
Hybridization of Curcumin Analogues with Cinnamic Acid Derivatives as Multi-Target Agents Against Alzheimer’s Disease Targets


Abstract
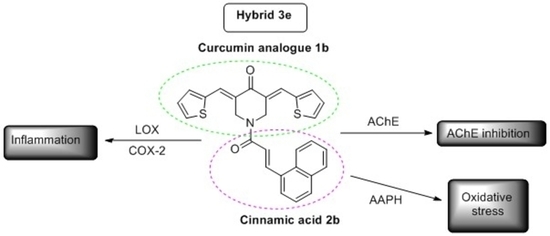
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Physicochemical Studies
2.2.1. Experimental Determination of Lipophilicity as RM Values
2.2.2. In Silico Determination of Drug-Likeness
2.3. Biological Evaluation
2.4. Computational Studies—Docking Simulation Soybean Lipoxygenase
2.4.1. Putative Binding Modes of Curcumin Analogues 1k (Blue), 1l (Purple), 1m (Green), and 1o (Pink) in Soybean LOX.
2.4.2. Molecular Modeling of the Synthesized Derivatives in Soybean LOX
2.4.3. Molecular Docking Studies of the Novel Derivatives on AChE
3. Experimental Section
3.1. Materials and Instruments
3.2. Chemistry General Procedures
3.2.1. Synthesis of Curcumin Analogues 1a–q
General Method A
- (3E,5E)-3,5-bis(naphthalen-1-ylmethylene)piperidin-4-one (1a) [40].
- (3E,5E)-3,5-bis(thiophen-2-ylmethylene)piperidin-4-one (1b) [40].
- (3E,5E)-3,5-bis((1H-indol-3-yl)methylene)piperidin-4-one (1c) [40].
- (3E,5E)-3,5-bis((1H-indol-5-yl)methylene)piperidin-4-one (1d) [40].
- (3E,5E)-3,5-bis(4-((4-bromophenoxy)methyl)benzylidene)piperidin-4-one (1e) [40].
- (3E,5E)-3,5-bis((1H-imidazol-2-yl)methylene)piperidin-4-one (1f) [40].
- (3E,5E)-3,5-dibenzylidenepiperidin-4-one (1g) [40].
- (3E,5E)-3,5-bis((3-methylthiophen-2-yl)methylene)piperidin-4-one (1h) [40].
- (3E,5E)-3,5-bis((5-methylthiophen-2-yl)methylene)piperidin-4-one (1i) [40].
General Method B—Synthesis of Curcumin Analogues 1k–n Using Microwave (MW) Irradiation
General Method C—Synthesis of Curcumin Analogues 1o–p
3.2.2. Synthesis of Acids 2a–c [37]
General Method D—Synthetic Method for the Synthesis of Hybrids of Curcumin Analogues 3a–3h
3.3. Physicochemical Studies
Determination of RM Values
3.4. Biological In Vitro Assays
3.4.1. Inhibition of Linoleic Acid Peroxidation
3.4.2. Soybean Lipoxygenase Inhibition Study
3.4.3. AChE Inhibition Study
3.4.4. Cyclooxygenase 2 Inhibition Study
3.5. Computational Methods.
3.5.1. Molecular Docking Studies on Soybean Lipoxygenase
3.5.2. Molecular Docking Studies on AChE
3.5.3. In Silico Determination of Drug-Likeness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Attitudes to Dementia-World Alzheimer Report 2019. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2019.Pdf (accessed on 26 January 2020).
- Song, J.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.; Tong, B.C.; Iyaswamy, A.; Shang, W.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A Small Molecule Transcription Factor EB Activator Ameliorates beta-amyloid Precursor Protein and Tau Pathology in Alzheimer’s Disease Models. Aging Cell 2019, 19, e13069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulya, A.; İlhami, G. Potent Acetylcholinesterase Inhibitors: Potential Drugs for Alzheimer’s Disease. Mini–Rev. Med. Chem. 2020, 20, 1–13. [Google Scholar]
- Brufani, M.; Filocamo, L. Rational Design of New Acetylcholinesterase Inhibitors. In Alzheimer Disease; Springer Science and Business Media LLC/Birkhauser: Boston, MA, USA, 1997; pp. 171–177. [Google Scholar] [CrossRef]
- Gao, X.-H.; Liu, L.-B.; Liu, H.-R.; Tang, J.-J.; Kang, L.; Wu, H.; Cui, P.; Yan, J. Structure–activity Relationship Investigation of Benzamide and Picolinamide Derivatives Containing Dimethylamine Side Chain As Acetylcholinesterase Inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 33, 110–114. [Google Scholar] [CrossRef]
- Dhull, D.K.; Jindal, A.; Dhull, R.K.; Aggarwal, S.; Bhateja, D.; Padi, S.S.V. Neuroprotective Effect of Cyclooxygenase Inhibitors in ICV-STZ Induced Sporadic Alzheimer’s Disease in Rats. J. Mol. Neurosci. 2011, 46, 223–235. [Google Scholar] [CrossRef]
