Weitzsch, C.; Hornberg, M.; Kirchner, H. Investigations of cytokine production in whole blood cultures of paranoid and residual schizophrenic patients. Eur. Arch. Psychiatry Clin. Neurosci. 1996, 246, 279–284. [Google Scholar] [CrossRef]
- Müller, N.; Riedel, M.; Ackenheil, M.; Schwarz, M.J. Cellular and humoral immune system in schizophrenia: A conceptual re-evaluation. World J. Biol. Psychiatry 2000, 1, 173–179. [Google Scholar] [CrossRef]
- Petitto, J.M.; McCarthy, D.B.; Rinker, C.M.; Huang, Z.; Getty, T. Modulation of behavioral and neurochemical measures of forebrain dopamine function in mice by species-specific interleukin-2. J. Neuroimmunol. 1997, 73, 183–190. [Google Scholar] [CrossRef]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Attari, A.; Mojdeh, A.; Soltani, F.A.S.K.; Najarzadegan, M.R. Aspirin inclusion in antipsychotic treatment on severity of symptoms in schizophrenia: A randimized clinical trial. Iran. J. Psychiatry Behav. Sci. 2017, 11, e5848. [Google Scholar]
- Müller, N.; Schwarz, M.J. The immunological basis of glutamatergic disturbance in schizophrenia: Towards an integrated view. J. Neural Transm. 2007, 72, 269–280. [Google Scholar]
- Genius, J.; Geiger, J.; Dölzer, A.L.; Benninghoff, J.; Giegling, I.; Hartmann, A.M.; Möller, H.J.; Rujescu, D. Glutamatergic dysbalance and oxidative stress in in vivo and in vitro models of psychosis based on chronic NMDA receptor antagonism. PLoS ONE 2013, 8, e59395. [Google Scholar] [CrossRef] [Green Version]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Weidinger, E.; Leitner, B.; Schwarz, M.J. The role of inflammation in schizophrenia. Front. Neurosci. 2015, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jarskog, L.F.; Xiao, H.; Wilkie, M.B.; Lauder, J.M.; Gilmore, J.H. Cytokine regulation of embryonic rat dopamine and serotonin neuronal survival in vitro. Int. J. Dev. Neurosci. 1997, 15, 711–716. [Google Scholar] [CrossRef]
- Kabiersch, A.; Del Rey, H.F.A.; Besedovsky, H.O. Administration of interleukin-1 at birth affects dopaminergic neurons in adult mice. Ann. N. Y. Acad. Sci. 1998, 840, 123–127. [Google Scholar] [CrossRef]
- Ling, Z.D.; Potter, E.D.; Lipton, J.W.; Carvey, P.M. Differentiation of mesencephalic progenitor cells into dopaminergic neurons by cytokines. Exp. Neurol. 1998, 149, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Potter, E.D.; Ling, Z.D.; Carvey, P.M. Cytokine-induced conversion of mesencephalic-derived progenitor cells into dopamine neurons. Cell Tissue Res. 1999, 296, 235–246. [Google Scholar] [CrossRef]
- Holden, J.M.; Meyers-Manor, J.E.; Overmier, J.B.; Gahtan, E.; Sweeney, W.; Miller, H. Lipopolysaccharide-induced immune activation impairs attention but has little effect on short-term working memory. Behav. Brain Res. 2008, 194, 138–145. [Google Scholar] [CrossRef]
- Möller, H.J. Clinical evaluation of negative symptoms in schizophrenia. Eur. Psychiatry 2007, 22, 380–386. [Google Scholar] [CrossRef]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr. Res. 2009, 110, 1–3. [Google Scholar] [CrossRef]
- Muller, N.; Myint, A.-M.; Schwarz, M.J. Immunological treatment options for schizophrenia. Curr. Pharm. Biotechnol. 2012, 82, 210–219. [Google Scholar] [CrossRef]
- Schmidt, L.; Ceglarek, U.; Kortz, L.; Hoop, M.; Kirkby, K.; Thiery, J.; Himmerich, H. Mechanisms of involvement of eicosanoids and their precursors in the pathophysiology and treatment of schizophrenia. Med. Chem. 2013, 9, 763–773. [Google Scholar] [CrossRef]
