
A Review of Molecular Imaging of Glutamate Receptors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
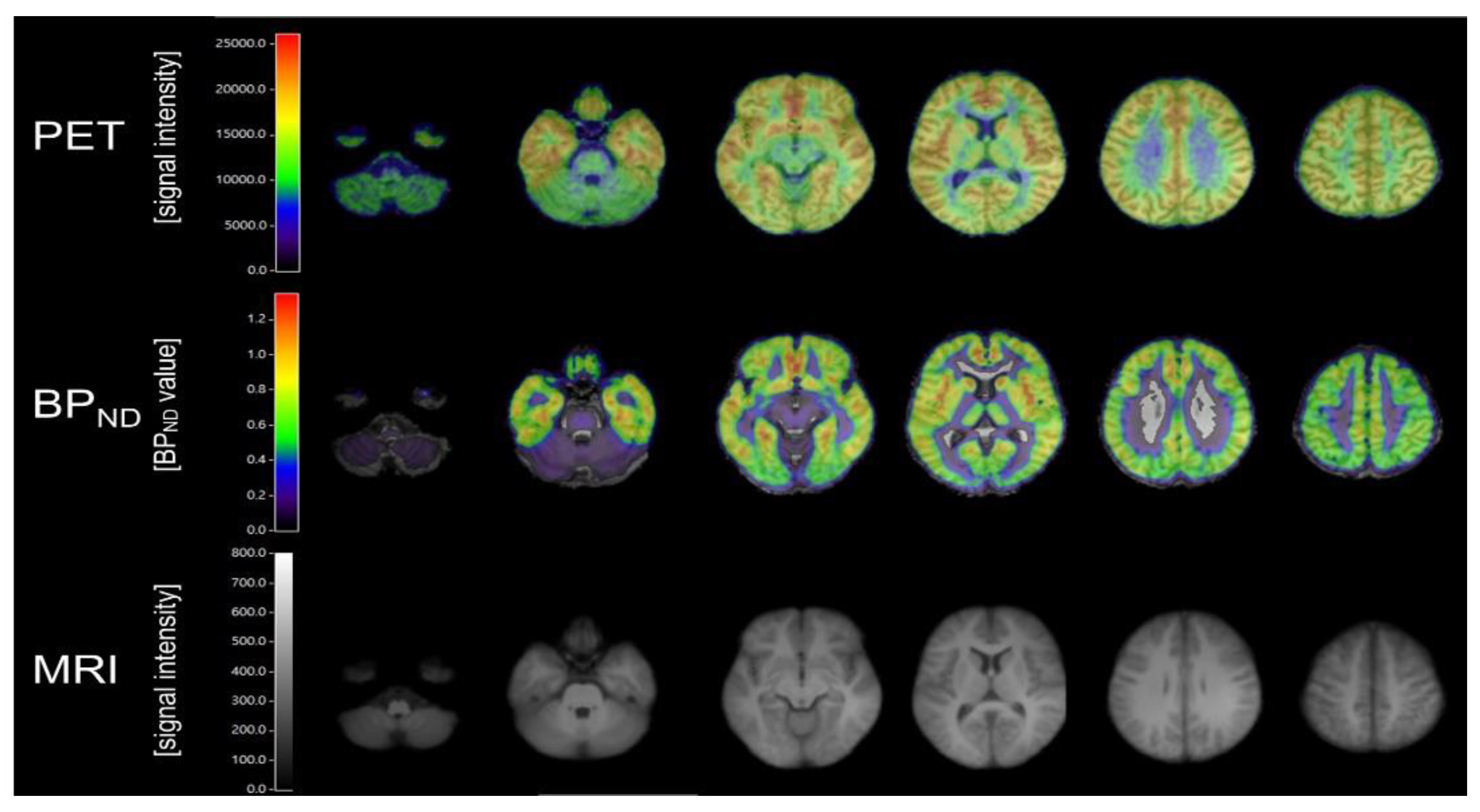{kind=link}
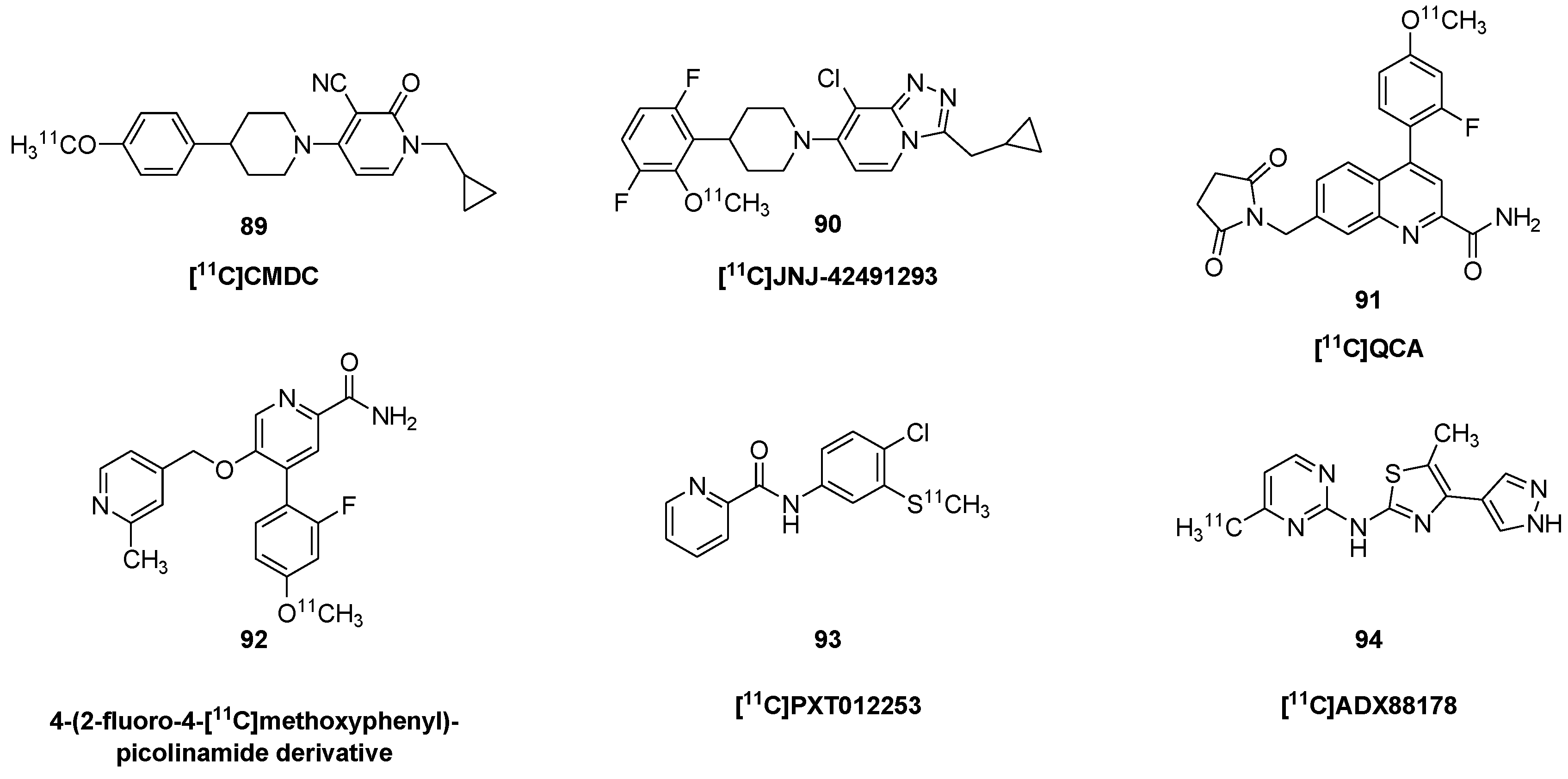{kind=link}
Abstract
:1. Introduction
A Brief Note on the Various Endpoints and Units of Binding Studies
2. Glutamate Receptors
2.1. Ionotropic Glutamate Receptors (iGluR)
2.1.1. Pharmacology of NMDA Receptor Ligands
Ligands for NMDA Receptor Ion Channels (PCP/MK801 Binding Site)
Ligands for GluN2B Sites of the NMDA Receptor
Glycine Binding Site on the GluN1 Subunit of NMDA Receptors
NMDA Allosteric Modulators
2.1.2. AMPA Receptors
2.1.3. Kainate Receptors
2.2. Metabotropic Glutamate Receptors (mGluRs)
2.2.1. Group I (mGluR1) Metabotropic Glutamate Receptors
2.2.2. Group I (mGluR5) Metabotropic Glutamate Receptors
2.2.3. Group II (mGluR2 and mGluR3) Metabotropic Glutamate Receptors
2.2.4. Group III (mGluR4, mGluR6, mGluR7 and mGluR8) Metabotropic Glutamate Receptors
3. Conclusions and Outlook
Funding
Conflicts of Interest
Abbreviations: Acronym
| Compound | No. | Name: Synonyms |
| [11C]ABP688 | 82 | 3-(6-methyl-pyridin-2-ylethynyl)-cyclohex-2-enone-O-[11C]methyl-oxime |
| [11C]ADX88178 | 94 | 5-methyl-N-(4-[11C]methylpyrimidin-2-yl)-4-(1H-pyrazol-4-yl) thiazol-2-amine |
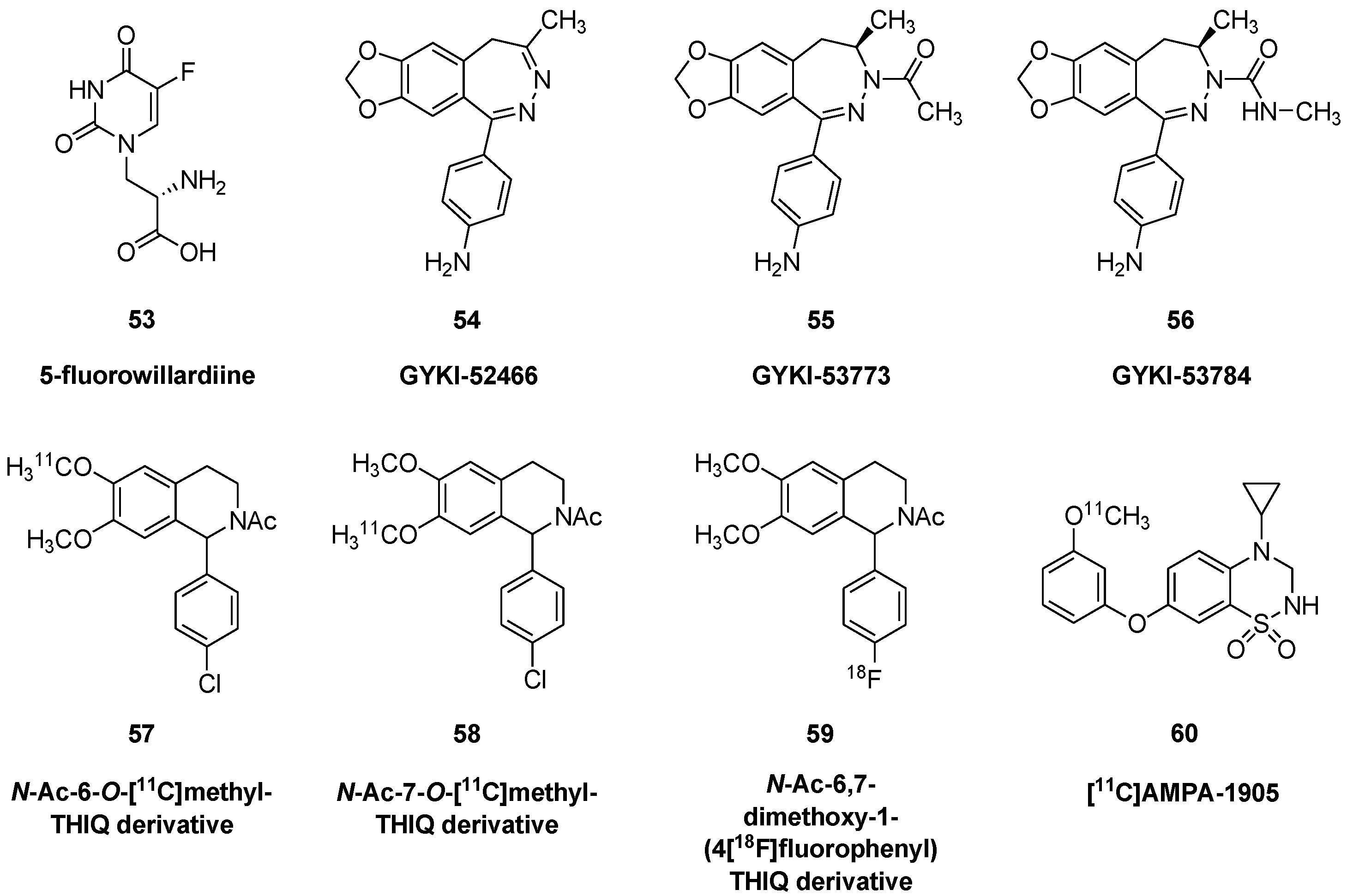
| [11C]AMPA-1905 | 60 | 4-cyclopropyl-7-(3-[11C]methoxyphenoxy)-3,4-dihydro-2H-benzo[e][1,2,4] thiadiazine 1,1-dioxide |
| BIII 277 CL | (−)-(1R,5S,2″R)-3′-Hydroxy-N-(2″-methoxypropyl)-5,9,9-trimethyl-6,7-benzomorphan | |
| Bmax | maximal binding capacity | |
| Boc | t-Boc, tert-butoxycarbonyl group | |
| BPND | binding potential relative to the no displaceable compartment | |
| Bq | Becquerel | |
| cAMP | cyclic adenosine monophosphate | |
| [11C]CBA | 45 | N-(2-[11C]methoxybenzyl) cinnamamidine |
| [11C]CMD | 89 | 1-(cyclopropylmethyl)-4-(4-[11C]methoxyphenyl)-piperidin-1-yl-2-oxo- 1,2-dihydropyridine-3-carbonitrile |
| [*I]CNS1261 | 37 | N-(1-napthyl)-N′-(3-[*I]-iodophenyl)-N-methylguanidine ([*I]CNS-1261 |
| [11C]CNS-5161 | 36 | N-(2-chloro-5-methylthiophenyl)-N′-(3-methylthio-phenyl)-N′-[11C]methylguanidine |
| Dextrometorphan | 19 | (+)-3-methoxy-N-methyl-morphinan, (9S,13S,14S)-dextrometorphan, CAS RN: [125-71-3] |
| Dextrorphan | 20 | (+)-3-hydroxy-N-methyl-morphinan, (9S,13S,14S)-dextrorphan, CAS RN: [125-73-5] |
| EEAs | excitatory amino acids | |
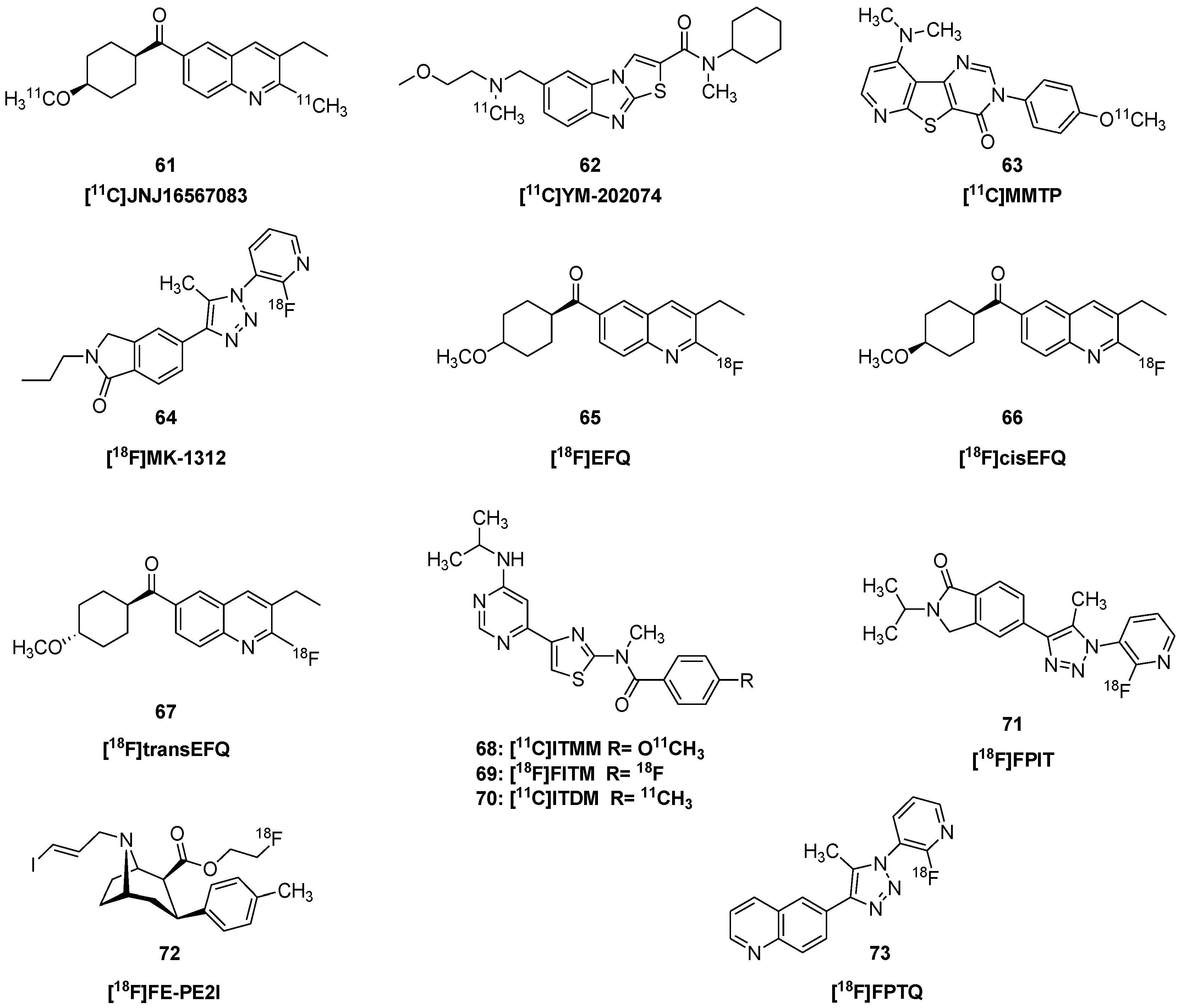
| [18F]EFQ | 65 | 3-ethyl-2-[18F]fluoroquinolin- 6-yl-(4-methoxycyclohexyl)methanone |
| [18F]FE-PE2I | 72 | (E)-N-(3-iodoprop-2-enyl)-2β-carbo-[18F]fluoroethoxy-3β-(p-tolyl)-nortropane |
| [18F]FDG | 2-deoxy-2-[18F]fluoro-D-glucose, CAS RN: [86783-82-6] | |
| [18F]FITM | 69 | 4-[18F]fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]- N-methylbenzamide |
| [18F]FMEM | 32 | 1-amino-3-[18F]fluoromethyl-5-methyl-adamantane, [18F]-memantine, |
| [18F]FNM | 33 | [18F]fluoroethylnormemantine |
| EOS | end of synthesis | |
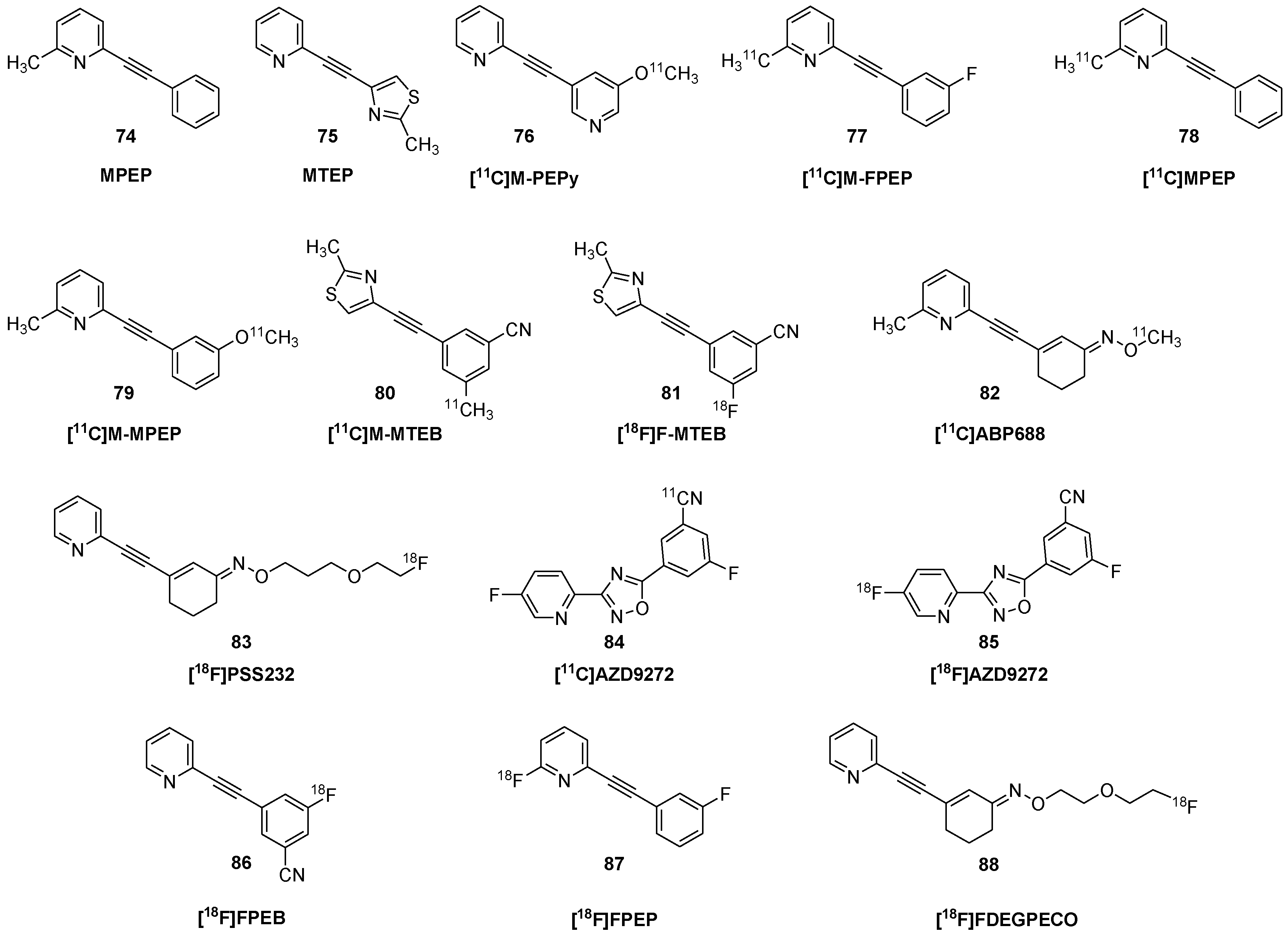
| [18F]FPEB | 86 | 3-[18F]fluoro-5-[(pyridin-3-yl)ethynyl]benzonitrile |
| [18F]FPEP | 87 | 2-[18F]fluoro-6-[(3-fluorophenyl)ethynyl]pyridine |