- Qin, W.-P.; Eho, L.; Pompl, P.N.; Peng, Y.; Zhao, Z.; Xiang, Z.; Robakis, N.K.; Shioi, J.; Suh, J.; Pasinetti, G.M. Cyclooxygenase (COX)-2 and COX-1 Potentiate β-Amyloid Peptide Generation through Mechanisms That Involve γ-Secretase Activity. J. Biol. Chem. 2003, 278, 50970–50977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannopoulos, P.F.; Joshi, Y.B.; Praticò, D. Novel Lipid Signaling Pathways in Alzheimer’s Disease Pathogenesis. Biochem. Pharmacol. 2013, 88, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firuzi, O.; Zhou, J.; Chinnici, C.M.; Wisniewski, T.; Praticò, D. 5- Lipoxygenase Gene Disruption Reduces Amyloid-Beta Pathology in a Mouse Model of Alzheimer’s Disease. FASEB J. 2008, 22, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Joshi, Y.B.; Praticã, D.; Praticò, D. The 5-Lipoxygenase Pathway: Oxidative and Inflammatory Contributions to the Alzheimer’s Disease Phenotype. Front. Cell. Neurosci. 2015, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shekhar, S.; Yadav, S.K.; Rai, N.; Kumar, R.; Tripathi, M.; Dey, A.B. 5-LOX in Alzheimer’s Disease: Potential Serum Marker and In Vitro Evidences for Rescue of Neurotoxicity by Its Inhibitor YWCS. Mol. Neurobiol. 2017, 55, 2754–2762. [Google Scholar] [CrossRef]
- Chainoglou, E.; Hadjipavlou-Litina, D. Curcumin in Health and Diseases: Alzheimer’s Disease and Curcumin Analogues, Derivatives, and Hybrids. Int. J. Mol. Sci. 2020, 21, 1975. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Gou, S.; Liu, X.; Cao, F.; Cheng, L. Design, Synthesis and Anti-Alzheimer Properties of Dimethylaminomethyl-Substituted Curcumin Derivatives. Bioorganic Med. Chem. Lett. 2014, 24, 40–43. [Google Scholar] [CrossRef]
- Venigalla, M.; Sonego, S.; Gyengési, E.; Sharman, M.J.; Münch, G. Novel Promising Therapeutics Against Chronic Neuroinflammation and Neurodegeneration in Alzheimer’s Disease. Neurochem. Int. 2016, 95, 63–74. [Google Scholar] [CrossRef]
- Swarit, J.; Ye, H.; Jurgen, B. How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J. Med. Chem. 2017, 60, 3879–3886. [Google Scholar]
- Chainoglou, E.; Hadjipavlou-Litina, D.; Chainoglou, E. Curcumin Analogues and Derivatives With Anti-Proliferative and Anti-Inflammatory Activity: Structural Characteristics and Molecular Targets. Expert Opin. Drug Discov. 2019, 14, 821–842. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Morón, E.; Calderón-Montaño, J.M.; Salvador, J.; Robles, A.; López-Lázaro, M. The Dark Side of Curcumin. Int. J. Cancer 2010, 126, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Exploration and Synthesis of Curcumin Analogues With Improved Structural Stability Both in Vitro and in Vivo As Cytotoxic Agents. Bioorganic Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Shetty, D.; Kim, Y.J.; Shim, H.; Snyder, J.P. Eliminating the Heart from the Curcumin Molecule: Monocarbonyl Curcumin Mimics (MACs). Molecumel 2014, 20, 249–292. [Google Scholar] [CrossRef] [Green Version]
- Yadav, B.; Taurin, S.; Rosengren, R.J.; Schumacher, M.; Diederich, M.; Somers-Edgar, T.J.; Larsen, L. Synthesis and Cytotoxic Potential of Heterocyclic Cyclohexanone Analogues of Curcumin. Bioorganic Med. Chem. 2010, 1, 6701–6707. [Google Scholar] [CrossRef]
- Das, U.; Sharma, R.; Dimmock, J.R. 1,5-Diaryl-3-Oxo-1,4-Pentadienes: A Case for Antineoplastics With Multiple Targets. Curr. Med. Chem. 2009, 16, 2001–2020. [Google Scholar] [CrossRef] [Green Version]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and Biological Evaluation of Novel Curcumin Analogues As Anti-Cancer and Anti-Angiogenesis Agents. Bioorg. Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef]
- Lagisetty, P.; Powell, D.R.; Awasthi, V. Synthesis and Structural Determination of 3,5-bis(2-Fluorobenzylidene)-4-Piperidone Analogues of Curcumin. J. Mol. Struct. 2009, 936, 23–28. [Google Scholar] [CrossRef]
- Lagisetty, P.; Vilekar, P.; Sahoo, K.; Anant, S.; Awasthi, V. CLEFMA-An Antiproliferative Curcuminoid from Structure-Activity Relationship Studies on 3,5-bis(benzylidene)-4-Piperidones. Bioorg. Med. Chem. 2010, 18, 6109–6120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.; Duffield, K.M. 3,5-Bis(arylmethylene)-4-Piperidone Derivatives As Novel Anticancer Agents. Indian J. Chem. 2007, 38, 2313–2320. [Google Scholar] [CrossRef]
- Makarov, M.V.; Leonova, E.S.; Rybalkina, E.Y.; Tongwa, P.; Khrustalev, V.N.; Timofeeva, T.V.; Odinets, I.L. Synthesis, Characterization and structure–activity Relationship of Novel N-Phosphorylated E,E-3,5-bis(thienylidene)piperid-4-Ones. Eur. J. Med. Chem. 2010, 45, 992–1000. [Google Scholar] [CrossRef]
