
The Budesonide-Hydroxypropyl-β-Cyclodextrin Complex Attenuates ROS Generation, IL-8 Release and Cell Death Induced by Oxidant and Inflammatory Stress. Study on A549 and A-THP-1 Cells

Abstract
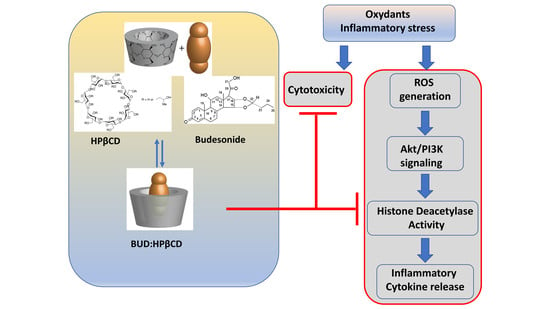
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
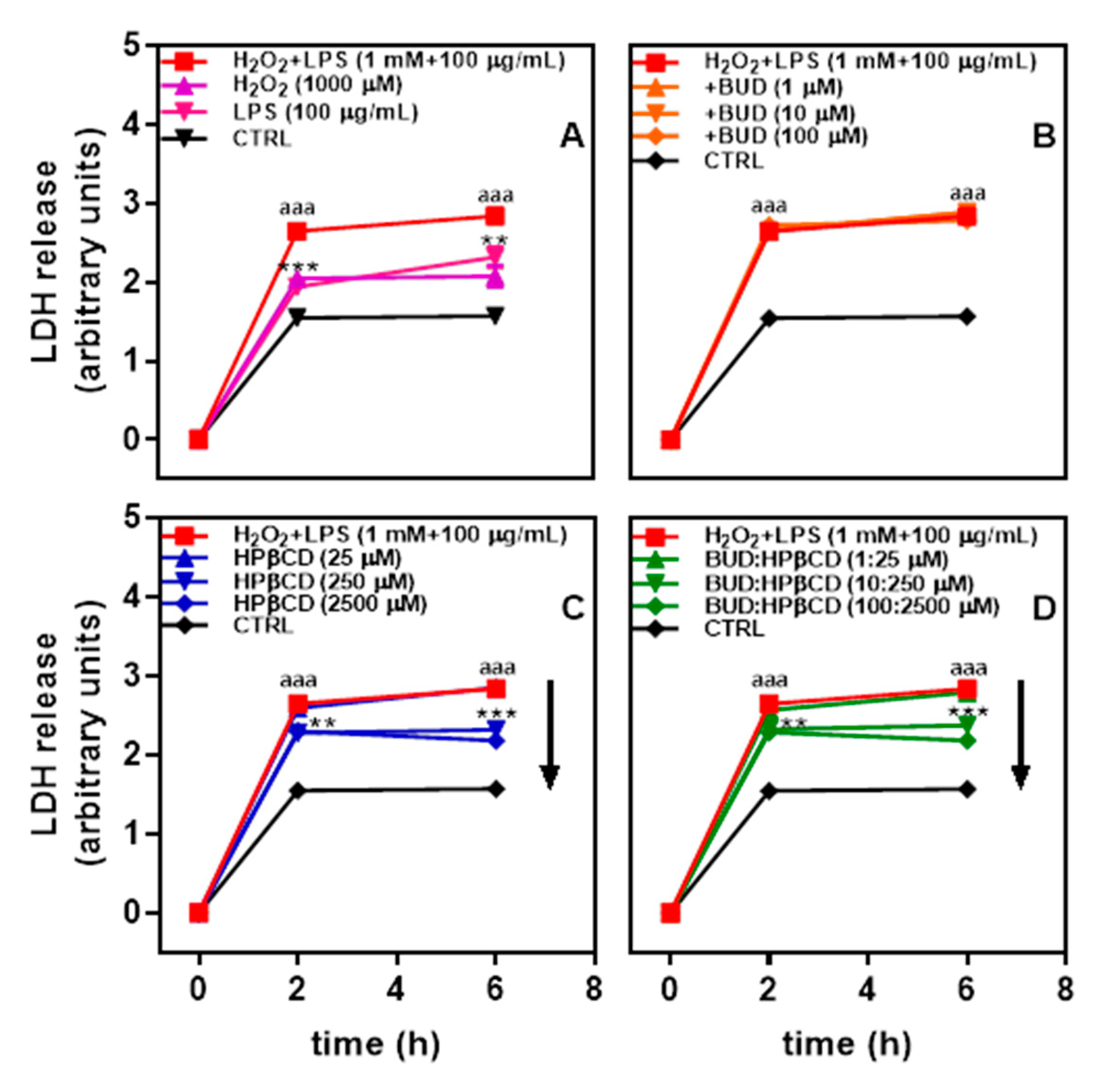
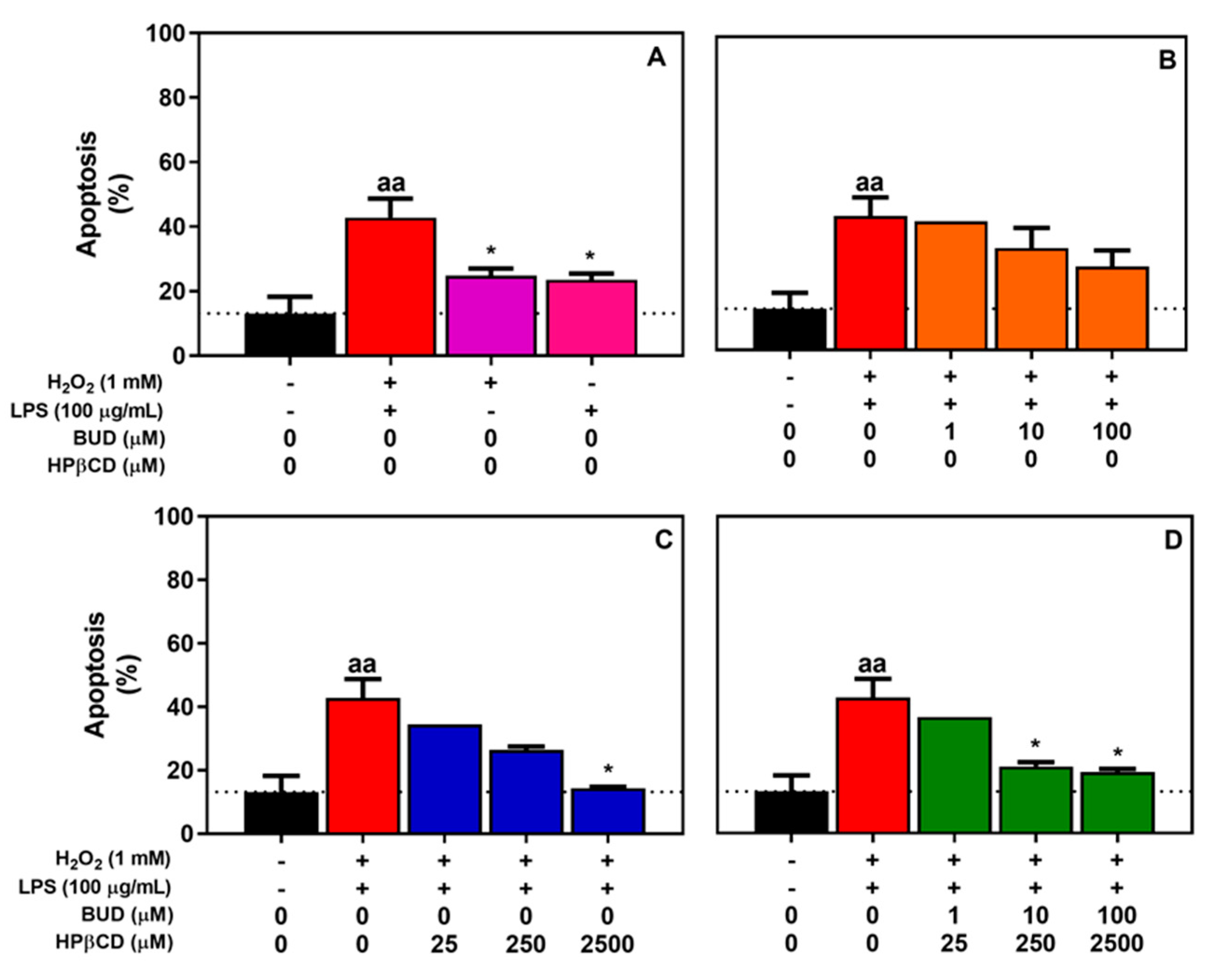
2.1. BUD:HPβCD Complex and HPβCD Attenuate H2O2 + LPS-Induced Cytotoxicity
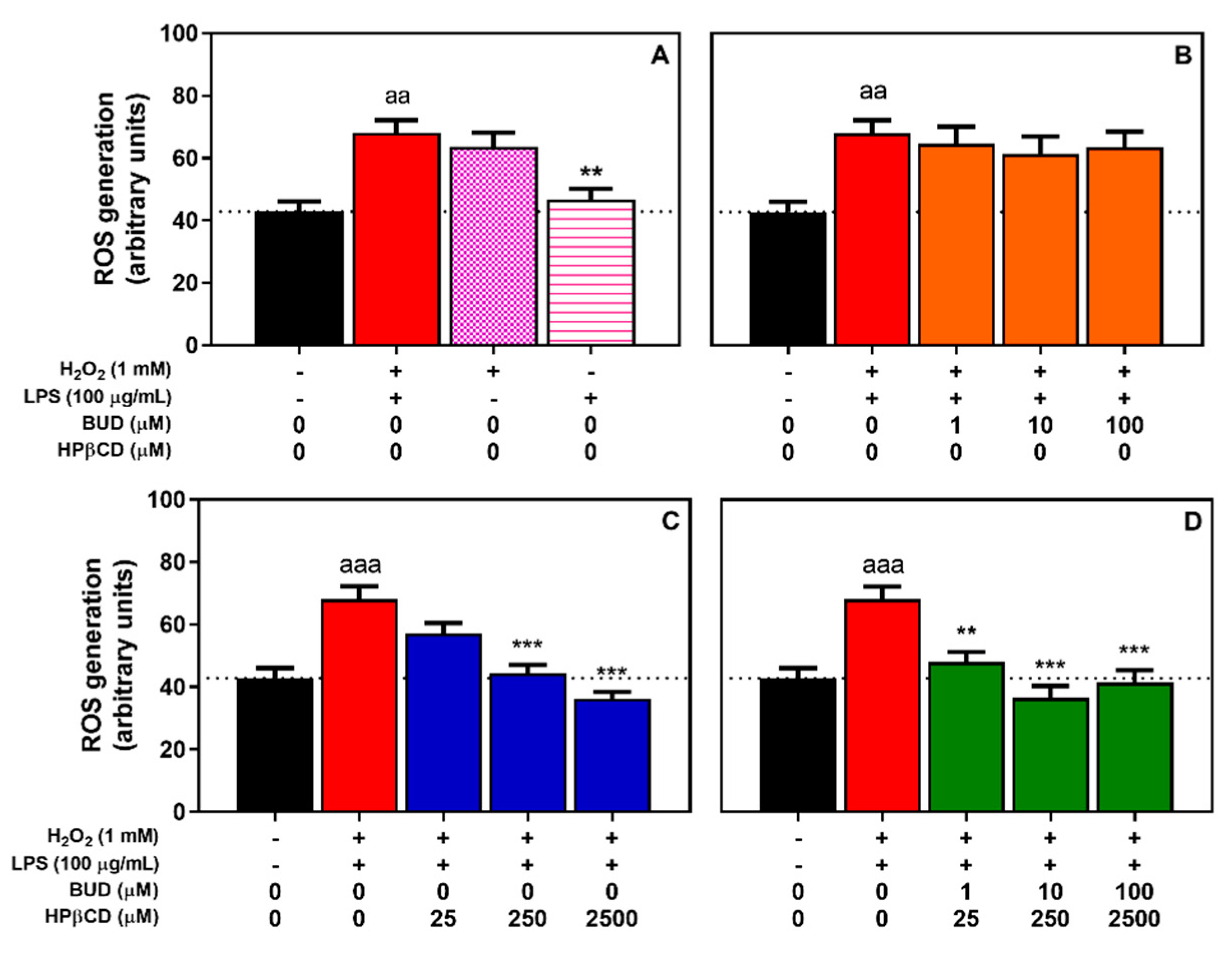
2.2. BUD:HPβCD Complex and HPβCD Protect against H2O2 + LPS-Induced Oxidative Stress in A549 Cells: Dose and Time-Dependent Effects
2.3. BUD:HPβCD Complex and HPβCD Attenuate H2O2 + LPS-Induced Phosphoinositide-3-Kinase/Akt Signaling in A549 Cells
2.4. Cholesterol Might Limit the Effects of BUD:HPβCD Complex and HPβCD in ROS Generation and PI3K/Akt Signaling Induced by H2O2 + LPS
2.4.1. Cholesterol Content Might Influence the Effects of the BUD:HPβCD Complex and HPβCD in ROS Generation Induced by H2O2 + LPS
2.4.2. Cholesterol Limits the Effects of the BUD:HPβCD Complex and HPβCD in PI3K/Akt Signaling Induced by H2O2 + LPS
2.5. BUD:HPβCD Complex and HPβCD Protect against H2O2 + LPS-Induced Increase in HDAC2 Phosphorylation in A549 Cells
2.6. BUD:HPβCD Complex and HPβCD Attenuate H2O2 + LPS-Induced Inflammatory Response in THP-1 Cells
3. Discussion
4. Material and Methods
4.1. Material
4.2. BUD:HPβCD Complex Stock Solution Preparation and Characterization
4.3. Cell Handling
4.4. Cytotoxicity Studies
4.4.1. Lactate Dehydrogenase Assay
4.4.2. HOECHST Nuclear Staining
4.5. DCF Assay for Determining ROS Generation
4.6. Evaluation of Protein Quantity by Western Blotting
4.7. Evaluation of Inflammatory Cytokine (IL-8) Expression by Sandwich ELISA
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cazzola, M.; Rogliani, P.; Stolz, D.; Matera, M.G. Pharmacological treatment and current controversies in COPD. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nici, L.; Mammen, M.J.; Charbek, E.; Alexander, P.E.; Au, D.H.; Boyd, C.M.; Criner, G.J.; Donaldson, G.C.; Dreher, M.; Fan, V.S.; et al. Pharmacologic Management of Chronic Obstructive Pulmonary Disease. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit Care Med. 2020, 201, e56–e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marwick, J.A.; Adcock, I.M.; Chung, K.F. Overcoming reduced glucocorticoid sensitivity in airway disease: Molecular mechanisms and therapeutic approaches. Drugs 2010, 70, 929–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Glucocorticosteroids. Handb. Exp. Pharmacol. 2017, 237, 93–115. [Google Scholar]
- Mei, D.; Tan, W.S.D.; Wong, W.S.F. Pharmacological strategies to regain steroid sensitivity in severe asthma and COPD. Curr. Opin. Pharmacol. 2019, 46, 73–81. [Google Scholar] [CrossRef]
- Bi, J.; Min, Z.; Yuan, H.; Jiang, Z.; Mao, R.; Zhu, T.; Liu, C.; Zeng, Y.; Song, J.; Du, C.; et al. PI3K inhibitor treatment ameliorates the glucocorticoid insensitivity of PBMCs in severe asthma. Clin. Transl. Med. 2020, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Pelaia, G.; Vatrella, A.; Busceti, M.T.; Fabiano, F.; Terracciano, R.; Matera, M.G.; Maselli, R. Molecular and cellular mechanisms underlying the therapeutic effects of budesonide in asthma. Pulm. Pharmacol. Ther. 2016, 40, 15–21. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Lipworth, B.; Brattsand, R. Benefit: Risk Profile of Budesonide in Obstructive Airways Disease. Drugs 2019, 79, 1757–1775. [Google Scholar] [CrossRef] [Green Version]
- Janson, C. Treatment with inhaled corticosteroids in chronic obstructive pulmonary disease. J. Thorac. Dis. 2020, 12, 1561–1569. [Google Scholar] [CrossRef]
- Dufour, G.; Bigazzi, W.; Wong, N.; Boschini, F.; de Tullio, P.; Piel, G.; Cataldo, D.; Evrard, B. Interest of cyclodextrins in spray-dried microparticles formulation for sustained pulmonary delivery of budesonide. Int. J. Pharm. 2015, 495, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Saokham, P.; Sa Couto, A.R. Self-association of cyclodextrins and cyclodextrin complexes in aqueous solutions. Int. J. Pharm. 2019, 560, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Crini, G. Review: A history of cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef]
