
Lysosomotropic Features and Autophagy Modulators among Medical Drugs: Evaluation of Their Role in Pathologies



Abstract
:1. Introduction
2. Low-Molecular-Weight Weak Bases in Experimental and Clinical Medicine
2.1. Chloroquine
2.2. Antitumor Drugs with Lysosomotropism
2.3. Autophagy/Lipophagy and Proteases
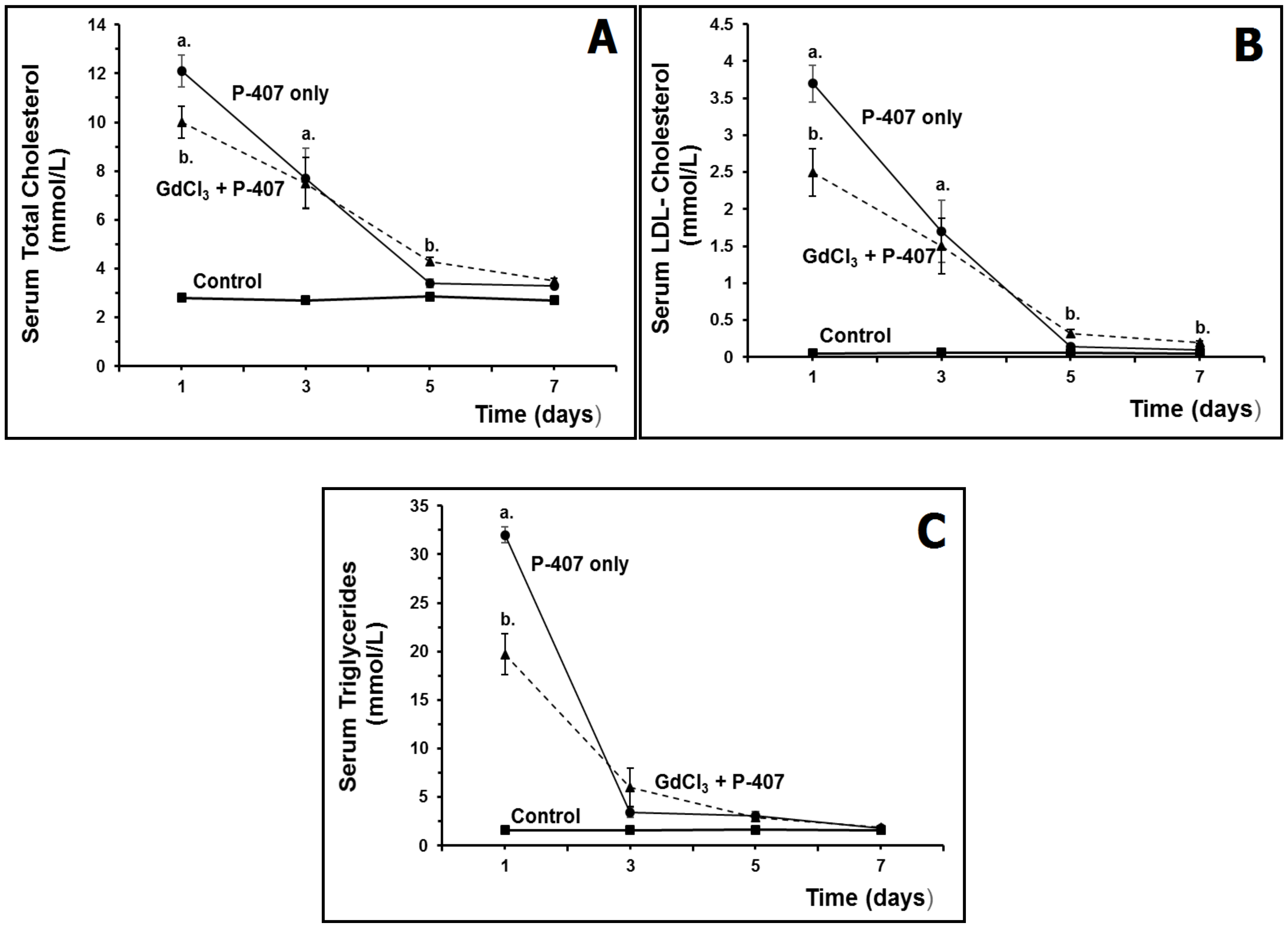
2.4. Lysosomotropic Gadolinium Compounds and Suppression of Liver Macrophages
Effect of Macrophage Suppression on Serum Lipids in Hyperlipidemic Mice
3. High-Molecular-Weight Lysosomotropic Agents
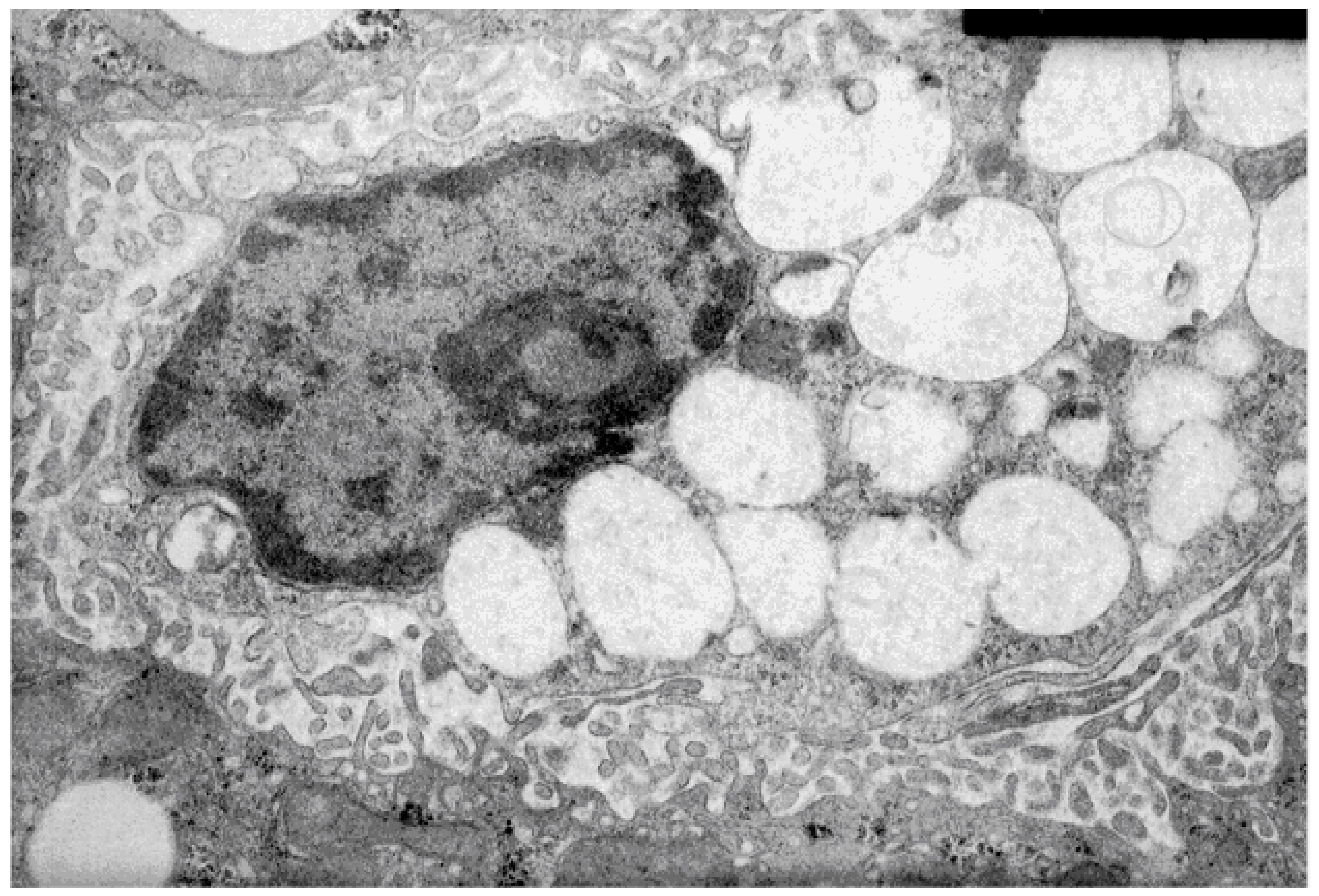
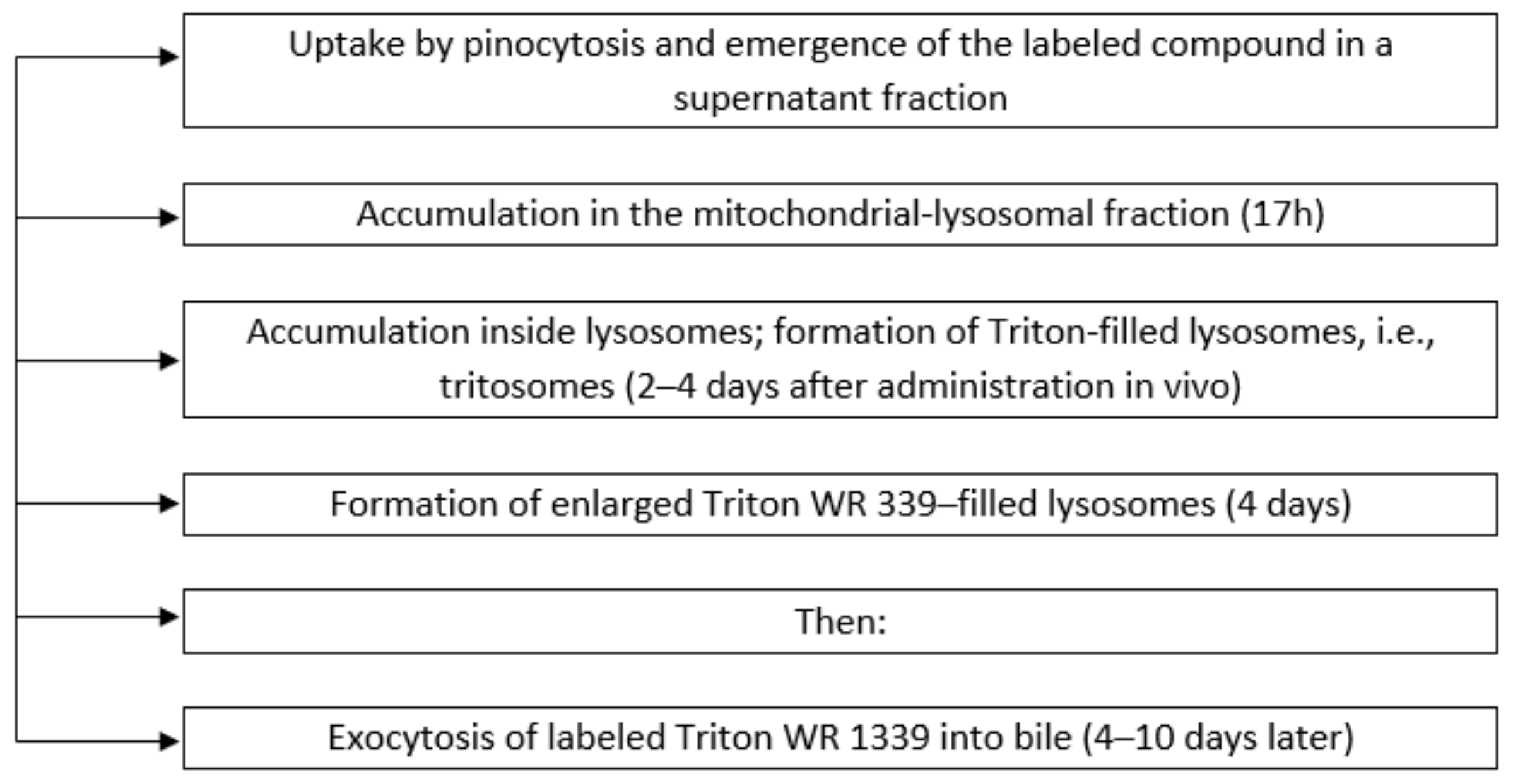
3.1. Characteristics of the Membranes of Liver Lysosomes of Animals with Intralysosomal Storage Syndrome Induced by Triton WR 1339
3.2. Characteristics of the Hyperlipidemia Induced by Triton WR 1339
3.3. Autophagy Induction
3.4. Autophagy Regulation
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- de Duve, C.; de Barsy, T.; Poole, B.; Trouet, A.; Tulkens, P.; Van Hoof, F. Commentary. Lysosomotropic agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar] [CrossRef]
- Pisonero-Vaquero, S.; Medina, D.L. Lysosomotropic drugs: Pharmacological tools to study lysosomal function. Curr. Drug Metab. 2017, 18, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Pourahmad, J.; Hosseini, M.J.; Eskandari, M.R.; Rahmani, F. Involvement of four different intracellular sites in chloroacetaldehyde- induced oxidative stress cytotoxicity. Iran. J. Pharm. Res. 2012, 11, 265–276. [Google Scholar] [PubMed]
- Kuzu, O.F.; Toprak, M.; Noory, M.A.; Robertson, G.P. Effect of lysosomotropic molecules on cellular homeostasis. Pharmacol. Res. 2017, 117, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Korolenko, T.A.; Busch, U. A review of drug-induced lysosomal disorders of the liver in man and laboratory animals. Microsc. Res. Tech. 1997, 36, 253–275. [Google Scholar] [CrossRef]
- Carriere, F.; Longhi, S.; Record, M. The endosomal lipid bis(monoacylglycero) phosphate as a potential key player in the mechanism of action of chloroquine against SARS-COV-2 and other enveloped viruses hijacking the endocytic pathway. Biochimie 2020. [Google Scholar] [CrossRef] [PubMed]
- Blaess, M.; Kaiser, L.; Sauer, M.; Csuk, R.; Deigner, H.P. COVID-19/SARS-CoV-2 Infection: Lysosomes and Lysosomotropism Implicate New Treatment Strategies and Personal Risks. Int. J. Mol. Sci. 2020, 21, 4953. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U.; Tuck, A.; Norgren, K.; Munic Kos, V. Existing highly accumulating lysosomotropic drugs with potential for repurposing to target COVID-19. Biomed. Pharmacother. 2020, 130, 110582. [Google Scholar] [CrossRef]
- Plattner, H.; Henning, R.; Brauser, B. Formation of triton WR 1339-filled rat liver lysosomes. II. Involvement of autophagy and of pre-existing lysosomes. Exp. Cell Res. 1975, 94, 377–391. [Google Scholar] [CrossRef]
- Panagiotidis, G.; Salehi, A.A.; Lundquist, I. Effect of the lysosomotropic drug suramin on islet lysosomal enzyme activities and the insulin-secretory response induced by various secretagogues. Pharmacology 1991, 43, 163–168. [Google Scholar] [CrossRef]
- Constantopoulos, G.; Rees, S.; Cragg, B.G.; Barranger, J.A.; Brady, R.O. Experimental animal model for mucopolysaccharidosis: Suramin-induced glycosaminoglycan and sphingolipid accumulation in the rat. Proc. Natl. Acad. Sci. USA 1980, 77, 3700–3704. [Google Scholar] [CrossRef] [Green Version]
- Korolenko, T.A.; Pupyshev, A.B.; Malygin, A.E. Heterophagic function and rate of intralysosomal proteolysis during lysosomotropic agents administration. Acta Biol. Med. Ger. 1981, 40, 1613–1617. [Google Scholar] [PubMed]
- De Santi, M.; Baldelli, G.; Diotallevi, A.; Galluzzi, L.; Schiavano, G.F.; Brandi, G. Metformin prevents cell tumorigenesis through autophagy-related cell death. Sci. Rep. 2019, 9, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, T.; Ribeiro, D.; Proenca, C.; Chiste, R.C.; Fernandes, E.; Freitas, M. Size-dependent cytotoxicity of silver nanoparticles in human neutrophils assessed by multiple analytical approaches. Life Sci. 2016, 145, 247–254. [Google Scholar] [CrossRef]