- Aboul-Fadl, T.; El-Shorbagi, A.N.; AHozien, Z.; Sarhan, A.W. Investigation of Alkylating, Antineoplastic and Anti-HIV Potentials of the Chalcones: 2-(3-arylpropenoyl)benzimidazole and Their Corresponding N1-Methyl Derivatives. Boll. Chim. Farm. 2001, 139, 228–234. [Google Scholar]
- Pontiki, E.; Hadjipavlou-Litina, D. Antioxidant and Anti-Inflammatory Activity of Aryl-Acetic and Hydroxamic Acids As Novel Lipoxygenase Inhibitors. Med. Chem. 2006, 2, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Geromichalos, G.; Papageorgiou, A.; Hadjipavlou-Litina, D. Anticancer Activity and Quantitative-Structure Activity Relationship (QSAR) Studies of a Series of Antioxidant/Anti-Inflammatory Aryl-Acetic and Hydroxamic Acids. Chem. Biol. Drug Des. 2009, 74, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Hadjipavlou-Litina, D. Synthesis and Pharmacochemical Evaluation of Novel Aryl-Acetic Acid Inhibitors of Lipoxygenase, Antioxidants, and Anti-Inflammatory Agents. Bioorganic Med. Chem. 2007, 15, 5819–5827. [Google Scholar] [CrossRef]
- Naz, S.; Ahmad, S.; Rasool, S.A.; Sayeed, S.A.; Siddiqi, R. Antibacterial Activity Directed Isolation of Compounds from Onosma Hispidum. Microbiol. Res. 2006, 161, 43–48. [Google Scholar] [CrossRef]
- Nardini, M.; D’Aquino, M.; Tomassi, G.; Gentili, V.; Di Felice, M.; Scaccini, C. Inhibition of Human Low-Density Lipoprotein Oxidation by Caffeic Acid and Other Hydroxycinnamic Acid Derivatives. Free. Radic. Biol. Med. 1995, 19, 541–552. [Google Scholar] [CrossRef]
- Yasuko, K.; Tomohiro, N.; Sei-Itsu, M.; Ai-Na, L.; Yasuo, F.; Takashi, T. Caffeic Acid Is a Selective Inhibitor for Leukotriene Biosynthesis. Biochim. Biophys. Acta (BBA)–Lipids Lipid Metab. 1984, 792, 92–97. [Google Scholar] [CrossRef]
- Szwajgier, D.; Borowiec, K.; Pustelniak, K. The Neuroprotective Effects of Phenolic Acids: Molecular Mechanism of Action. Nutrients 2017, 9, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, L.-Y.; Cosma, G.; Gardner, H.; Shi, X.; Castranova, V.; Vallyathan, V. Effect of Antioxidant Protection by P-Coumaric Acid on Low-Density Lipoprotein Cholesterol Oxidation. Am. J. Physiol. Physiol. 2000, 279, C954–C960. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Tang, J.; Liu, H.; Liu, L.; Kang, L.; Chen, W. Structure–activity Relationship Investigation of Tertiary Amine Derivatives of Cinnamic Acid As Acetylcholinesterase and Butyrylcholinesterase Inhibitors: Compared With That of Phenylpropionic Acid, Sorbic Acid and Hexanoic Acid. J. Enzym. Inhib. Med. Chem. 2018, 33, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Peperidou, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Voulgari, E.; Avgoustakis, K. Multifunctional Cinnamic Acid Derivatives. Molecules 2017, 22, 1247. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Peperidou, A.; Fotopoulos, I.; Hadjipavlou-Litina, D.J. Cinnamate Hybrids: A Unique Family of Compounds With Multiple Biological Activities. Curr. Pharm. Biotechnol. 2019, 19, 1019–1048. [Google Scholar] [CrossRef]
- Noureddin, S.A.; El-Shishtawy, R.M.; Al-Footy, K.O. Curcumin Analogues and Their Hybrid Molecules As Multifunctional Drugs. Eur. J. Med. Chem. 2019, 182, 111631. [Google Scholar] [CrossRef]
- Katsori, A.-M.; Chatzopoulou, M.; Dimas, K.; AKontogiorgis, C.; Patsilinakos, A.; Trangas, T.; Hadjipavloulitina, D. Curcumin Analogues As Possible Anti-Proliferative & Anti-Inflammatory Agents. Eur. J. Med. Chem. 2011, 46, 2722–2735. [Google Scholar] [CrossRef]
- Katsori, A.-M.; Palagani, A.; Bougarne, N.; Hadjipavlou-Litina, D.; Haegeman, G.; Berghe, W.V. Inhibition of the NF-κB Signaling Pathway by a Novel Heterocyclic Curcumin Analogue. Molecules 2015, 20, 863–878. [Google Scholar] [CrossRef] [Green Version]
- Abdulrahman, A.; Raju, K.; Farzana, B.; Amir, S.; Hasnah, O.; Rusli, I.; Tan, C.; Brian, S.; Kellen, M.; Alaa, N.; et al. Facile, Regio- and Diastereoselective Synthesis of Spiro-Pyrrolidine and Pyrrolizine Derivatives and Evaluation of Their Antiproliferative Activities. Molecules 2014, 19, 10033–10055. [Google Scholar]