- Braga, S.S. Cyclodextrins: Emerging Medicines of the New Millennium. Biomolecules 2019, 9, 801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [Green Version]
- di Cagno, M.P. The Potential of Cyclodextrins as Novel Active Pharmaceutical Ingredients: A Short Overview. Molecules 2016, 22, 1. [Google Scholar] [CrossRef] [Green Version]
- Gould, S.; Scott, R.C. 2-Hydroxypropyl-beta-cyclodextrin (HP-beta-CD): A toxicology review. Food Chem. Toxicol. 2005, 43, 1451–1459. [Google Scholar] [CrossRef]
- Malanga, M.; Szeman, J.; Fenyvesi, E.; Puskas, I.; Csabai, K.; Gyemant, G.; Fenyvesi, F.; Szente, L. “Back to the Future”: A New Look at Hydroxypropyl Beta-Cyclodextrins. J. Pharm. Sci. 2016, 105, 2921–2931. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Role of HDAC2 in the pathophysiology of COPD. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasserre, R.; Guo, X.J.; Conchonaud, F.; Hamon, Y.; Hawchar, O.; Bernard, A.M.; Soudja, S.M.; Lenne, P.F.; Rigneault, H.; Olive, D.; et al. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nat. Chem. Biol. 2008, 4, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Calay, D.; Vind-Kezunovic, D.; Frankart, A.; Lambert, S.; Poumay, Y.; Gniadecki, R. Inhibition of Akt signaling by exclusion from lipid rafts in normal and transformed epidermal keratinocytes. J. Investig. Dermatol. 2010, 130, 1136–1145. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, J. Spatiotemporal analysis of differential Akt regulation in plasma membrane microdomains. Mol. Biol. Cell. 2008, 19, 4366–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, A.G.; Bayiha, J.C.; Dufour, G.; Cataldo, D.; Evrard, B.; Silva, L.C.; Deleu, M.; Mingeot-Leclercq, M.P. Changes in membrane biophysical properties induced by the Budesonide/Hydroxypropyl-beta-cyclodextrin complex. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1930–1940. [Google Scholar] [CrossRef]
- Nakayama, T.; Church, D.F.; Pryor, W.A. Quantitative analysis of the hydrogen peroxide formed in aqueous cigarette tar extracts. Free Radic. Biol. Med. 1989, 7, 9–15. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kagota, S.; Haginaka, J.; Kunitomo, M. Peroxynitrite-generating species: Good candidate oxidants in aqueous extracts of cigarette smoke. Jpn. J. Pharmacol. 2000, 82, 78–81. [Google Scholar] [CrossRef] [Green Version]