- Yamamoto, M.; Suzuki, S.O.; Himeno, M. Resveratrol-induced autophagy in human U373 glioma cells. Oncol. Lett. 2010, 1, 489–493. [Google Scholar] [CrossRef]
- Evans, T.D.; Jeong, S.J.; Zhang, X.; Sergin, I.; Razani, B. TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy 2018, 14, 724–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belzile, J.P.; Sabalza, M.; Craig, M.; Clark, E.; Morello, C.S.; Spector, D.H. Trehalose, an mTOR-independent inducer of autophagy, inhibits human cytomegalovirus infection in multiple cell types. J. Virol. 2016, 90, 1259–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.S.; Cho, E.D.; Jung Ahn, W.; Won Lee, K.; Lee, S.J.; Lee, H.J. Is trehalose an autophagic inducer? Unraveling the roles of non-reducing disaccharides on autophagic flux and alpha-synuclein aggregation. Cell Death Dis. 2017, 8, e3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justice, M.J.; Bronova, I.; Schweitzer, K.S.; Poirier, C.; Blum, J.S.; Berdyshev, E.V.; Petrache, I. Inhibition of acid sphingomyelinase disrupts LYNUS signaling and triggers autophagy. J. Lipid Res. 2018, 59, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Gulbins, A.; Schumacher, F.; Becker, K.A.; Wilker, B.; Soddemann, M.; Boldrin, F.; Muller, C.P.; Edwards, M.J.; Goodman, M.; Caldwell, C.C.; et al. Antidepressants act by inducing autophagy controlled by sphingomyelin-ceramide. Mol. Psychiatry 2018, 23, 2324–2346. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Li, X.; Umetani, M.; Boini, K.M.; Li, P.L.; Zhang, Y. Tricyclic antidepressant amitriptyline inhibits autophagic flux and prevents tube formation in vascular endothelial cells. Basic Clin. Pharmacol. Toxicol. 2019, 124, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Oh, S.; Choi, Y.J.; Oh, S.H.; Yang, Y.S.; Yang, M.J.; Lee, K.; Lee, B.H. Activation of autophagy rescues amiodarone-induced apoptosis of lung epithelial cells and pulmonary toxicity in rats. Toxicol. Sci. 2013, 136, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Easwaranathan, A.; Inci, B.; Ulrich, S.; Brunken, L.; Nikiforova, V.; Norinder, U.; Swanson, S.; Munic Kos, V. Quantification of Intracellular Accumulation and Retention of Lysosomotropic Macrocyclic Compounds by High-Throughput Imaging of Lysosomal Changes. J. Pharm. Sci. 2019, 108, 652–660. [Google Scholar] [CrossRef]
- Halliwell, W.H. Cationic amphiphilic drug-induced phospholipidosis. Toxicol. Pathol. 1997, 25, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Derendorf, H. Excessive lysosomal ion-trapping of hydroxychloroquine and azithromycin. Int. J. Antimicrob. Agents 2020, 55, 106007. [Google Scholar] [CrossRef] [PubMed]
- Salata, C.; Calistri, A.; Parolin, C.; Baritussio, A.; Palu, G. Antiviral activity of cationic amphiphilic drugs. Expert Rev. Anti Infect. Ther. 2017, 15, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Renna, M.; Schaffner, C.; Brown, K.; Shang, S.; Tamayo, M.H.; Hegyi, K.; Grimsey, N.J.; Cusens, D.; Coulter, S.; Cooper, J.; et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J. Clin. Investig. 2011, 121, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Mukai, S.; Moriya, S.; Hiramoto, M.; Kazama, H.; Kokuba, H.; Che, X.F.; Yokoyama, T.; Sakamoto, S.; Sugawara, A.; Sunazuka, T.; et al. Macrolides sensitize EGFR-TKI-induced non-apoptotic cell death via blocking autophagy flux in pancreatic cancer cell lines. Int. J. Oncol. 2016, 48, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Carlier, M.B.; Zenebergh, A.; Tulkens, P.M. Cellular uptake and subcellular distribution of roxithromycin and erythromycin in phagocytic cells. J. Antimicrob. Chemother. 1987, 20 (Suppl. B), 47–56. [Google Scholar] [CrossRef]