- Singaram, K.; Marimuthu, D.; Baskaran, S.; Ramaswamy, V. Synthesis and Antimicrobial Activity of New 3,5-Diarylidene-4-Piperidone Derivatives. J. Serb. Chem. Soc. 2016, 81, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Du, Z.; Zheng, X.; Cui, X.-X.; Conney, A.H.; Zhang, K. Synthesis and Evaluation of Curcumin-Related Compounds for Anticancer Activity. Eur. J. Med. Chem. 2012, 53, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Nehmedo, G.; Fawzy Siva, P.; Walid, F.; El Sayed, S.; Aladdin, S.; Adel, G. Synthesis, Human Topoisomerase IIa Inhibitory Properties and Molecular Modeling Studies of Antiproliferative Curcumin Mimics. RSC Adv. 2019, 9, 33761. [Google Scholar]
- Xie, X.; Tu, J.; You, H.; Hu, B. Design, Synthesis, and Biological Evaluation of Novel EF24 and EF31 Analogs As Potential IκB Kinase β Inhibitors for the Treatment of Pancreatic Cancer. Drug Des. Dev. Ther. 2017, 11, 1439–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmock, J.; Padmanilayam, M.P.; Puthucode, R.N.; Nazarali, A.J.; Motaganahalli, N.L.; Zello, G.A.; Quail, J.W.; Oloo, E.O.; Kraatz, H.-B.; Prisciak, J.S.; et al. A Conformational and Structure−Activity Relationship Study of Cytotoxic 3,5-Bis(arylidene)-4-Piperidones and RelatedN-Acryloyl Analogues. J. Med. Chem. 2001, 44, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bai, H.; Han, L.; Zhang, H.; Xu, B.; Cui, J.; Wang, X.; Ge, Z.; Li, R. Synthesis and Biological Evaluation of Curcumin Derivatives Modified With α-Amino Boronic Acid As Proteasome Inhibitors. Bioorganic Med. Chem. Lett. 2018, 28, 2459–2464. [Google Scholar] [CrossRef]
- Almansour, A.I.; Suresh Kumar, R.; Arumugam, N.; Basiri, A.; Kia, Y.; Ashraf Ali, M. An Expedient Synthesis, Acetylcholinesterase Inhibitory Activity, and Molecular Modeling Study of Highly Functionalized Hexahydro-1,6-Naphthyridines. BioMed Res. Int. 2015, 965987. [Google Scholar] [CrossRef]
- Yalda, K.; Hasnah, O.; Raju, K.; Alireza, B.; Vikneswaran, M. Synthesis and Discovery of Highly Functionalized Mono- and Bis-Spiro-Pyrrolidines As Potent Cholinesterase Enzyme Inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 1815–1819. [Google Scholar]
- Dimmock, J.; Jha, A.; Zello, G.A.; Quail, J.W.; Oloo, E.O.; Nienaber, K.H.; Kowalczyk, E.S.; Allen, T.S.; Santos, C.L.; De Clercq, L.; et al. Cytotoxic N-[4-(3-Aryl-3-Oxo-1-propenyl)phenylcarbonyl]-3,5- bis(phenylmethylene)-4-Piperidones and Related Compounds. Eur. J. Med. Chem. 2002, 37, 961–972. [Google Scholar] [CrossRef]
- Dimmock, J.R.; Arora, V.K.; Wonko, S.L.; Hamon, N.W.; Quail, J.W.; Jia, Z.; Warrington, R.C.; Fang, W.D.; Lee, J.S. 3,5-Bis-Benzylidene-4-Piperidones and Related Compounds With High Activity towards P388 Leukemia Cells. Drug Des. Deliv. 1990, 6, 183–194. [Google Scholar]
- Hossain, M.; Das, S.; Das, U.; Doroudi, A.; Zhu, J.; Dimmock, J.R. Novel Hybrid Molecules of 3,5-bis(benzylidene)-4-Piperidones and Dichloroacetic Acid Which Demonstrate Potent Tumour-Selective Cytotoxicity. Bioorganic Med. Chem. Lett. 2020, 30, 126878. [Google Scholar] [CrossRef] [PubMed]
- Saeed Abaee, M.; Mojtahedi, M.; Mehdi Zahedi, M.; Mohammad, S. First Highly Efficient Synthesis of bis(Arylmethylidene)pyranones. Mediated by Lithium Perchlorate.Lithium Perchlorate. Synth. Commun. 2006, 36, 199–206. [Google Scholar] [CrossRef]
- Saeed Abaee, M.; Mohammad, M.; Mehdi Zahedi, M.; Sharifi, R.A. Highly Efficient Method for Solvent-Free Synthesis of Bisarylmethylidenes of Pyranonesand Thiopyranones. Heteroat. Chem. 2007, 18, 1. [Google Scholar]
- Madhusdana Reddy, M.B.; Nizam, A.; Pasha, M.A. Molecular Iodine: An Efficient and Environment Friendly Catalyst for the Synthesis of 3,5-Bis-(arylmethylidene)- Tetrahydropyran-4-Ones. Synth. Commun. 2010, 40, 3728–3733. [Google Scholar] [CrossRef]
- Nelson, J.; Debabrata, C. 7-Pyrones by Isomerizaton. Substituted 3,5-Dibenzyl-4H-Pyran-4-Ones. J. Amer. Chem. Soc. 1957, 79, 1474–1482. [Google Scholar]
- Mardyukov, A.; Niedek, D.; Schreiner, P.R. Unravelling Lawesson’s Reagent: The Structure of Monomeric (4-methoxyphenyl)phosphine Disulfide. Chem. Commun. 2018, 54, 2715–2718. [Google Scholar] [CrossRef] [PubMed]
- Version 2016.10. Available online: https://www.molinspiration.com (accessed on 25 October 2020).