- Müller, N.; Riedel, M.; Scheppach, C.; Brandstätter, B.; Sokullu, S.; Krampe, K.; Ulmschneider, M.; Engel, R.R.; Möller, H.J.; Schwarz, M.J.; et al. Beneficial antipsychotic effects of celecoxib add-on therapy compared to risperidone alone in schizophrenia. Am. J. Psychiatry 2002, 159, 1029–1034. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Tabatabaee, M.; Amini, H.; Ahmadi Abhari, S.A.; Abbasi, S.H.; Behnam, B. Celecoxib as adjunctive therapy in schizophrenia: A double-blind, randomized and placebo-controlled trial. Schizophr. Res. 2007, 90, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Krause, D.; Dehning, S.; Musil, R.; Schennach-Wolff, R.; Obermeier, M.; Möller, H.J.; Klauss, V.; Schwarz, M.J.; Riedel, M. Celecoxib treatment in an early stage of schizophrenia: Results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr. Res. 2010, 121, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Cai, D.-B.; Yang, X.H.; Ungvari, G.S.; Ng, C.H.; Müller, N.; Ning, Y.P.; Xiang, Y.T. Adjunctive celecoxib for schizophrenia: A meta-analysis of randomized, double-blind, placebo-controlled trials. J. Psychiatr. Res. 2017, 92, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Laan, W.; Grobbee, D.E.; Selten, J.P.; Heijnen, C.J.; Kahn, R.S.; Burger, H. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: Results from a randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 2010, 71, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institute of Mental Health. Available online: https://www.nimh.nih.gov/health/topics/bipolar-disorder/index.shtml (accessed on 8 September 2020).
- Kupfer, D.J. The increasing medical burden in bipolar disorder. JAMA 2005, 293, 2528–2530. [Google Scholar] [CrossRef] [PubMed]
- Fagiolini, A.; Forgione, R.; Maccari, M.; Cuomo, A.; Morana, B.; Dell’Osso, M.C.; Pellegrini, F.; Rossi, A. Prevalence, chronicity, burden and borders of bipolar disorder. J. Affect. Disord. 2013, 148, 161–169. [Google Scholar] [CrossRef]
- Rosenblat, J.D.; Kakar, R.; Berk, M.; Kessing, L.V.; Vinberg, M.; Baune, B.T.; Mansur, R.B.; Brietzke, E.; Goldstein, B.I.; Mcintyre, R.S. Anti-inflammatory agents in the treatment of bipolar depression: A systematic review and meta-analysis. Bipolar Disord. 2016, 18, 89–101. [Google Scholar] [CrossRef]
- Gershon, S.; Soares, J.C. Current therapeutic profile of lithium. Arch. Gen. Psychiatry 1997, 54, 16–20. [Google Scholar] [CrossRef]
- Soares, J.C. Recent advances in the treatment of bipolar mania, depression, mixed states, and rapid cycling. Int. Clin. Psychopharmacol. 2000, 15, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.G.; Monkul, E.S.; Hatch, J.P.; Fonseca, M.; Zunta-Soares, G.B.; Frey, B.N.; Bowden, C.L.; Soares, J.C. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: A double-blind, randomized, placebo-controlled study. Hum. Psychopharmacol. 2008, 23, 87–94. [Google Scholar] [CrossRef]
- Arabzadeh, S.; Ameli, N.; Zeinoddini, A.; Rezaei, F.; Farokhnia, M.; Mohammadinejad, P.; Ghaleiha, A.; Akhondzadeh, S. Celecoxib adjunctive therapy for acute bipolar mania: A randomized, double-blind, placebo-controlled trial. Bipolar Disord. 2015, 17, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, I.G.; Bauer, M.E.; MacHado-Vieira, R.; Teixeira, A.L. Cytokines in bipolar disorder: Paving the way for neuroprogression. Neural Plast. 2014, 2014, 360841. [Google Scholar] [CrossRef] [PubMed]