| [18F]FPIT | 71 | 1-(2-[18F] fluoro-3-pyridyl)-4-(2-isopropyl-1-oxo-isoindoline-5-yl)- 5-methyl- 1H-1,2,3-triazole |
| [18F]FPTQ | 73 | 6-[1-(2-[18F]fluoro-3-pyridyl)-5-methyl-1H-1,2,3-triazol-4-yl]quinolone |
| [11C]GMOM | 34 | N-(2-chloro-5-thiomethylphenyl)-N′-(3-[11C]methoxy-phenyl)-N′-methylguanidine |
| GYKI-52466 | 54 | 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine, CAS RN: [192065-56-8, as hydrochloride] |
| GYKI-53773 | 55 | (R)-(−)-1-(4-aminophenyl)-3-acetyl-4-methyl-7,8-methylenedioxy-3,4,dihydro-5H- benzodiazepine, talampanel, LY-300164, CAS RN: [161832-65-1] |
| GYKI-53784 | 56 | (R)-(−)-1-(4-aminophenyl)-3-methylcarbamoyl-4-methyl-7,8-methylenedioxy- 3,4,dihydro-5H-benzodiazepine, LY-303070, CAS RN: [161832-71-9] |
| [11C]HACH242 | 49 | N-((5-(4-fluoro-2-[11C]methoxyphenyl)pyridin-3-yl)methyl)cyclopentanamine |
| IC50 | inhibition constant required for displacement of 50% of radioligand | |
| ID | injected dose | |
| iGluR | ionotropic glutamate receptors | |
| [11C]ITDM | 70 | N-[4-[6-(isopropylamino)-pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methyl- 4-[11C]-methylbenzamide |
| [11C]ITMM | 68 | N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-4-[11C]methoxy- N-methyl-benzamide |
| [11C]JNJ16259685 | 61 | (3-ethyl-2-[11C]methyl-6-quinolinyl)(cis- 4-methoxycyclohexyl)methanone |
| KD | dissociation constant or half-saturation concentration | |
| Ki | inhibition constant in vitro | |
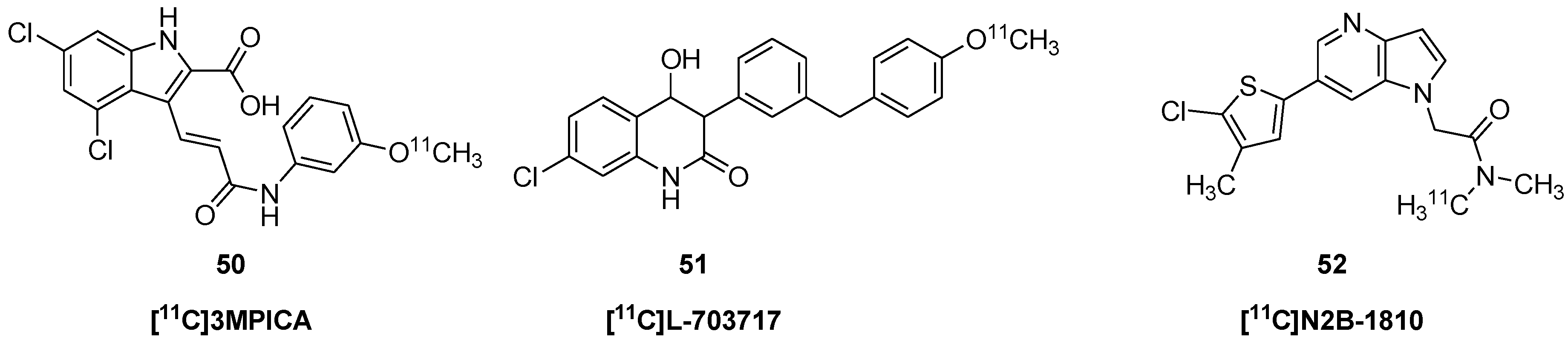
| [11C]L-703717 | 51 | 7-chloro-4-hydroxy-3-[3-(4-[11C]methoxybenzyl)phenyl-2-(1H)-quinolone |
| L-AP4 | L-2-amino-4-phosphonobutyrate | |
| Levorphan | 18 | levorphanol, (−)-3-hydroxy-N-methyl-morphinan, (9R,13R,14R)-levorphan, CAS RN: [77-07-6] |
| MDD | major depressive disorder | |
| mGluR | metabotropic glutamate receptor | |
| [11C]methyl-BIII 277 CL | 39 | (−)-(1R,5S,2″R)-3′-[11C]methoxy-N-(2″-methoxypropyl)-5,9,9-trimethyl-6,7-benzomorphan; [2R-[2α,3(R), 6α]-1,2,3,4,5,6-hexahydro-3-(2-methoxypropyl)-6,11,11-trimethyl-2,6-methano-9-[11C]methoxy-3-benzazocine |
| [11C]M-FPEP | 77 | 2-[11C]methyl-6-(3′-fluoro-phenylethynyl)-pyridine |
| [11C]Me-NB1 | 42a | 7-[11C]methoxy-3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepine-1-ol |
| [18F]MK-1312 | 64 | 1-(2-[18F]fluoro-3-pyridyl)-4-(2-propyl-1-oxo-isoindoline-5-yl)-5-methyl-1H-1,2,3-triazole |
| [11C]M-MPEP | 79 | 2-(2-(3-[11C]methoxyphenyl)ethynyl)pyridine |
| [11C]MMTP | 63 | dimethylamino-3(4-[11C]methoxyphenyl)-3H-pyrido[3′,2′:4,5]thieno- [3,2-d]pyrimidin-4-one |
| [11C]MPEP | 78 | 2-[11C]methyl-6-(2-phenylethynyl)pyridine |
| [11C]3MPICA | 50 | 3-[2-[(3-[11C]methoxyphenylamino)carbonyl]ethenyl]-4,6-dichloroindole-2-carboxylic acid |
| NAAG | N-acetylaspartylglutamic acid, CAS RN: [3106-85-2] | |
| [18F]N2B-0518 | 48 | 2-((1-(4-[18F]fluoro-3-methylphenyl)-1H-1,2,3-triazol-4-yl)methoxy)-5-methoxypyrimidine |
| NB1 | 3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepine-1,7-diol | |
| [11C]NBA | 46 | N-(2-[11C]methoxybenzyl)-2-naphthamidine |
| NMDA | N-methyl-D-aspartic acid, CAS RN: [6384-92-5] | |
| NMDAR | N-methyl-D-aspartate receptor | |