- Lin, X.; Han, L.; Weng, J.; Wang, K.; Chen, T. Rapamycin inhibits proliferation and induces autophagy in human neuroblastoma cells. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 2016, 1861, 269–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergin, I.; Evans, T.D.; Zhang, X.; Bhattacharya, S.; Stokes, C.J.; Song, E.; Ali, S.; Dehestani, B.; Holloway, K.B.; Micevych, P.S.; et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017, 8, 15750. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Liu, Y.; Zhang, J. Review: The role and mechanisms of macrophage autophagy in sepsis. Inflammation 2019, 42, 6–19. [Google Scholar] [CrossRef]
- Seebacher, F.; Zeigerer, A.; Kory, N.; Krahmer, N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Yang, N.; Shen, H.M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef]
- Kumar, M.; Ojha, S.; Rai, P.; Joshi, A.; Kamat, S.S.; Mallik, R. Insulin activates intracellular transport of lipid droplets to release triglycerides from the liver. J. Cell Biol. 2019, 218, 3697–3713. [Google Scholar] [CrossRef] [Green Version]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Li, Y.; Liu, R.; Wu, J.; Li, X. Self-eating: Friend or foe? The emerging role of autophagy in fibrotic diseases. Theranostics 2020, 10, 7993–8017. [Google Scholar] [CrossRef]
- Escamilla-Ramirez, A.; Castillo-Rodriguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseno, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a potential therapy for malignant glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.J.; Lee, M.N.; Oh, G.T. The role of macrophage lipophagy in reverse cholesterol transport. Endocrinol. Metab. 2017, 32, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging roles of lipophagy in health and disease. Front. Cell Dev. Biol. 2019, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Silwal, P.; Kim, S.Y.; Yoshimori, T.; Jo, E.K. Autophagy-activating strategies to promote innate defense against mycobacteria. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Stark, M.; Silva, T.F.D.; Levin, G.; Machuqueiro, M.; Assaraf, Y.G. The lysosomotropic activity of hydrophobic weak base drugs is mediated via their intercalation into the lysosomal membrane. Cells 2020, 9, 1082. [Google Scholar] [CrossRef]
- Melo, F.R.; Lundequist, A.; Calounova, G.; Wernersson, S.; Pejler, G. Lysosomal membrane permeabilization induces cell death in human mast cells. Scand. J. Immunol. 2011, 74, 354–362. [Google Scholar] [CrossRef]
- Villamil Giraldo, A.M.; Appelqvist, H.; Ederth, T.; Ollinger, K. Lysosomotropic agents: Impact on lysosomal membrane permeabilization and cell death. Biochem. Soc. Trans. 2014, 42, 1460–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Korolenko, T.A.; Rukavishnikova, E.V.; Safina, A.F.; Dushkin, M.I.; Mynkina, G.I. Endocytosis by liver cells during suppression of intralysosomal proteolysis. Biol. Chem. Hoppe Seyler 1992, 373, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Raley, M.J.; Schwacha, M.G.; Loegering, D.J. Lysosomotropic agents ameliorate macrophage dysfunction following the phagocytosis of IgG-coated erythrocytes: A role for lipid peroxidation. Inflammation 1997, 21, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Dielschneider, R.F.; Henson, E.S.; Gibson, S.B. Lysosomes as oxidative targets for cancer therapy. Oxid. Med. Cell Longev. 2017, 2017, 3749157. [Google Scholar] [CrossRef] [Green Version]
- Parks, A.; Marceau, F. Lysosomotropic cationic drugs induce cytostatic and cytotoxic effects: Role of liposolubility and autophagic flux and antagonism by cholesterol ablation. Toxicol. Appl. Pharmacol. 2016, 305, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Zhitomirsky, B.; Yunaev, A.; Kreiserman, R.; Kaplan, A.; Stark, M.; Assaraf, Y.G. Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. 2018, 9, 1191. [Google Scholar] [CrossRef]
- Giuliano, S.; Dufies, M.; Ndiaye, P.D.; Viotti, J.; Borchiellini, D.; Parola, J.; Vial, V.; Cormerais, Y.; Ohanna, M.; Imbert, V.; et al. Resistance to lysosomotropic drugs used to treat kidney and breast cancers involves autophagy and inflammation and converges in inducing CXCL5. Theranostics 2019, 9, 1181–1199. [Google Scholar] [CrossRef]