- Canavan, N. FDA and Drug Companies Alike Want ADME-Tox Testing Performed Earlier and Earlier in a Drug’s Life Cycle. Drug Discov. Dev. 2007, 10, 34–36. [Google Scholar]
- ICM v3.8-5, 2016. Available online: https://www.molsoft.com (accessed on 25 October 2020).
- 2013. Available online: https://cyprules.cmdm.Tw (accessed on 25 October 2020).
- Version 2.0, 2008/10. Available online: https://preadmet.bmdrc.kr/ (accessed on 25 October 2020).
- 2017. Available online: http://www.swissadme.ch (accessed on 25 October 2020).
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Kavetsou, E.; Katopodi, A.; Argyri, L.; Chainoglou, E.; Pontiki, E.; Hadjipavlou-Litina, D.; Chroni, A.; Detsi, A. Novel 3-aryl-5-substituted-coumarin Analogues: Synthesis and Bioactivity Profile. Drug Dev. Res. 2020, 81, 456–469. [Google Scholar] [CrossRef]
- Chu, J.; Praticò, D. 5-Lipoxygenase As an Endogenous Modulator of Amyloid Beta Formation in Vivo. Ann. Neurol. 2010, 69, 34–46. [Google Scholar] [CrossRef]
- Praticò, D. Oxidative Stress Hypothesis in Alzheimer’s Disease: A Reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef]
- Chu, J.; Li, J.-G.; Ceballos-Diaz, C.; Golde, T.; Praticò, D. The Influence of 5-Lipoxygenase on Alzheimer’s Disease-Related Tau Pathology: In Vivo and In Vitro Evidence. Biol. Psychiatry 2013, 74, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrzypczak-Jankun, E.; Zhou, K.; Jankun, J. Inhibition of Lipoxygenase by (-)-Epigallocatechin Gallate: X-Ray Analysis at 2.1 A Reveals Degradation of EGCG and Shows Soybean LOX-3 Complex With EGC Instead. Int. J. Mol. Med. 2003, 12, 415–420. [Google Scholar] [CrossRef]
- Moody, T.W.; Leyton, J.; Martínez, A.; Hong, S.; Malkinson, A.; Mulshine, J.L. Lipoxygenase Inhibitors Prevent Lung Carcinogenesis and Inhibit Non-Small Cell Lung Cancer Growth. Exp. Lung Res. 1998, 24, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.M.; Rojas, C.J.; Learn, K.S.; Perrone, M.H.; Bilder, G.E. Characterization and Inhibition of 15-Lipoxygenase in Human Monocytes: Comparison With Soybean 15-Lipoxygenase. Am. J. Physiol. Physiol. 1995, 268, C1301–C1307. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, D.; Ciofani, G.; Pierdomenico, S.D.; Giamberardino, M.A.; Cuccurullo, F. Dihydrolipoic Acid Inhibits 15-Lipoxygenase-Dependent Lipid Peroxidation. Free. Radic. Biol. Med. 2003, 35, 1203–1209. [Google Scholar] [CrossRef]
- Nuhn, P.; Büge, A.; Köhler, T.; Lettau, H.; Schneider, R. Trends in the Development of Lipoxygenase inhibitors. Die Pharmazie 1991, 46, 81–88. [Google Scholar] [PubMed]
- Maccarrone, M.; Van Aarle, P.G.; AVeldink, G.; Vliegenthart, J.F. In Vitro Oxygenation of Soybean Biomembranes by Lipoxygenase-2. Biochim. Biophys. Acta (BBA)–Biomembr. 1994, 1190, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Pontiki, E.; Hadjipavlou-Litina, D. Lipoxygenase Inhibitors: A Comparative QSAR Study Review and Evaluation of New QSARs. Med. Res. Rev. 2007, 28, 39–117. [Google Scholar] [CrossRef]
- Liargkova, T.; Hadjipavlou-Litina, D.; Koukoulitsa, C.; Voulgari, E.; Avgoustakis, C. Simple Chalcones and Bis-Chalcones Ethers As Possible Pleiotropic Agents. J. Enzym. Inhib. Med. Chem. 2015, 31, 302–313. [Google Scholar] [CrossRef]
- Mintzer, J.; Armstrong, D.; Faison, W.E. Combination Pharmacotherapy in Alzheimer’s Disease. Neurocircuitry 2003, 5, 299–305. [Google Scholar]
- Pontiki, E.; Hadjipavlou-Litina, D. Multi-Target Cinnamic Acids for Oxidative Stress and Inflammation: Design, Synthesis, Biological Evaluation and Modeling Studies. Molecules 2018, 24, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Thompson, J.D.; Higgins, D.G.; Lopez, R.; McWilliam, H.; et al. A Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 11, 539. [Google Scholar] [CrossRef]
- Liargkova, T.; Eleftheriadis, N.; Dekker, F.J.; Voulgari, E.; Avgoustakis, K.; Sagnou, M.; Mavroidi, B.; Pelecanou, M.; Hadjipavlou-Litina, D. Small Multitarget Molecules Incorporating the Enone Moiety. Molecules 2019, 24, 199. [Google Scholar] [CrossRef] [Green Version]
- Pontiki, E.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolotti, O.; Carotti, A. Design, Synthesis and Pharmacobiological Evaluation of Novel Acrylic Acid Derivatives Acting As Lipoxygenase and Cyclooxygenase-1 Inhibitors With Antioxidant and Anti-Inflammatory Activities. Eur. J. Med. Chem. 2011, 46, 191–200. [Google Scholar] [CrossRef]
- Fiser, A.; Šali, A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comp. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE–AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 950–1958. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking With a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt. A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://ambermd.org (accessed on 25 October 2020).