| [11C]N2B-1810 | 52 | 2-(6-(5-chloro-4-methylthiophen-2-yl)-1H-pyrrolo[3,2-b]pyridin-1-yl)-N-(λ1-methyl-11C)-N-methyl-acetamide |
| [11C]NR2B-SMe | 44 | [S-methyl-11C](±)-7-methoxy-3-(4-(4-(methylthio)phenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol |
| [18F]OF-NB1 | 42b | 3-(4-(2-[18F]fluoro-phenyl)butyl)-2,3,4,5-tetrahydro-1H-3-benzazepine-1,7-diol |
| [18F]PF-NB1 | 42c | 3-(4-(4-[18F]phenyl)butyl)-2,3,4,5-tetrahydro-1H-3-benzazepine-1,7-diol |
| PCP | 23 | phencyclidine, CAS RN: [77-10-1] |
| [18F]FPDEGPECO | 88 | (E)-3-(pyridin-2-ylethynyl)cyclohex-2-enone O-2-(2-[18]F-fluoroethoxy)ethyl oxime |
| PET | positron emission tomography | |
| [18F]PK-209 | 35 | ([3-(2-chloro-5-(methylthio)phenyl)-1-(3-([18F]fluoromethoxy)phenyl)- 1-methylguanidine] |
| [18F]PSS232 | 83 | (E)-3-(pyridine-2-yl-etheynyl-1)-cyclohex-2-enon-O-(3-(2-[18F]-fluoroethoxy)propyl) oxime |
| PTSD | post-traumatic stress disorder | |
| [11C]PTX012253 | 93 | N-(4-chloro-3-([11C]methylthio)phenyl)picolinamide, [11C]KALB012 |
| [11C]QBA | 47 | N-(2-[11C]methoxybenzyl)quinoline-3-carboxamidine |
| [11C]QCA | 91 | 7-((2,5-dioxopyrrolidin-1-yl)methyl)-4-(2-fluoro- 4-[11C]methoxyphenyl) quinoline-2-carboxamide |
| [11C]Ro 04-5595 | 43 | 1-(2-(4-chlorophenyl)ethyl)-7-hydroxy-6-methoxy-2-[11C]methyl-1,2,3,4-tetrahydro- isoquinoline |
| SCAAs | sulfur-containing amino acids | |
| SKF10,047 | N-allyl-normetazocine; 2′-hydroxy-5,9-dimethyl-N2-allyl-6,7-benzomorphan; CAS: [825594-24-9] | |
| SPECT | single photon emission computed tomography | |
| SUV | standard uptake value | |
| TBAOH | tetrabutylammonium hydroxide, CAS RN: [2052-49-5] | |
| TCP | N-(1-[thienyl]cyclohexyl)3,4-piperidine, tenocyclidine, CAS RN: [2150098-1] | |
| Versidyne | 1-(2-(4-chlorophenyl)ethyl)-6,7-dimethoxy-2-methyl-1,2,3,4-tetrahydro-isoquinoline, methopholin, NIH-7672, CAS RN: [2154-02-1] | |
| VT | total distribution volume | |
| [11C]YM-202074 | 62 | N-cyclohexyl-6-{[N-(2-methoxyethyl)-N-[11C]methylamino]methyl}-N-methyl- thiazolo[3,2-a]-benz imidazole-2-carboxamide |
References
- Kerkut, G.A.; Walker, R.J. The effect of L-glutamate, acetylcholine and gamma-aminobutyric acid on the miniature end-plate potentials and contractures of the coxal muscles of the cockroach, Periplaneta Americana. Comp. Biochem. Physiol. 1966, 17, 435–454. [Google Scholar] [CrossRef]
- Gerschenfeld, H.M. Chemical transmission in invertebrate central nervous systems and neuromuscular junctions. Physiol. Rev. 1973, 53, 1–119. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. Transport and metabolism of glutamate and GABA in neurons and glial cells. In International Review of Neurobiolog; Smythies, J.R., Bradley, R.J., Eds.; Academic Press: Cambridge, MA, USA, 1981; Volume 22, pp. 1–45. [Google Scholar]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural. Transm. (Vienna) 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Jiang, X.; Zu, Y.; Yang, Y.; Liu, Y.; Sun, X.; Xu, Z.; Ding, H.; Zhao, Q. A comprehensive description of GluN2B-selective N-methyl-D-aspartate (NMDA) receptor antagonists. Eur. J. Med. Chem. 2020, 200, 112447. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Ann. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L. Molecular pharmacology of metabotropic receptors targeted by neuropsychiatric drugs. Nat. Struct. Mol. Biol. 2019, 26, 535–544. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, S.; Fu, P.; Zhang, Z.; Lin, K.; Ko, J.K.-S.; Yung, K.K.-L. Roles of glutamate receptors in Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 4391. [Google Scholar] [CrossRef] [Green Version]