- Papanagnou, P.; Papadopoulos, G.E.; Stivarou, T.; Pappas, A. Toward fully exploiting the therapeutic potential of marketed pharmaceuticals: The use of octreotide and chloroquine in oncology. Onco Targets Ther. 2019, 12, 319–339. [Google Scholar] [CrossRef] [Green Version]
- Lubow, C.; Bockstiegel, J.; Weindl, G. Lysosomotropic drugs enhance pro-inflammatory responses to IL-1beta in macrophages by inhibiting internalization of the IL-1 receptor. Biochem. Pharmacol. 2020, 175, 113864. [Google Scholar] [CrossRef]
- Lamoureux, F.; Thomas, C.; Crafter, C.; Kumano, M.; Zhang, F.; Davies, B.R.; Gleave, M.E.; Zoubeidi, A. Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin. Cancer Res. 2013, 19, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Thomas, S.; Golden, E.B.; Hofman, F.M.; Chen, T.C.; Petasis, N.A.; Schonthal, A.H.; Louie, S.G. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012, 326, 143–154. [Google Scholar] [CrossRef]
- Jacques, P.J. The selection and design of lysosomotropic drugs. Adv. Exp. Med. Biol. 1976, 73 Pt A, 289–313. [Google Scholar] [CrossRef]
- Jacques, P.J.; Lambert, F. Alterations of rat liver lysosomes after treatment with particulate beta 1-3 glucan from Saccharomyces cerevisiae. Adv. Exp. Med. Biol. 1979, 121, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Lu, J.H. Autophagy and macrophage functions: Inflammatory response and phagocytosis. Cells 2020, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1178–1187. [Google Scholar] [CrossRef]
- Hazari, Y.; Bravo-San Pedro, J.M.; Hetz, C.; Galluzzi, L.; Kroemer, G. Autophagy in hepatic adaptation to stress. J. Hepatol. 2020, 72, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Umahara, T.; Uchihara, T.; Hirao, K.; Shimizu, S.; Hashimoto, T.; Kohno, M.; Hanyu, H. Essential autophagic protein Beclin 1 localizes to atherosclerotic lesions of human carotid and major intracranial arteries. J. Neurol. Sci. 2020, 414, 116836. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Fernández, Á.F.; López-Otín, C. The functional and pathologic relevance of autophagy proteases. J. Clin. Investig. 2015, 125, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Weiss-Sadan, T.; Maimoun, D.; Oelschlagel, D.; Kaschani, F.; Misiak, D.; Gaikwad, H.; Ben-Nun, Y.; Merquiol, E.; Anaki, A.; Tsvirkun, D.; et al. Cathepsins drive anti-inflammatory activity by regulating autophagy and mitochondrial dynamics in macrophage foam cells. Cell Physiol. Biochem. 2019, 53, 550–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Cui, W.; Sathyanarayan, A.; Lopresti, M.; Aghajan, M.; Chen, C.; Mashek, D.G. Lipophagy-derived fatty acids undergo extracellular efflux via lysosomal exocytosis. Autophagy 2020, 1–16. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, Z.; Chen, Y.; Qian, L.; Jiang, S.; Zhou, J.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Lipophagy and liver disease: New perspectives to better understanding and therapy. Biomed. Pharmacother. 2018, 97, 339–348. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Singh, R. Autophagy and lipid droplets in the liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef]
- Shi, L.; Wang, K.; Deng, Y.; Wang, Y.; Zhu, S.; Yang, X.; Liao, W. Role of lipophagy in the regulation of lipid metabolism and the molecular mechanism. Nan Fang Yi Ke Da Xue Xue Bao 2019, 39, 867–874. [Google Scholar] [CrossRef]
- Goeritzer, M.; Vujic, N.; Schlager, S.; Chandak, P.G.; Korbelius, M.; Gottschalk, B.; Leopold, C.; Obrowsky, S.; Rainer, S.; Doddapattar, P.; et al. Active autophagy but not lipophagy in macrophages with defective lipolysis. Biochim. Biophys. Acta 2015, 1851, 1304–1316. [Google Scholar] [CrossRef] [Green Version]