- Munkholm, K.; Braüner, J.V.; Kessing, L.V.; Vinberg, M. Cytokines in bipolar disorder vs. healthy control subjects: A systematic review and meta-analysis. J. Psychiatr. Res. 2013, 47, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Modabbernia, A.; Taslimi, S.; Brietzke, E.; Ashrafi, M. Cytokine alterations in bipolar disorder: A meta-analysis of 30 studies. Biol. Psychiatry 2013, 74, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Leza, J.C.; Bueno, B.; Bioque, M.; Arango, C.; Parellada, M.; Do, K.; O’Donnell, P.; Bernardo, M. Inflammation in schizophrenia: A question of balance. Neurosci. Biobehav. Rev. 2015, 55, 612–626. [Google Scholar] [CrossRef]
- Barbosa, I.G.; Machado-Vieira, R.; Soares, J.C.; Teixeira, A.L. The immunology of bipolar disorder. Neuroimmunomodulation 2014, 21, 117–122. [Google Scholar] [CrossRef]
- Barbosa, I.G.; Rocha, N.P.; Bauer, M.E.; De Miranda, A.S.; Huguet, R.B.; Reis, H.J.; Zunszain, P.A.; Horowitz, M.A.; Pariante, C.M.; Teixeira, A.L. Chemokines in bipolar disorder: Trait or state? Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 159–165. [Google Scholar] [CrossRef]
- Munkholm, K.; Vinberg, M.; Vedel Kessing, L. Cytokines in bipolar disorder: A systematic review and meta-analysis. J. Affect. Disord. 2013, 144, 16–27. [Google Scholar] [CrossRef]
- Munkholm, K.; Weikop, P.; Kessing, L.V.; Vinberg, M. Elevated levels of IL-6 and IL-18 in manic and hypomanic states in rapid cycling bipolar disorder patients. Brain Behav. Immun. 2015, 43, 205–213. [Google Scholar] [CrossRef]
- Brietzke, E.; Kauer-Sant’Anna, M.; Teixeira, A.L.; Kapczinski, F. Abnormalities in serum chemokine levels in euthymic patients with bipolar disorder. Brain Behav. Immun. 2009, 23, 1079–1082. [Google Scholar] [CrossRef]
- Duncan, R.E.; Bazinet, R.P. Brain arachidonic acid uptake and turnover: Implications for signaling and bipolar disorder. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 130–138. [Google Scholar] [CrossRef]
- Rao, J.S.; Rapoport, S.I. Mood-stabilizers target the brain arachidonic acid cascade. Curr. Mol. Pharm. 2009, 2, 207–214. [Google Scholar] [CrossRef]
- Bazinet, R.P. Is the brain arachidonic acid cascade a common target of drugs used to manage bipolar disorder? Biochem. Soc. Trans. 2009, 37, 1104–1109. [Google Scholar] [CrossRef]
- Rapoport, S.I.; Basselin, M.; Kim, H.W.; Rao, J.S. Bipolar disorder and mechanisms of action of mood stabilizers. Brain Res. Rev. 2009, 61, 185–209. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, S.I. Brain arachidonic and docosahexaenoic acid cascades are selectively altered by drugs, diet and disease. Prostaglandins Leukot. Essent. Fat. Acids 2008, 79, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.S.; Lee, H.J.; Rapoport, S.I.; Bazinet, R.P. Mode of action of mood stabilizers: Is the arachidonic acid cascade a common target? Mol. Psychiatry 2008, 13, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Van Strater, A.C.P.; Bouvy, P.F. Omega-3 fatty acids in the treatment of affective disorders: An overview of the literature. Tijdschr. Psychiatr. 2007, 49, 85–94. [Google Scholar]
- Quiroz, J.A.; Gould, T.D.; Manji, H.K. Molecular effects of lithium. Mol. Interv. 2004, 4, 259–272. [Google Scholar] [CrossRef]
- Diagnostic and Statistical Manual of Mental Disorders: DSM-5; American Psychiatric Association: Arlington, VA, USA, 2013.