- Fern, R.; Matute, C. Glutamate receptors and white matter stroke. Neurosci. Lett. 2019, 694, 86–92. [Google Scholar] [CrossRef]
- Hanada, T. Ionotropic glutamate receptors in epilepsy: A review focusing on AMPA and NMDA receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levite, M. Glutamate receptor antibodies in neurological diseases: Anti-AMPA-GluR3 antibodies, anti-NMDA-NR1 antibodies, anti-NMDA-NR2A/B antibodies, anti-mGluR1 antibodies or anti-mGluR5 antibodies are present in subpopulations of patients with either: Epilepsy, encephalitis, cerebellar ataxia, systemic lupus erythematosus (SLE) and neuropsychiatric SLE, Sjogren’s syndrome, schizophrenia, mania or stroke. These autoimmune anti-glutamate receptor antibodies can bind neurons in few brain regions, activate glutamate receptors, decrease glutamate receptor’s expression, impair glutamate-induced signaling and function, activate blood brain barrier endothelial cells, kill neurons, damage the brain, induce behavioral/psychiatric/cognitive abnormalities and Ataxia in animal models, and can be removed or silenced in some patients by immunotherapy. J. Neural Transm. (Vienna) 2014, 121, 1029–1075. [Google Scholar] [PubMed]
- Balu, D.T. The NMDA receptor and schizophrenia: From pathophysiology to treatment. In Advances in Pharmacology; Schwarcz, R., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 76, pp. 351–382. [Google Scholar]
- Dean, B.; Gibbons, A.S.; Boer, S.; Uezato, A.; Meador-Woodruff, J.; Scarr, E.; McCullumsmith, R.E. Changes in cortical N-methyl-D-aspartate receptors and post-synaptic density protein 95 in schizophrenia, mood disorders and suicide. Aust. N. Z. J. Psychiatry 2016, 50, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksymetz, J.; Moran, S.P.; Conn, P.J. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 2017, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Murrough, J.; Abdallah, C.; Mathew, S. Targeting glutamate signalling in depression: Progress and prospects. Nat. Rev. Drug Discov. 2017, 16, 472–486. [Google Scholar] [CrossRef]
- Kalmoe, M.C.; Janski, A.M.; Zorumski, C.F.; Nagele, P.; Palanca, B.J.; Conway, C.R. Ketamine and nitrous oxide: The evolution of NMDA receptor antagonists as antidepressant agents. J. Neurol. Sci. 2020, 412, 116778. [Google Scholar] [CrossRef]
- Ueno, F.; Suzuki, T.; Nakajima, S.; Matsushita, S.; Mimura, M.; Miyazaki, T.; Takahashi, T.; Uchida, H. Alteration in AMPA receptor subunit expression and receptor binding among patients with addictive disorders: A systematic review of human postmortem studies. Neuropsychopharmacol. Rep. 2019, 39, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, L.; Schubiger, P.A.; Ametamey, S.M. Radioligands for the PET imaging of metabotropic glutamate receptor subtype 5 (mGluR5). Curr. Top. Med. Chem. 2010, 10, 1558–1568. [Google Scholar] [CrossRef]