- Carr, R.M.; Ahima, R.S. Pathophysiology of lipid droplet proteins in liver diseases. Exp. Cell Res. 2016, 340, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Khaddaj, R.; Cottier, S.; Stradalova, V.; Jacob, C.; Schneiter, R. Mature lipid droplets are accessible to ER luminal proteins. J. Cell Sci. 2016, 129, 3803–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrick, M.J.; Lieberman, A.P. Autophagic dysfunction in a lysosomal storage disorder due to impaired proteolysis. Autophagy 2013, 9, 234–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet. J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleat, D.E.; Wiseman, J.A.; Sohar, I.; El-Banna, M.; Zheng, H.; Moore, D.F.; Lobel, P. Proteomic analysis of mouse models of Niemann-Pick C disease reveals alterations in the steady-state levels of lysosomal proteins within the brain. Proteomics 2012, 12, 3499–3509. [Google Scholar] [CrossRef] [Green Version]
- Cluzeau, C.V.; Watkins-Chow, D.E.; Fu, R.; Borate, B.; Yanjanin, N.; Dail, M.K.; Davidson, C.D.; Walkley, S.U.; Ory, D.S.; Wassif, C.A.; et al. Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet. 2012, 21, 3632–3646. [Google Scholar] [CrossRef] [Green Version]
- Meyers, A.; Weiskittel, T.M.; Dalhaimer, P. Lipid droplets: Formation to breakdown. Lipids 2017, 52, 465–475. [Google Scholar] [CrossRef]
- Jakos, T.; Pislar, A.; Pecar Fonovic, U.; Kos, J. Lysosomal peptidases in innate immune cells: Implications for cancer immunity. Cancer Immunol. Immunother. 2020, 69, 275–283. [Google Scholar] [CrossRef]
- Chen, N.; Ou, Z.; Zhang, W.; Zhu, X.; Li, P.; Gong, J. Cathepsin B regulates non-canonical NLRP3 inflammasome pathway by modulating activation of caspase-11 in Kupffer cells. Cell Prolif. 2018, 51, e12487. [Google Scholar] [CrossRef] [Green Version]
- Sleyster, E.C.; Knook, D.L. Relation between localization and function of rat liver Kupffer cells. Lab. Investig. 1982, 47, 484–490. [Google Scholar]
- Ferland, G.; Perea, A.; Audet, M.; Tuchweber, B. Characterization of liver lysosomal enzyme activity in hepatocytes, Kupffer and endothelial cells during aging: Effect of dietary restriction. Mech. Ageing Dev. 1990, 56, 143–154. [Google Scholar] [CrossRef]
- Ansorge, S.; Wiederanders, B.; Riemann, S.; Brouwer, A.; Knook, D.L. Distribution of thiol-protein disulfide oxidoreductase, insulin-glucagon proteinase and cathepsin D in different cell types of the rat liver. Biomed. Biochim. Acta 1984, 43, 1213–1221. [Google Scholar] [PubMed]
- Shiozaki, M.; Hayakawa, N.; Shibata, M.; Koike, M.; Uchiyama, Y.; Gotow, T. Closer association of mitochondria with lipid droplets in hepatocytes and activation of Kupffer cells in resveratrol-treated senescence-accelerated mice. Histochem. Cell Biol. 2011, 136, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Hardonk, M.J.; Dijkhuis, F.W.; Hulstaert, C.E.; Koudstaal, J. Heterogeneity of rat liver and spleen macrophages in gadolinium chloride-induced elimination and repopulation. J. Leukoc. Biol. 1992, 52, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Guo, W.; Fang, H.; Cao, S.; Yan, B.; Chen, S.; Zhang, K.; Zhang, S. Kupffer cell depletion by gadolinium chloride aggravates liver injury after brain death in rats. Mol. Med. Rep. 2018, 17, 6357–6362. [Google Scholar] [CrossRef] [Green Version]
- Akai, S.; Uematsu, Y.; Tsuneyama, K.; Oda, S.; Yokoi, T. Kupffer cell-mediated exacerbation of methimazole-induced acute liver injury in rats. J. Appl. Toxicol. 2016, 36, 702–715. [Google Scholar] [CrossRef]
- Liu, C.; Yang, Z.; Wang, L.; Lu, Y.; Tang, B.; Miao, H.; Xu, Q.; Chen, X. Combination of sorafenib and gadolinium chloride (GdCl3) attenuates dimethylnitrosamine(DMN)-induced liver fibrosis in rats. BMC Gastroenterol. 2015, 15, 159. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Zhong, W.; Zhang, J.; Zhang, X.; Wang, Y.; McClain, C.J. Kupffer cell depletion protects against the steatosis, but not the liver damage, induced by marginal-copper, high-fructose diet in male rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G934–G945. [Google Scholar] [CrossRef] [Green Version]