- Valenca, S.S.; Silva, B.F.; Lopes, A.A.; Romana-Souza, B.; Marinho Cavalcante, M.C.; Lima, A.B.; Goncalves Koatz, V.L.; Porto, L.C. Oxidative stress in mouse plasma and lungs induced by cigarette smoke and lipopolysaccharide. Environ. Res. 2008, 108, 199–204. [Google Scholar] [CrossRef]
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marwick, J.A.; Caramori, G.; Stevenson, C.S.; Casolari, P.; Jazrawi, E.; Barnes, P.J.; Ito, K.; Adcock, I.M.; Kirkham, P.A.; Papi, A. Inhibition of PI3Kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am. J. Respir. Crit Care Med. 2009, 179, 542–548. [Google Scholar] [CrossRef] [Green Version]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Hammoud, Z.; Khreich, N.; Auezova, L.; Fourmentin, S.; Elaissari, A.; Greige-Gerges, H. Cyclodextrin-membrane interaction in drug delivery and membrane structure maintenance. Int. J. Pharm. 2019, 564, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Revuelta, A.; Sanchez-Gallego, J.I.; Hernandez-Hernandez, A.; Sanchez-Yague, J.; Llanillo, M. Membrane cholesterol contents influence the protective effects of quercetin and rutin in erythrocytes damaged by oxidative stress. Chem. Biol. Interact. 2006, 161, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, F.; Chen, J.; Huang, S.; Chen, J.; Huang, J.; Li, N.; Sun, S.; Chu, X.; Zha, L. Soyasaponin Bb inhibits the recruitment of toll-like receptor 4 (TLR4) into lipid rafts and its signaling pathway by suppressing the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent generation of reactive oxygen species. Mol. Nutr. Food Res. 2016, 60, 1532–1543. [Google Scholar] [CrossRef]
- Wu, J.; Liu, C.; Zhang, L.; Qu, C.H.; Sui, X.L.; Zhu, H.; Huang, L.; Xu, Y.F.; Han, Y.L.; Qin, C. Histone deacetylase-2 is involved in stress-induced cognitive impairment via histone deacetylation and PI3K/AKT signaling pathway modification. Mol. Med. Rep. 2017, 16, 1846–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.J.; Li, Z.H.; Zhang, Y.; Zhong, X.N.; He, Z.Y.; Zhou, J.H.; Chen, S.N.; Feng, Y. Theophylline and dexamethasone in combination reduce inflammation and prevent the decrease in HDAC2 expression seen in monocytes exposed to cigarette smoke extract. Exp. Ther. Med. 2020, 19, 3425–3431. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Farkas, L.; Wolf, K.; Kratzel, K.; Eissner, G.; Pfeifer, M. Differences in LPS-induced activation of bronchial epithelial cells (BEAS-2B) and type II-like pneumocytes (A-549). Scand. J. Immunol. 2002, 56, 294–302. [Google Scholar] [CrossRef]
- Bosshart, H.; Heinzelmann, M. THP-1 cells as a model for human monocytes. Ann. Transl. Med. 2016, 4, 438. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Piao, J.; Chen, L.; Quan, T.; Li, L.; Quan, C.; Piao, Y.; Jin, T.; Lin, Z. Superior efficacy of co-treatment with the dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A against NSCLC. Oncotarget 2016, 7, 60169–60180. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.S.; McCartney, M.M.; Falcon, A.K.; Linderholm, A.L.; Ebeler, S.E.; Kenyon, N.J.; Harper, R.H.; Schivo, M.; Davis, C.E. Modeling cellular metabolomic effects of oxidative stress impacts from hydrogen peroxide and cigarette smoke on human lung epithelial cells. J. Breath Res. 2019, 13, 036014. [Google Scholar] [CrossRef]
- Hasday, J.D.; Bascom, R.; Costa, J.J.; Fitzgerald, T.; Dubin, W. Bacterial endotoxin is an active component of cigarette smoke. Chest 1999, 115, 829–835. [Google Scholar] [CrossRef]
- Volobuef, C.; Moraes, C.M.; Nunes, L.A.; Cereda, C.M.; Yokaichiya, F.; Franco, M.K.; Braga, A.F.; De, P.E.; Tofoli, G.R.; Fraceto, L.F.; et al. Sufentanil-2-hydroxypropyl-beta-cyclodextrin inclusion complex for pain treatment: Physicochemical, cytotoxicity, and pharmacological evaluation. J. Pharm. Sci. 2012, 101, 3698–3707. [Google Scholar] [CrossRef]
- Lachowicz, M.; Stanczak, A.; Kolodziejczyk, M. Characteristic of Cyclodextrins: Their role and use in the pharmaceutical technology. Curr. Drug Targets 2020. [Google Scholar] [CrossRef] [PubMed]