- Majo, V.J.; Prabhakaran, J.; Mann, J.J.; Kumar, J.S. PET and SPECT tracers for glutamate receptors. Drug Discov. Today 2013, 18, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Gruber, S.; Ametamey, S.M. Imaging the glutamate receptor subtypes-Much achieved, and still much to do. Drug Discov. Today Technol. 2017, 25, 27–36. [Google Scholar] [CrossRef]
- Fu, H.; Chen, Z.; Josephson, L.; Li, Z.; Liang, S.H. Positron Emission Tomography (PET) Ligand Development for Ionotropic Glutamate Receptors: Challenges and Opportunities for Radiotracer Targeting N-Methyl-D-aspartate (NMDA), α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA), and Kainate Receptors. J. Med. Chem. 2018, 62, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, N.G.; Siegler Retchless, B.; Johnson, J.W. Molecular bases of NMDA receptor subtype-dependent properties. J Physiol. 2015, 593, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, D.J.A.; Livesey, M.R.; Hardingham, G.E. Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 2013, 74, 4–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, M.; Yong, X.L.H.; Roche, K.W.; Anggono, V. Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J. Neurochem. 2020, 154, 121–143. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Xu, Y.; Cheng, X.; Chen, X.; Xie, Y.; Zhang, L.; Wang, L.; Hu, J.; Gao, Z. The differences between GluN2A and GluN2B signaling in the brain. J. Neuro. Res. 2018, 96, 1430–1443. [Google Scholar] [CrossRef]
- Sapkota, K.; Mao, Z.; Synowicki, P.; Lieber, D.; Liu, M.; Ikezu, T.; Gautam, V.; Monaghan, D.T. GluN2D N-Methyl-D-aspartate receptor subunit contribution to the stimulation of brain activity and gamma oscillations by ketamine: Implications for schizophrenia. J. Pharm. Exp. Ther. 2016, 356, 702–711. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.; Pullan, L.M.; Romano, C.; Powel, R.J.; Salama, A.I.; Molinoff, P.B. An antagonist/partial agonist at the polyamine recognition site of the N-methyl-D-aspartate receptor that alters the properties of the glutamate recognition site. J. Pharm. Exp. Ther. 1992, 262, 539–544. [Google Scholar]
- Casy, A.F.; Parfitt, R.T. Opioid Analgesics: Chemistry and Receptors; Plenum Press: New York, NY, USA, 1986; ISBN 0-306-42130-5. [Google Scholar]
- Mohacsi, E.; Leimgruber, W.; Baruth, H. Synthesis and pharmacology of metabolically stable tert-butyl ethers of morphine and levorphanol. J. Med. Chem. 1982, 25, 1264–1266. [Google Scholar] [CrossRef]
- Wong, E.H.F.; Knight, A.R.; Woodruff, G.N. [3H]MK-801 Labels a Site on the N-Methyl-D-Aspartate Receptor Channel Complex in Rat Brain Membranes. J. Neurochem. 1988, 50, 274–281. [Google Scholar] [CrossRef]
- Bowery, N.; Wong, E.; Hudson, A. Quantitative autoradiography of [3H]-MK-801 binding sites in mammalian brain. Br. J. Pharmacol. 1988, 93, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Blin, J.; Denis, A.; Yamaguchi, T.; Crouzel, C.; MacKenzie, E.T.; Baron, J.C. PET studies of [18F]methyl-MK-801, a potential NMDA receptor complex radioligand. Neurosci. Lett. 1991, 121, 183–186. [Google Scholar] [CrossRef]