- Kiris, I.; Okutan, H.; Savas, C.; Yonden, Z.; Delibas, N. Gadolinium chloride attenuates aortic occlusion-reperfusion-induced myocardial injury in rats. Saudi Med. J. 2007, 28, 347–352. [Google Scholar]
- Zhu, F.; Li, X.; Jiang, Y.; Zhu, H.; Zhang, H.; Zhang, C.; Zhao, Y.; Luo, F. GdCl3 suppresses the malignant potential of hepatocellular carcinoma by inhibiting the expression of CD206 in tumorassociated macrophages. Oncol. Rep. 2015, 34, 2643–2655. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Ballabio, A. Lysosome: Regulator of lipid degradation pathways. Trends Cell Biol. 2014, 24, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Johnston, T.P.; Korolenko, T.A.; Sahebkar, A. P-407-induced mouse model of dose-controlled hyperlipidemia and atherosclerosis: 25 years later. J. Cardiovasc. Pharmacol. 2017, 70, 339–352. [Google Scholar] [CrossRef]
- Winchester, B. Lysosomal diseases: Diagnostic update. J. Inherit. Metab. Dis. 2014, 37, 599–608. [Google Scholar] [CrossRef]
- Winchester, B.; Vellodi, A.; Young, E. The molecular basis of lysosomal storage diseases and their treatment. Biochem. Soc. Trans. 2000, 28, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norinder, U.; Munic Kos, V. QSAR models for predicting five levels of cellular accumulation of lysosomotropic macrocycles. Int. J. Mol. Sci. 2019, 20, 5938. [Google Scholar] [CrossRef] [Green Version]
- Leighton, F.; Poole, B.; Beaufay, H.; Baudhuin, P.; Coffey, J.W.; Fowler, S.; De Duve, C. The large-scale separation of peroxisomes, mitochondria, and lysosomes from the livers of rats injected with triton WR-1339. Improved isolation procedures, automated analysis, biochemical and morphological properties of fractions. J. Cell Biol. 1968, 37, 482–513. [Google Scholar] [CrossRef] [Green Version]
- Wattiaux, R.; Wibo, M.; Baudhuin, P. Influence of the injection of Triton WR-1339 on the properties of rat liver lysosomes. In Ciba Foundation Symposium: Lysosomes; de Reuck M.Sc, A.V.S., Margrate, P., Cameron, M.A., Eds.; John and Wiley and Sons: London, UK, 1963; pp. 176–200. [Google Scholar]
- Hayashi, H.; Niinobe, S.; Matsumoto, Y.; Suga, T. Effects of Triton WR-1339 on lipoprotein lipolytic activity and lipid content of rat liver lysosomes. J. Biochem. 1981, 89, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.S.; Cromley, D.A.; McCoy, M.G.; Rader, D.J.; Billheimer, J.T. Determining hepatic triglyceride production in mice: Comparison of poloxamer 407 with Triton WR-1339. J. Lipid Res. 2005, 46, 2023–2028. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12, e0173771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdou, H.M.; Yousef, M.I.; Newairy, A.A. Triton WR-1339-induced hyperlipidemia, DNA fragmentation, neurotransmitters inhibition, oxidative damage, histopathological and morphometric changes: The protective role of soybean oil. J. Basic Appl. Zool. 2018, 79, 51. [Google Scholar] [CrossRef]
- Korolenko, T.A.; Johnston, T.P.; Machova, E.; Bgatova, N.P.; Lykov, A.P.; Goncharova, N.V.; Nescakova, Z.; Shintyapina, A.B.; Maiborodin, I.V.; Karmatskikh, O.L. Hypolipidemic effect of mannans from C. albicans serotypes a and B in acute hyperlipidemia in mice. Int. J. Biol. Macromol. 2018, 107, 2385–2394. [Google Scholar] [CrossRef]
- Serrano-Puebla, A.; Boya, P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem. Soc. Trans. 2018, 46, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.H.; Jan, M.S.; Chiu, H.W. Cytotoxic properties of tyloxapol. Pharm. Res. 2006, 23, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Huterer, S.; Phillips, M.J.; Wherrett, J.R. Effects of prolonged administration of triton WR-1339 to the rat on morphology and phospholipids of liver. Lab. Investig. 1975, 33, 305–310. [Google Scholar] [PubMed]