- Anraku, M.; Iohara, D.; Wada, K.; Taguchi, K.; Maruyama, T.; Otagiri, M.; Uekama, K.; Hirayama, F. Antioxidant and renoprotective activity of 2-hydroxypropyl-beta-cyclodextrin in nephrectomized rats. J. Pharm. Pharmacol. 2016, 68, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, S.; Grebe, A.; Bakke, S.S.; Bode, N.; Halvorsen, B.; Ulas, T.; Skjelland, M.; De, N.D.; Labzin, L.I.; Kerksiek, A.; et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci. Transl. Med. 2016, 8, 333ra50. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Nicolas, J.M.; Rodriguez-Bonilla, P.; Garcia-Carmona, F. Cyclodextrins and antioxidants. Crit Rev. Food Sci. Nutr. 2014, 54, 251–276. [Google Scholar] [CrossRef]
- Gross, N.J.; Barnes, P.J. New Therapies for Asthma and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit Care Med. 2017, 195, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; de Vries, A.H.; Marrink, S.J. Computational microscopy of cyclodextrin mediated cholesterol extraction from lipid model membranes. Sci. Rep. 2013, 3, 2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K.; Nishimoto, Y.; Kimura, G.; Masuko, T.; Barnes, P.J.; Ito, K.; Kizawa, Y. Repeated lipopolysaccharide exposure causes corticosteroid insensitive airway inflammation via activation of phosphoinositide-3-kinase delta pathway. Biochem. Biophys. Rep. 2016, 7, 367–373. [Google Scholar] [PubMed]
- Zheng, X.; Zhang, W.; Hu, X. Different concentrations of lipopolysaccharide regulate barrier function through the PI3K/Akt signalling pathway in human pulmonary microvascular endothelial cells. Sci. Rep. 2018, 8, 9963. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- George, S.M.; Gaylor, J.D.; Leadbitter, J.; Grant, M.H. The effect of betacyclodextrin and hydroxypropyl betacyclodextrin incorporation into plasticized poly(vinyl chloride) on its compatibility with human U937 cells. J. Biomed. Mater. Res. B Appl. Biomater. 2011, 96, 310–315. [Google Scholar] [CrossRef]
- Matassoli, F.L.; Leao, I.C.; Bezerra, B.B.; Pollard, R.B.; Lutjohann, D.; Hildreth, J.E.K.; Arruda, L.B. Hydroxypropyl-Beta-Cyclodextrin Reduces Inflammatory Signaling from Monocytes: Possible Implications for Suppression of HIV Chronic Immune Activation. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishi, M.; Ozasa, K.; Kobiyama, K.; Ohata, K.; Kitano, M.; Taniguchi, K.; Homma, T.; Kobayashi, M.; Sato, A.; Katakai, Y.; et al. Hydroxypropyl-beta-cyclodextrin spikes local inflammation that induces Th2 cell and T follicular helper cell responses to the coadministered antigen. J. Immunol. 2015, 194, 2673–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higham, A.; Karur, P.; Jackson, N.; Cunoosamy, D.M.; Jansson, P.; Singh, D. Differential anti-inflammatory effects of budesonide and a p38 MAPK inhibitor AZD7624 on COPD pulmonary cells. Int. J. Chronic Obstruct. Pulmon. Dis. 2018, 13, 1279–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, V.J.; Rao, V.M.; Zannou, E.A.; Zia, V. Mechanisms of drug release from cyclodextrin complexes. Adv. Drug Deliv. Rev. 1999, 36, 3–16. [Google Scholar] [CrossRef]
- Dahlstrom, K.; Thorsson, L.; Larsson, P.; Nikander, K. Systemic availability and lung deposition of budesonide via three different nebulizers in adults. Ann. Allergy Asthma Immunol. 2003, 90, 226–232. [Google Scholar] [CrossRef]
- Thorsson, L.; Edsbacker, S.; Conradson, T.B. Lung deposition of budesonide from Turbuhaler is twice that from a pressurized metered-dose inhaler P-MDI. Eur. Respir. J. 1994, 7, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.; Vanbever, R. Preclinical models for pulmonary drug delivery. Expert Opin. Drug Deliv. 2009, 6, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Dufour, G.; Evrard, B.; de Tullio, P. 2D-Cosy NMR Spectroscopy as a Quantitative Tool in Biological Matrix: Application to Cyclodextrins. AAPS J. 2015, 17, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Lemaire, S.; Mingeot-Leclercq, M.P.; Tulkens, P.M.; Van Bambeke, F. Study of macrophage functions in murine J774 cells and human activated THP-1 cells exposed to oritavancin, a lipoglycopeptide with high cellular accumulation. Antimicrob. Agents Chemother. 2014, 58, 2059–2066. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, S.L.; Albert, M.; Paquot, A.; Muccioli, G.G.; Tyteca, D.; Mingeot-Leclercq, M.P. Membrane cholesterol delays cellular apoptosis induced by ginsenoside Rh2, a steroid saponin. Toxicol. Appl. Pharmacol. 2018, 352, 59–67. [Google Scholar] [CrossRef]