- Maragos, W.F.; Penney, J.B.; Young, A.B. Anatomic correlation of NMDA and 3H-TCP-labeled receptors in rat brain. J. Neurosci. 1988, 2, 493–501. [Google Scholar] [CrossRef]
- Ferrarese, C.; Guidotti, A.; Costa, E.; Miletich, R.S.; Rice, K.C.; de Costa, B.R.; Fulham, M.J.; Di Chiro, G. In vivo study of NMDA-sensitive glutamate receptor by fluorothienylcyclohexylpiperidine, a possible ligand for positron emission tomography. Neuropharmacology 1991, 30, 899–905. [Google Scholar] [CrossRef]
- Ouyang, X.; Mukherjee, J.; Yang, Z.-Y. Synthesis, radiosynthesis, and biological evaluation of fluorinated thienylcyclohexyl piperidine derivatives as potential radiotracers for the NMDA receptor-linked calcium ionophore. Nucl. Med. Biol. 1996, 23, 315–324. [Google Scholar] [CrossRef]
- Roger, G.; Lagnel, B.; Besret, L.; Bramoullé, Y.; Coulon, C.; Ottaviani, M.; Kassiou, M.; Bottlaender, M.; Valette, H.; Dollé, F. Synthesis, radiosynthesis and In vivo evaluation of 5-[3-(4-Benzylpiperidin-1-yl)prop-1-ynyl]-1,3-dihydrobenzoimidazol-2-[11C]one, as a potent NR1A/2B subtype selective NMDA PET radiotracer. Bioorg. Med. Chem. 2003, 11, 5401–5408. [Google Scholar] [CrossRef] [PubMed]
- Shiue, C.-Y.; Vallabhahosula, S.; Wolf, A.P.; Dewey, S.L.; Fowler, J.S.; Schlyer, D.J.; Arnett, C.A.; Zhou, Y.-G. Carbon-11 labelled ketamine-synthesis, distribution in mice and PET studies in baboons. Nucl. Med. Biol. 1997, 24, 145–150. [Google Scholar] [CrossRef]
- Hartvig, P.; Valtysson, J.; Antoni, G.; Westerberg, G.; Långström, B.; Moberg, E.R.; Øye, I. Brain kinetics of (R)- and (S)-[N-methyl-11C]ketamine in the rhesus monkey studied by positron emission tomography (PET). Nucl. Med. Biol. 1994, 21, 927–934. [Google Scholar] [CrossRef]
- Salabert, A.-S.; Fonta, C.; Fontan, C.; Adel, D.; Alonso, M.; Pestourie, C.; Belhadij-Tahar, H.; Tafani, M.; Payoux, P. Radiolabeling of [18F]fluoroethylnormemantine and initial in vivo evaluation of this innovative PET tracer for imaging the PCP sites of NMDA receptors. Nucl. Med. Biol. 2015, 42, 643–653. [Google Scholar] [CrossRef]
- Salabert, A.-S.; Mora-Ramirez, E.; Beaurain, M.; Alonso, M.; Fontan, C.; Tahar, H.B.; Boizeau, M.L.; Tafani, M.; Bardiès, M.; Payoux, P. Evaluation of [18F]FNM biodistribution and dosimetry based on whole-body PET imaging of rats. Nucl. Med. Biol. 2018, 59, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ametamey, S.M.; Samnick, S.; Leenders, K.L.; Vontobel, P.; Quack, G.; Parsons, C.G.; Schubiger, P.A. Fluorine-18 radiolabelling, biodistribution studies and preliminary PET evaluation of a new memantine derivative for imaging the NMDA receptor. J. Recept. Signal Transduct. Res. 1999, 19, 129–141. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Bruehlmeier, M.; Kneifel, S.; Kokic, M.; Honer, M.; Arigoni, M.; Buck, A.; Burger, C.; Samnick, S.; Quack, G.; et al. PET studies of 18F-memantine in healthy volunteers. Nucl. Med. Biol. 2002, 29, 227–231. [Google Scholar] [CrossRef]