- Russo, M.; Russo, G.L. Autophagy inducers in cancer. Biochem. Pharmacol. 2018, 153, 51–61. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Remmerie, A.; Martens, L.; Scott, C.L. Macrophage subsets in obesity, aligning the liver and adipose tissue. Front. Endocrinol. 2020, 11, 259. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Lee, G.C.; Lin, C.H.; Tao, Y.C.; Yang, J.M.; Hsu, K.C.; Huang, Y.J.; Huang, S.H.; Kung, P.J.; Chen, W.L.; Wang, C.M.; et al. The potential of lactulose and melibiose, two novel trehalase-indigestible and autophagy-inducing disaccharides, for polyQ-mediated neurodegenerative disease treatment. Neurotoxicology 2015, 48, 120–130. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, N.; Rocchi, A.; Zhang, W.; Vassar, R.; Zhou, Y.; He, C. Identification of natural products with neuronal and metabolic benefits through autophagy induction. Autophagy 2017, 13, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarelli, R.; Roccheri, M.C. Heavy metals and metalloids as autophagy inducing agents: Focus on cadmium and arsenic. Cells 2012, 1, 597–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takanezawa, Y.; Nakamura, R.; Kusaka, T.; Ohshiro, Y.; Uraguchi, S.; Kiyono, M. Significant contribution of autophagy in mitigating cytotoxicity of gadolinium ions. Biochem. Biophys. Res. Commun. 2020, 526, 206–212. [Google Scholar] [CrossRef] [PubMed]
- la Fuente, F.P.; Quezada, L.; Sepulveda, C.; Monsalves-Alvarez, M.; Rodriguez, J.M.; Sacristan, C.; Chiong, M.; Llanos, M.; Espinosa, A.; Troncoso, R. Exercise regulates lipid droplet dynamics in normal and fatty liver. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 158519. [Google Scholar] [CrossRef]
- Lee, H.J.; Yoon, Y.S.; Lee, S.J. Mechanism of neuroprotection by trehalose: Controversy surrounding autophagy induction. Cell Death Dis. 2018, 9, 712. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and microglia: Novel partners in neurodegeneration and aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef] [Green Version]
- Mo, Y.; Sun, Y.Y.; Liu, K.Y. Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 2020, 15, 1388–1396. [Google Scholar] [CrossRef]
- Gopinath, A.; Collins, A.; Khoshbouei, H.; Streit, W. Microglia and other myeloid cells in CNS health and disease. J. Pharmacol. Exp. Ther. 2020. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Hashemiaghdam, A.; Mroczek, M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J. Neuroimmunol. 2020, 341, 577185. [Google Scholar] [CrossRef] [PubMed]
- Giaime, E.; Tong, Y.; Wagner, L.K.; Yuan, Y.; Huang, G.; Shen, J. Age-dependent dopaminergic neurodegeneration and impairment of the autophagy-lysosomal pathway in LRRK-deficient mice. Neuron 2017, 96, 796–807.e6. [Google Scholar] [CrossRef] [Green Version]
- Pupyshev, A.B.; Korolenko, T.A.; Akopyan, A.A.; Amstislavskaya, T.G.; Tikhonova, M.A. Suppression of autophagy in the brain of transgenic mice with overexpression of capital A, Cyrillic53capital TE, Cyrillic-mutant alpha-synuclein as an early event at synucleinopathy progression. Neurosci. Lett. 2018, 672, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Pupyshev, A.B.; Tikhonova, M.A.; Akopyan, A.A.; Tenditnik, M.V.; Dubrovina, N.I.; Korolenko, T.A. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name and Basic Functions of the Drug or Compound | Biological Effects on Lysosomes | References |
|---|---|---|
| Chloroquine, antimalarial, antiparasitic, anti-inflammatory effects; antiviral activities in vitro against viruses, including coronaviruses, dengue virus and the biosafety level 4 Nipah and Hendra paramyxoviruses. The in vivo efficacy in the treatment of COVID-19 is currently a matter of debate. | Intralysosomal pH alteration; lysosomal membrane permeabilization; inhibitor of autophagic flux. Inhibition of acidic proteases involved in the maturation of virus fusion protein. | Schneider et al., 1997 [5]; Pisonero-Vaquero, Medina, 2017 [2] Carrière et al., 2020 [6] Blaess et al., 2020 [7] Norinder et al., 2020 [8] |
| Hydroxychloroquine, antiviral effect in vitro, similar to chloroquine. The in vivo efficacy of hydroxychloroquine in the treatment of COVID-19 is currently a matter of debate. | Similar to chloroquine (but no phospholipidosis). | Carrière et al., 2020 [6] Blaess et al., 2020 [7] Norinder et al., 2020 [8] |