- National Institute of Mental Health. Available online: https://www.nimh.nih.gov/health/topics/autism-spectrum-disorders/index.shtml (accessed on 20 August 2020).
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The genetics of autism. Pediatrics 2004, 113, e472–486. [Google Scholar] [CrossRef] [Green Version]
- Goines, P.; Van De Water, J. The immune system’s role in the biology of autism. Curr. Opin. Neurol. 2010, 23, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Chiba, K. Involvement of neuroinflammation during brain development in social cognitive deficits in autism spectrum disorder and schizophrenia. J. Pharmacol. Exp. Ther. 2016, 358, 504. [Google Scholar] [CrossRef] [Green Version]
- Onore, C.; Careaga, M.; Ashwood, P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 2012, 26, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Petrelli, F.; Pucci, L.; Bezzi, P. Astrocytes and microglia and their potential link with autism spectrum disorders. Front. Cell. Neurosci. 2016, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.S. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev. Neurobiol. 2012, 72, 1272–1276. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.Y.; Wang, S.; Huang, N.; Yeh, H.H.; Chen, C.Y. Prenatal infection and autism spectrum disorders in childhood: A population-based case-control study in Taiwan. Paediatr. Perinat. Epidemiol. 2015, 29, 307–316. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Xu, L.-L.; Shao, L.; Xia, R.-M.; Yu, Z.-H.; Ling, Z.-X.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef]
- Ziats, M.N.; Rennert, O.M. Expression profiling of autism candidate genes during human brain development implicates central immune signaling pathways. PLoS ONE 2011, 6, e24691. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.K.; Essa, M.M.; de Paula Martins, R.; Lovejoy, D.B.; Bilgin, A.A.; Waly, M.I.; Al-Farsi, Y.M.; Al-Sharbati, M.; Al-Shaffae, M.A.; Guillemin, G.J. Altered kynurenine pathway metabolism in autism: Implication for immune-induced glutamatergic activity. Autism Res. 2016, 9, 621–631. [Google Scholar] [CrossRef]
- Croonenberghs, J.; Bosmans, E.; Deboutte, D.; Kenis, G.; Maes, M. Activation of the inflammatory response system in autism. Neuropsychobiology 2002, 45, 1–6. [Google Scholar] [CrossRef]
- Brigida, A.L.; Schultz, S.; Cascone, M.; Antonucci, N.; Siniscalco, D. Endocannabinod signal dysregulation in autism spectrum disorders: A correlation link between inflammatory state and Neuro-Immune alterations. Int. J. Mol. Sci. 2017, 18, 1425. [Google Scholar] [CrossRef] [Green Version]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Heuer, L.; Ashwood, P.; Schauer, J.; Goines, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Croen, L.A.; Pessah, I.N.; Van De Water, J. Reduced levels of immunoglobulin in children with autism correlateswith behavioral symptoms. Autism Res. 2008, 1, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Wills, S.; Cabanlit, M.; Bennett, J.; Ashwood, P.; Amaral, D.; Van De Water, J. Autoantibodies in autism spectrum disorders (ASD). Ann. N. Y. Acad. Sci. 2007, 1107, 79–91. [Google Scholar] [CrossRef]