- Dumont, F.; Sultana, A.; Waterhouse, R.N. Synthesis and in vitro evaluation of N,N′-diphenyl and N-naphthyl-N′-phenylguanidines as N-methyl-D-aspartate receptor ion-channel ligands. Bioorg. Med. Chem. Lett. 2002, 12, 1583–1586. [Google Scholar] [CrossRef]
- Naumiec, G.R.; Jenko, K.J.; Zoghbi, S.S.; Innis, R.B.; Cai, L.; Pike, V.W. N’-3-(Trifluoromethyl)phenyl Derivatives of N-Aryl-N’-methylguanidines as Prospective PET Radioligands for the Open Channel of the N-Methyl-D-aspartate (NMDA) Receptor: Synthesis and Structure-Affinity Relationships. J. Med. Chem. 2015, 58, 9722–9730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, R.N.; Slifstein, M.; Dumont, F.; Zhao, J.; Chang, R.C.; Sudo, Y.; Sultana, A.; Balter, A.; Laruelle, M. In vivo evaluation of [11C]N-(2-chloro-5-thiomethylphenyl)-N′- (3-methoxy-phenyl)-N′-methylguanidine ([11C]GMOM) as a potential PET radiotracer for the PCP/NMDA receptor. Nucl. Med. Biol. 2004, 31, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.J.; Schuit, R.C.; Metaxas, A.; Christiaans, J.A.M.; Kooijman, E.; Lammertsma, A.A.; van Berckel, B.N.M.; Windhorst, A.D. Synthesis, radiolabeling and preclinical evaluation of a [11C]GMOM derivative as PET radiotracer for the ion channel of the N-methyl-D-aspartate receptor. Nucl. Med. Biol. 2017, 51, 25–32. [Google Scholar] [CrossRef]
- Golla, S.S.V.; Klein, P.J.; Bakker, J.; Schuit, R.C.; Christiaans, J.A.M.; van Geest, L.; Kooijman, E.J.M.; Oropeza-Seguias, G.M.; Langermans, J.A.M.; Leysen, J.E.; et al. Preclinical evaluation of [18F]PK-209, a new PET ligand for imaging the ion-channel site of NMDA receptors. Nucl. Med. Biol. 2015, 42, 205–212. [Google Scholar] [CrossRef] [Green Version]
- van der Aart, J.; Golla, S.S.V.; van der Pluijm, M.; Schwarte, L.A.; Schuit, R.C.; Klein, P.J.; Metaxas, A.; Windhorst, A.D.; Boellaard, R.; Lammertsma, A.A.; et al. First in human evaluation of [18F]PK-209, a PET ligand for the ion channel binding site of NMDA receptors. EJNMMI Res. 2018, 8, 69. [Google Scholar] [CrossRef]
- Biegon, A.; Gibbs, A.; Alvarado, M.; Ono, M.; Taylor, S. In vitro and in vivo characterization of [3H]CNS-5161—A use-dependent ligand for the N-methyl-D-aspartate receptor in rat brain. Synapse 2007, 61, 577–586. [Google Scholar] [CrossRef]
- Dhawan, V.; Robeson, W.; Bjelke, D.; Chaly, T.; Graf, K.; Hellman, M.; Zhuo, L.; Mackay, M.; Eidelberg, D. Human radiation dosimetry for the N-Methyl-D-Aspartate receptor radioligand 11C-CNS5161. J. Nucl. Med. 2015, 56, 869–872. [Google Scholar] [CrossRef] [Green Version]
- Owens, J.; Tebbutt, A.A.; McGregor, A.L.; Kodama, K.; Magar, S.S.; Perlman, M.E.; Robins, D.J.; Durant, G.J.; McCulloch, J. Synthesis and binding characteristics of N-(1-naphthyl)-N′-(3-[125I]- iodophenyl)-N′-methylguanidine ([125I]-CNS 1261): A potential SPECT agent for imaging NMDA receptor activation. Nucl. Med. Biol. 2000, 27, 557–564. [Google Scholar] [CrossRef]
- Erlandsson, K.; Bressan, R.A.; Mulligan, R.S.; Gunn, R.N.; Cunningham, V.J.; Owens, J.; Wyper, D.; Ell, P.J.; Pilowsky, L.S. Kinetic modelling of [123I]CNS 1261—A potential SPET tracer for the NMDA receptor. Nucl. Med. Biol. 2003, 30, 441–454. [Google Scholar] [CrossRef]