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.; Case, D. Antechamber, An Accessory Software Package, For Molecular Mechanical Calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring With Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | RM ± S.D. | Milog P a |
|---|---|---|
| 1k | 0.872 ± 0.039 | 6.42 |
| 1l | 0091 ± 0.036 | 3.91 |
| 1m | 0.577 ± 0.027 | 5.87 |
| 1n | 0.131 ± 0.142 | 6.41 |
| 1o | −0.848 ± 0.19 | 4.62 |
| 1p | −0.679 ± 0.035 | 6.58 |
| 1q | −0.019 ± 0.143 | 5.39 |
| 2a | −0.485 ± 0.044 | 1.91 |
| 2b | −0.576 ± 0.077 | 2.89 |
| 2c | −0.852 ± 0.059 | 0.94 |
| 3a | 0.599 ± 0.024 | 5.88 |
| 3b | 0.509 ± 0.062 | 7.58 |
| 3c | −0.658 ± 0.061 | 6.60 |
| 3d | 0.237 ± 0.06 | 8.45 |
| 3e | 0.514 ± 0.039 | 5.95 |
| 3f | 0.564 ± 0.045 | 6.04 |
| 3g | 0.235 ± 0.039 | 6.93 |
| 3h | −0.657 ± 0.013 | 5.62 |
| Curcumin | 0.175 ± 0.029 | 2.30 |
| Compounds | Milog P a | TPSA b | No Atoms | NoO,N c | No OH, NH d | No Violations | No Rotational Bonds e | Volume f | MW g | logBB h |
|---|---|---|---|---|---|---|---|---|---|---|
| 1k | 6.42 | 17.07 | 28 | 1 | 0 | 1 | 2 | 340.01 | 360.46 | 0.660 |
| 1l | 3.91 | 26.30 | 21 | 2 | 0 | 0 | 2 | 261.02 | 276.33 | 0.179 |
| 1m | 5.87 | 26.30 | 29 | 2 | 0 | 1 | 2 | 349.00 | 376.45 | 0.483 |
| 1n | 6.41 | 17.07 | 29 | 1 | 0 | 1 | 2 | 358.14 | 392.52 | 0.659 |
| 1o | 4.62 | 12.03 | 21 | 1 | 1 | 0 | 2 | 273.31 | 291.42 | 0.760 |
| 1p | 6.58 | 12.03 | 29 | 1 | 1 | 1 | 2 | 361.30 | 391.54 | 1.064 |
| 1q | 5.39 | 38.31 | 27 | 3 | 1 | 1 | 2 | 324.43 | 371.46 | 0.616 |
| 2a | 1.91 | 37.30 | 11 | 2 | 1 | 0 | 2 | 138.46 | 148.16 | 0.087 |
| 2b | 2.89 | 37.30 | 15 | 2 | 1 | 0 | 2 | 182.45 | 198.22 | 0.239 |
| 2c | 0.94 | 77.75 | 13 | 4 | 3 | 0 | 2 | 154.50 | 180.16 | −0,468 |
| 3a | 5.88 | 37.38 | 33 | 3 | 0 | 1 | 3 | 399.51 | 429.52 | 0.702 |
| 3b | 7.58 | 37.38 | 39 | 3 | 0 | 2 | 4 | 470.61 | 505.62 | 0.637 |
| 3c | 6.60 | 37.38 | 35 | 3 | 0 | 1 | 4 | 426.62 | 455.56 | 0.485 |
| 3d | 8.45 | 37.38 | 43 | 3 | 0 | 2 | 4 | 514.60 | 555.68 | 0.772 |
| 3e | 5.95 | 37.38 | 33 | 3 | 0 | 1 | 4 | 392.49 | 441.48 | 0.384 |
| 3f | 6.04 | 37.38 | 33 | 3 | 0 | 1 | 4 | 408.04 | 467.62 | 0.398 |
| 3g | 6.93 | 37.38 | 37 | 3 | 0 | 1 | 4 | 436.48 | 491.54 | 0.536 |
| 3h | 5.62 | 37.38 | 31 | 3 | 0 | 1 | 4 | 382.62 | 405.50 | 0.384 |
| Curcumin | 2.30 | 93.07 | 27 | 6 | 2 | 0 | 8 | 332.18 | 368.38 | −0.410 |
| Comp. | CYP1A2 | CYP2C19 | CYP3A4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CypRules | Swiss ADME | CypRules | Swiss ADME | CypRules | Swiss ADME | CypRules | Swiss ADME | CypRules | Swiss ADME | |
| 1a | No | No | No | No | No | No | No | No | No | No |
| 1b | No | Inh. | No | Inh. | No | Inh. | No | No | No | Inh. |
| 1c | No | No | No | Inh. | No | Inh. | No | No | No | No |
| 1d | No | Inh. | No | No | No | No | No | No | No | Inh. |
| 1e | No | No | No | No | No | No | No | Inh. | No | No |
| 1f | No | Inh. | No | No | No | No | No | No | No | No |
| 1g | No | No | No | No | No | Inh. | No | Inh. | No | No |
| 1h | No | Inh. | No | Inh. | No | Inh. | No | No | No | Inh. |
| 1i | No | Inh. | No | Inh. | No | Inh. | No | No | No | Inh. |
| 1j | No | Inh. | No | No | No | Inh. | No | Inh. | No | No |
| 2a | No | No | No | No | No | No | No | No | No | No |
| 2b | No | No | No | No | No | No | No | No | No | No |
| 2c | No | No | No | No | No | No | No | No | No | No |
| 3a | No | No | No | No | No | No | No | No | No | Inh. |
| 3b | No | No | No | No | No | No | No | No | No | Inh. |
| 3c | No | No | No | No | No | No | No | No | No | No |
| 3d | No | No | No | Inh. | No | Inh. | No | No | No | Inh. |
| 3e | No | No | No | Inh. | No | No | No | No | No | Inh. |
| 3f | No | No | No | No | No | No | No | No | No | Inh. |