- Grigorenko, E.L.; Han, S.S.; Yrigollen, C.M.; Leng, L.; Mizue, Y.; Anderson, G.M.; Mulder, E.J.; De Bildt, A.; Minderaa, R.B.; Volkmar, F.R.; et al. Macrophage migration inhibitory factor and autism spectrum disorders. Pediatrics 2008, 122, e438–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.Z.; Wu, H.B.; Xu, S.Q.; Guo, X.R.; Yang, J.; Shen, X.F. Macrophage migration inhibitory factor activates cyclooxygenase 2-prostaglandin E2 in cultured spinal microglia. Neurosci. Res. 2011, 71, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.J. Research on psychiatric disorders targets inflammation. JAMA 2014, 312, 474–476. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Careaga, M.; Schwartzer, J.; Ashwood, P. Inflammatory profiles in the BTBR mouse: HOW relevant are they to autism spectrum disorders? Brain Behav. Immun. 2015, 43, 11–16. [Google Scholar] [CrossRef] [Green Version]
- El-Ansary, A.; Al-Ayadhi, L. Neuroinflammation in autism spectrum disorders. J. Neuroinflammation 2012, 9, 265. [Google Scholar] [CrossRef] [Green Version]
- Masi, A.; Quintana, D.S.; Glozier, N.; Lloyd, A.R.; Hickie, I.B.; Guastella, A.J. Cytokine aberrations in autism spectrum disorder: A systematic review and meta-analysis. Mol. Psychiatry 2015, 20, 440–446. [Google Scholar] [CrossRef]
- Boris, M.; Kaiser, C.C.; Goldblatt, A.; Elice, M.W.; Edelson, S.M.; Adams, J.B.; Feinstein, D.L. Effect of pioglitazone treatment on behavioral symptoms in autistic children. J. Neuroinflammation 2007, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.J.; Kwon, K.J.; Park, J.Y.; Lee, S.H.; Moon, C.H.; Baik, E.J. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: Associated with iNOS and COX-2. Brain Res. 2002, 941, 1–2. [Google Scholar] [CrossRef]
- Sepanjnia, K.; Modabbernia, A.; Ashrafi, M.; Modabbernia, M.J.; Akhondzadeh, S. Pioglitazone adjunctive therapy for moderate-to-severe major depressive disorder: Randomized double-blind placebo-controlled trial. Neuropsychopharmacology 2012, 37, 2093–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asadabadi, M.; Mohammadi, M.R.; Ghanizadeh, A.; Modabbernia, A.; Ashrafi, M.; Hassanzadeh, E.; Forghani, S.; Akhondzadeh, S. Celecoxib as adjunctive treatment to risperidone in children with autistic disorder: A randomized, double-blind, placebo-controlled trial. Psychopharmacology 2013, 225, 51–59. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial | Patients | Therapy | Effects |
|---|---|---|---|
| Mendlewicz et al. | 24 | Fluoxetine + aspirin | Aspirin accelerates fluoxetine effect |
| Muller et al. | 40 | Reboxetine + celecoxib | Depressive symptom improvements |
| Akhondzadeh et al. | 40 | Fluoxetine + celecoxib | Depressive symptom improvements |
| Abbasi et al. | 40 | Sertraline + celecoxib | Reduction of IL-6 levels |
| Iyengar et al. | 1497 | Ibuprofen/naproxen/celecoxib * | Favor of NSAIDs |
| Majd et al. | 30 | Sertraline + celecoxib | Acceleration of sertraline therapeutic action onset/increase response and remission rate in depressive disorders |
| Fields et al. | 2528 | Celecoxib/naproxen * | Unfavorable results |
| Trial | Patients | Therapy | Effects |
|---|---|---|---|
| Muller et al. | 50 | Risperidone + celecoxib | Beneficial effects on schizophrenia symptoms |
| Rapaport et al. | 38 | Olanzapine/risperidone + celecoxib | No clinical symptoms improvement |
| Akhondzadeh et al. | 60 | Risperidone + celecoxib | Positive symptoms improvement |
| Muller et al. | 49 | Amisulpride + celecoxib | Superior therapeutic effect |
| Zheng et al. | 40 | Risperidone + celecoxib | Superior therapeutic effect |