- Wang, H.; Joseph, J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Pick, E.; Keisari, Y. A simple colorimetric method for the measurement of hydrogen peroxide produced by cells in culture. J. Immunol. Methods 1980, 38, 161–170. [Google Scholar] [CrossRef]
- Bahorun, T.; Gressier, B.; Trotin, F.; Brunet, C.; Dine, T.; Luyckx, M.; Vasseur, J.; Cazin, M.; Cazin, J.C.; Pinkas, M. Oxygen species scavenging activity of phenolic extracts from hawthorn fresh plant organs and pharmaceutical preparations. Arzneimittelforschung 1996, 46, 1086–1089. [Google Scholar]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT-Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Matheus, N.; Hansen, S.; Rozet, E.; Peixoto, P.; Maquoi, E.; Lambert, V.; Noel, A.; Frederich, M.; Mottet, D.; de Tullio, P. An easy, convenient cell and tissue extraction protocol for nuclear magnetic resonance metabolomics. Phytochem. Anal. 2014, 25, 342–349. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. The complex has to be prepared each three months for sake of stability. |











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayiha, J.C.; Evrard, B.; Cataldo, D.; De Tullio, P.; Mingeot-Leclercq, M.-P. The Budesonide-Hydroxypropyl-β-Cyclodextrin Complex Attenuates ROS Generation, IL-8 Release and Cell Death Induced by Oxidant and Inflammatory Stress. Study on A549 and A-THP-1 Cells. Molecules 2020, 25, 4882. https://doi.org/10.3390/molecules25214882
Bayiha JC, Evrard B, Cataldo D, De Tullio P, Mingeot-Leclercq M-P. The Budesonide-Hydroxypropyl-β-Cyclodextrin Complex Attenuates ROS Generation, IL-8 Release and Cell Death Induced by Oxidant and Inflammatory Stress. Study on A549 and A-THP-1 Cells. Molecules. 2020; 25(21):4882. https://doi.org/10.3390/molecules25214882
Chicago/Turabian StyleBayiha, Jules César, Brigitte Evrard, Didier Cataldo, Pascal De Tullio, and Marie-Paule Mingeot-Leclercq. 2020. "The Budesonide-Hydroxypropyl-β-Cyclodextrin Complex Attenuates ROS Generation, IL-8 Release and Cell Death Induced by Oxidant and Inflammatory Stress. Study on A549 and A-THP-1 Cells" Molecules 25, no. 21: 4882. https://doi.org/10.3390/molecules25214882