| 3g | No | No | No | Inh. | No | Inh. | No | No | No | Inh. |
| 3h | No | No | No | No | No | No | No | No | No | No |
| 1k | No | No | No | No | No | No | No | No | No | No |
| 1l | No | No | No | Inh. | No | Inh. | No | Inh. | No | No |
| 1m | No | No | No | No | No | No | No | No | No | No |
| 1n | No | No | No | No | No | No | No | No | No | Inh. |
| 1o | No | No | No | Inh. | No | Inh. | No | No | No | Inh. |
| 1p | No | No | No | No | No | No | No | No | No | Inh. |
| 1q | No | No | No | Inh. | No | Inh. | No | No | No | Inh. |
| Curcumin (Keto) | No | No | No | Inh. | No | No | No | No | No | Inh. |
| Curcumin (Enol) | No | No | No | Inh. | No | No | No | No | No | Inh. |
| Ames test | Carcinogenicity | ||||||
|---|---|---|---|---|---|---|---|
| Comp. | Ames Test | TA100_10RLI | TA100_NA | TA1535_10RLI | TA1535_NA | Carcino_Mouse | Carcino_Rat |
| 1a | Mutagen | Negative | Negative | Negative | Negative | Positive | Negative |
| 1b | Mutagen | Negative | Positive | Positive | Positive | Negative | Negative |
| 1c | Mutagen | Negative | Negative | Negative | Positive | Positive | Positive |
| 1d | Mutagen | Negative | Negative | Negative | Negative | Positive | Negative |
| 1e | Mutagen | Negative | Negative | Negative | Negative | Negative | Positive |
| 1f | Mutagen | Negative | Positive | Negative | Positive | Negative | Negative |
| 1g | Mutagen | Negative | Positive | Negative | Positive | Negative | Negative |
| 1h | Mutagen | Negative | Positive | Positive | Positive | Negative | Negative |
| 1i | Mutagen | Negative | Positive | Negative | Positive | Negative | Negative |
| 1j | Mutagen | Negative | Negative | Positive | Negative | Negative | Negative |
| 2a | Mutagen | Positive | Negative | Negative | Negative | Negative | Negative |
| 2b | Mutagen | Positive | Negative | Positive | Negative | Negative | Negative |
| 2c | Mutagen | Negative | Positive | Negative | Positive | Negative | Positive |
| 3a | Mutagen | Positive | Negative | Negative | Negative | Negative | Positive |
| 3b | Mutagen | Positive | Negative | Negative | Negative | Negative | Positive |
| 3c | Mutagen | Positive | Negative | Negative | Negative | Negative | Positive |
| 3d | Non-Mutagen | Negative | Negative | Negative | Negative | Positive | Negative |
| 3e | Mutagen | Positive | Negative | Negative | Negative | Negative | Negative |
| 3f | Mutagen | Negative | Negative | Negative | Negative | Negative | Negative |
| 3g | Mutagen | Positive | Negative | Positive | Negative | Negative | Positive |
| 3h | Mutagen | Negative | Negative | Negative | Negative | Negative | Negative |
| 1k | Mutagen | Positive | Negative | Negative | Negative | Negative | Negative |
| 1l | Mutagen | Positive | Positive | Positive | Negative | Negative | Negative |
| 1m | Mutagen | Positive | Negative | Negative | Negative | Positive | Negative |
| 1n | Mutagen | Positive | Negative | Negative | Negative | Negative | Negative |
| 1o | Mutagen | Negative | Positive | Negative | Positive | Negative | Negative |
| 1p | Mutagen | Negative | Negative | Negative | Negative | Negative | Negative |
| 1q | Mutagen | Negative | Positive | Negative | Negative | Positive | Positive |
| Curcumin (Keto) | Non-Mutagen | Negative | Negative | Negative | Negative | Negative | Positive |
| Curcumin (Enol) | Mutagen | Negative | Negative | Negative | Negative | Positive | Positive |
| Drug-likeness | ||||||
|---|---|---|---|---|---|---|
| Comp. | Lipinski | Ghose | Veber | Egan | Muegge | Bioavailability Score |
| 1a | Yes * | Yes | Yes | Yes | No * | 0.55 |
| 1b | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1c | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1d | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1e | No ** | No *** | Yes | No * | No ** | 0.17 |