| Laan et al. | 70 | Aspirin/placebo | Schizophrenia symptoms reduction |
| Trial | Patients | Therapy | Effects |
|---|---|---|---|
| Nery et al. | 28 | Antipsychotics + celecoxib | Rapid-onset antidepressant effect |
| Kargar et al. | 48 | ECT + celecoxib | Reduction of TNF-α levels |
| Arabzadeh et al. | 46 | Sodium valproate + celecoxib | Positive adjuvant effect |
| Psychiatric Disorders | Title (NCT) | Treatments/Participants | Phase | Starting Date/Recruitment Status |
|---|---|---|---|---|
| Depression | Biomarkers of Neuroinflammation and Anti-Inflammatory Treatments in Major Depressive Disorder (NCT02362529) | Minocycline + celecoxib/115 | I | 2015/Completed |
| Late-Life Stress and Inflammation (S&I) (NCT02389465) | Escitalopram + celecoxib/150 | IV | 2014/Suspended (Study halted due to COVID-19 but it potentially will be resumed) | |
| Salicylic Augmentation in Depression (SAD) (NCT03152409) | Aspirin/74 | II | 2018/Recruiting | |
| Aspirin in Reducing Events in the Elderly (ASPREE) (NCT01038583) | Enteric-coated aspirin/19,114 | Not Reported | 2010/Active, not recruiting | |
| Schizophrenia | A Double-blind and Randomized Trial of Celecoxib Added to Risperidone in Treatment-naive First-episode Schizophrenia(NCT00686140) | Celecoxib/200 | NA | 2006/Completed a |
| Efficacy and Safety of celecoxib as Add-on Therapy to Risperidone Versus Risperidone Alone in Patients With Schizophrenia (NCT00639483) | Celecoxib/270 | II | 2003/Completed a | |
| Anti-inflammatories and Adolescent Schizophrenia (NCT04020588) | Celecoxib + minocycline/90 | IV | 2019/Recruiting | |
| Aspirin in Young Psychotic Patients (NCT02685748) | Aspirin + pantoprazole/60 | III | 2017/Completed a | |
| Randomized Controlled Trial of Aspirin vs. Placebo in the Treatment of Pre-psychosis (NCT02047539) | Aspirin/40 | Early I | 2015/Recruiting | |
| Bipolar disorder | Adjunctive Use of celecoxib in the Treatment of Bipolar Postpartum Depression (NCT02726659) | Celecoxib/56 | III | Recruiting |
| Minocycline and celecoxib as Adjunctive Treatments of Bipolar Depression (NCT02703363) | Celecoxib + minocycline/265 | III | Completed a | |
| Bipolar Depression and Inflammation (NCT01479829) | Escitalopram + celecoxib/88 | IV | Completed a | |
| Minocycline and Aspirin in the Treatment of Bipolar Depression (Minocycline) (NCT01429272) | Minocycline + aspirin/99 | III | Completed a | |
| N-Acetyl Cysteine and Aspirin as an Adjunctive Treatment for Bipolar Disorder (SMRI-Bipolar) (NCT01797575) | Aspirin + N-acetyl-cysteine/38 | II | Completed a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrone, M.G.; Centonze, A.; Miciaccia, M.; Ferorelli, S.; Scilimati, A. Cyclooxygenase Inhibition Safety and Efficacy in Inflammation-Based Psychiatric Disorders. Molecules 2020, 25, 5388. https://doi.org/10.3390/molecules25225388
Perrone MG, Centonze A, Miciaccia M, Ferorelli S, Scilimati A. Cyclooxygenase Inhibition Safety and Efficacy in Inflammation-Based Psychiatric Disorders. Molecules. 2020; 25(22):5388. https://doi.org/10.3390/molecules25225388
Chicago/Turabian StylePerrone, Maria Grazia, Antonella Centonze, Morena Miciaccia, Savina Ferorelli, and Antonio Scilimati. 2020. "Cyclooxygenase Inhibition Safety and Efficacy in Inflammation-Based Psychiatric Disorders" Molecules 25, no. 22: 5388. https://doi.org/10.3390/molecules25225388