| Triton WR 1339 (Tyloxapol), non-ionic surfactant, additive in pharmacy; PEGylated Tyloxapol niosomes for the controlled release of first-line anti-tuberculosis drugs. | Increased autophagy (induced by macrocyclon); lysosomal membrane permeabilization, lysosomal swelling | Plattner et al., 1975 [9] Schneider et al., 1997 [5] |
| Suramin (antiparasitic drug, to treat African sleeping sickness or trypanosomiasis. It works by inhibiting ATP signaling). | Accumulation in lysosomes; inhibitor of lysosomal enzymes, suppressing phagosome-lysosome fusion; is commonly employed as a tool for inducing experimental mucopolysaccharidosis and lipidosis. | Panagiotidis et al., 1991 [10] Schneider et al., 1997 [5] Constantopoulos et al., 1980 [11] Korolenko et al., 1981 [12] |
| Metformin (anti-aging; anti-diabetic; anti-tumor). | Prevents cell tumor through autophagy (by AMPK activation and inhibition of mTOR, major inhibitor of autophagic flux); able to block the autophagosome fusion with lysosomes. | De Santi et al., 2019 [13] |
| Gold sodium thiomalate, anti-inflammatory effect in rheumatoid arthritis, active treatment in severe cases (in children). | Effect on lysosomal enzyme activity in macrophages and lysosomal membrane, bacterial killing. | Schneider et al., 1997 [5] |
| PVP, for blood transfusion; (polyvinyl pyrrolidone)-coated AgNPs have an anti-leukemia effect against human myeloid leukemia cells. | Accumulation inside of lysosomes, lysosomal membrane stability changes; trigger of autophagy. | Soares et al., 2016 [14] |
| Resveratrol. | Enhancer of autophagy in human U373 glioma cells. | Yamamoto et al., 2010 [15] |
| Trehalose. | Disaccharide trehalose is a novel inducer of TFEB with atheroprotective effects. mTOR (the mammalian target of rapamycin)-independent inducer of autophagy. Potent blocker of the autophagic flux. | Evans et al., 2018 [16] Belzile et al., 2016 [17] Yoon et al., 2017 [18] |
| Imipramine (antidepressant), cationic amphiphilic drug. | Inhibition of lysosomal ceramide-producing enzyme, acid sphingomyelinase with imipramine or by various stresses are involved in cell death and has been implicated in autophagy. | Justice et al., 2018 [19] |
| Amitriptyline (antidepressant) Amiodarone (anti-arrhythmic), Strong cationic amphiphilic drug. | Autophagy inducer (neurons). Amiodarone inhibits autophagic flux and prevents tube formation in vascular endothelial cells. Amiodarone increased autophagic flux and apoptosis in H460 human lung epithelial cells and BEAS-2B normal human bronchial epithelial cells. | Gulbins et al., 2018 [20] Guan et al., 2019 [21] Lee et al., 2013 [22] Easwaranathan et al., 2019 [23] |
| Azithromycin (antibiotic) Strong cationic amphiphilic drug. | Excessive lysosomal ion-trapping. In chronic administration in vivo pharmacological blockade of autophagy. | Halliwell, 1997 [24] Derendorf, 2020 [25] Salata et al., 2013 [26] Renna et al., 2011 [27] Mukai et al., 2016 [28] |
| Erythromycin (antibiotic) Moderate cationic drug. | Accumulation inside of cells (in lysosomes of phagocytic cells). | Carlier et al., 1987 [29] |
| Rapamycin. | Induces autophagy in human neuroblastoma cells. | Lin et al. 2018 [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korolenko, T.A.; Johnston, T.P.; Vetvicka, V. Lysosomotropic Features and Autophagy Modulators among Medical Drugs: Evaluation of Their Role in Pathologies. Molecules 2020, 25, 5052. https://doi.org/10.3390/molecules25215052
Korolenko TA, Johnston TP, Vetvicka V. Lysosomotropic Features and Autophagy Modulators among Medical Drugs: Evaluation of Their Role in Pathologies. Molecules. 2020; 25(21):5052. https://doi.org/10.3390/molecules25215052
Chicago/Turabian StyleKorolenko, Tatiana A., Thomas P. Johnston, and Vaclav Vetvicka. 2020. "Lysosomotropic Features and Autophagy Modulators among Medical Drugs: Evaluation of Their Role in Pathologies" Molecules 25, no. 21: 5052. https://doi.org/10.3390/molecules25215052