| 1f | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1g | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1h | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1i | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1j | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 2a | Yes | No ** | Yes | Yes | No * | 0.56 |
| 2b | Yes | Yes | Yes | Yes | No * | 0.56 |
| 2c | Yes | Yes | Yes | Yes | No * | 0.56 |
| 3a | No ** | No *** | Yes | No * | No * | 0.17 |
| 3b | Yes * | No ** | Yes | Yes | No * | 0.55 |
| 3c | No ** | No *** | Yes | No * | No * | 0.17 |
| 3d | Yes * | No * | Yes | Yes | No * | 0.55 |
| 3e | Yes | No ** | Yes | Yes | No * | 0.55 |
| 3f | Yes * | No *** | Yes | No * | No * | 0.55 |
| 3g | Yes | Yes | Yes | Yes | No * | 0.55 |
| 3h | Yes * | No *** | Yes | No * | No * | 0.55 |
| 1k | Yes * | No * | Yes | No * | No ** | 0.55 |
| 1l | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1m | Yes * | No * | Yes | Yes | No * | 0.55 |
| 1n | Yes * | No * | Yes | No * | No * | 0.55 |
| 1o | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 1p | Yes * | No ** | Yes | Yes | No * | 0.55 |
| 1q | Yes | Yes | Yes | Yes | Yes | 0.55 |
| Curcumin (Keto) | Yes | Yes | Yes | Yes | Yes | 0.55 |
| Curcumin (Enol) | Yes | Yes | Yes | Yes | Yes | 0.56 |
| LOX IC50 (μM) or (% Inhibition at 100 μM) ± SEM | AAPH Inhibition (%, at 100 μΜ) | COX-2 IC50 (μM) or (% Inhibition at 100 μM) ± SEM | AChE IC50 (μM) or (% Inhibition at 100 μM) ± SEM | |
|---|---|---|---|---|
| 1a * | 37 μM ± 0.8 | |||
| 1b * | 330 μM ± 6.5 | |||
| 1c * | 380 μM ± 5.3 | |||
| 1d * | 410 μM ± 4.2 | |||
| 1f * | 36% ± 0.9 | |||
| 1g * | 8% ± 0.2 | |||
| 1j | 25.3% ± 0.4 | 36.2 ± 1.8 | n.a | 2.3% ± 0.01 |
| 1k | 14 μΜ ± 0.2 | 100 ± 2.2 | n.a | 36% ± 1.4 |
| 1l | 10% ± 0.3 | 42 ± 1.5 | n.a | 25% ± 0.7 |
| 1m | 50.5 μΜ ± 1.8 | 100 ± 2.8 | n.a | 32% ± 0.6 |
| 1n | 57.5 μΜ ± 0.9 | 100 ± 2.5 | n.a | 18% ± 0.3 |
| 1o | 10 μΜ ± 0.1 | 8 ± 0.01 | n.a | n.a |
| 1p | 35 μΜ ± 1.5 | 100 ± 3.3 | n.a | 26.60% ± 0.9 |
| 1q | 3% ± 0.0 | 13 ± 0.2 | n.a | 16.50% ± 0.4 |
| 2a | 56 μΜ ± 1.6 | 78 ±1.6 | n.a | 4.6% ± 0.2 |
| 2b | 27.5 μΜ ± 0.7 | 78 ± 0.7 | n.a | n.a |
| 2c | 10 μΜ ± 0.6 | 80 ± 2.2 | 57.5 μΜ ± 1.1 | 14% ± 0.1 |
| 3a | 50 μΜ ± 1.4 | 91 ± 1.9 | n.a | 85 μΜ ± 1.3 |
| 3b | 55 μΜ ± 1.1 | 90 ± 2.8 | n.a | 17% ± 0.6 |
| 3c | 37.5 μΜ ± 0.7 | 88 ± 2.0 | n.a | 29% ± 0.8 |
| 3d | 63.5 μΜ ± 1.5 | 84 ± 1.7 | n.a | 31% ± 0.5 |
| 3e | 52 μΜ ± 1.9 | 91 ± 2.5 | 19% ± 0.2 | 70 μΜ ± 3.1 |
| 3f | 55 μΜ ± 1.3 | 90 ± 2.4 | n.a | 24% ± 1.2 |
| 3g | 82 μΜ ± 2.2 | 53 ± 1.6 | n.a | 100 μΜ ± 2.5 |
| 3h | 34 μΜ ± 0.7 | 93 ± 2.2 | # | # |
| Curcumin | 38% ± 0.4 | 78 ± 1.8 | 19% ± 0.4 | 34% ± 1.3 |
| NDGA | 0.45 μΜ ± 0.03 | |||
| Trolox | 92 ± 2.1 | |||
| Indomethacin | 1.12 μΜ ± 0.1 | |||
| Tacrine | 98% ± 1.5/(0.03 μΜ ± 0.01) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chainoglou, E.; Siskos, A.; Pontiki, E.; Hadjipavlou-Litina, D. Hybridization of Curcumin Analogues with Cinnamic Acid Derivatives as Multi-Target Agents Against Alzheimer’s Disease Targets. Molecules 2020, 25, 4958. https://doi.org/10.3390/molecules25214958
Chainoglou E, Siskos A, Pontiki E, Hadjipavlou-Litina D. Hybridization of Curcumin Analogues with Cinnamic Acid Derivatives as Multi-Target Agents Against Alzheimer’s Disease Targets. Molecules. 2020; 25(21):4958. https://doi.org/10.3390/molecules25214958
Chicago/Turabian StyleChainoglou, Eirini, Argyris Siskos, Eleni Pontiki, and Dimitra Hadjipavlou-Litina. 2020. "Hybridization of Curcumin Analogues with Cinnamic Acid Derivatives as Multi-Target Agents Against Alzheimer’s Disease Targets" Molecules 25, no. 21: 4958. https://doi.org/10.3390/molecules25214958