- Stone, J.M.; Erlandsson, K.; Arstad, E.; Squassante, L.; Teneggi, V.; Bressan, R.A.; Krystal, J.H.; Ell, P.J.; Pilowsky, L.S. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: A [123I]CNS-1261 SPET study. Psychopharmacology 2008, 197, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Bressan, R.A.; Erlandsson, K.; Stone, J.M.; Mulligan, R.S.; Krystal, J.H.; Ell, P.J.; Pilowsky, L.S. Impact of schizophrenia and chronic antipsychotic treatment on [123I]CNS-1261 binding to N-methyl-D-aspartate receptors in vivo. Biol. Psychiatry 2005, 58, 41–46. [Google Scholar] [CrossRef]
- Pilowsky, L.S.; Bressan, R.A.; Stone, J.M.; Erlandsson, K.; Mulligan, R.S.; Krystal, J.H.; Ell, P.J. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry 2006, 11, 118–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robins, E.G.; Zhao, Y.; Khan, I.; Wilson, A.; Luthra, S.K.; Årstad, E. Synthesis and in vitro evaluation of 18F-labelled S-fluoroalkyl diarylguanidines: Novel high-affinity NMDA receptor antagonists for imaging with PET. Bioorg. Med. Chem. Lett. 2010, 20, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- McGinnity, C.J.; Hammers, A.; Riaño Barros, D.A.; Luthra, S.K.; Jones, P.A.; Trigg, W.; Micallef, C.; Symms, M.R.; Brooks, D.J.; Koepp, M.J.; et al. Initial evaluation of 18F-GE-179, a putative PET tracer for activated N-Methyl-D-Aspartate receptors. J. Nucl. Med. 2014, 55, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenberger, M.; Schroeder, F.A.; Placzek, M.S.; Carter, R.L.; Rosen, B.R.; Hooker, J.M.; Sander, C.Y. In vivo [18F]GE-179 brain signal does not show NMDA-specific modulation with drug challenges in rodents and nonhuman primates. ACS Chem. Neurosci. 2018, 9, 298–305. [Google Scholar] [CrossRef]
- McGinnity, C.J.; Årstad, E.; Beck, K.; Brooks, D.J.; Coles, J.P.; Duncan, J.S.; Galovic, M.; Hinz, R.; Hirani, E.; Howes, O.D.; et al. Comment on “In Vivo [18F]GE-179 Brain Signal Does Not Show NMDA-Specific Modulation with Drug Challenges in Rodents and Nonhuman Primates”. ACS Chem. Neurosci. 2019, 10, 768–772. [Google Scholar] [CrossRef] [Green Version]
- Vibholm, A.K.; Landau, A.M.; Alstrup, A.K.O.; Jacobsen, J.; Vang, K.; Munk, O.L.; Dietz, M.J.; Orlowski, D.; Sørensen, J.C.H.; Brooks, D.J. Activation of NMDA receptor ion channels by deep brain stimulation in the pig visualised with [18F]GE-179 PET. Brain Stimul. 2020, 13, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- López-Picón, F.; Snellman, A.; Shatillo, O.; Lehtiniemi, P.; Grönroos, T.J.; Marjamäki, P.; Trigg, W.; Jones, P.A.; Solin, O.; Pitkänen, A.; et al. Ex vivo tracing of NMDA and GABA-A receptors in rat brain after traumatic brain injury using 18F-GE-179 and 18F-GE-194 autoradiography. J. Nucl. Med. 2016, 57, 1442–1447. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.J.; Bechtel, W.D.; Grauert, M.; Harrison, P.; Merz, H.; Stransky, W. BIII 277 CL is a potent and specific ion-channel blocker of the NMDA receptor-channel complex. J. Pharm. Exp. Ther. 1995, 275, 1382–1389. [Google Scholar]
- Grauert, M.; Bechtel, W.D.; Ensinger, H.A.; Merz, H.; Carter, A.J. Synthesis and structure-activity relationships of 6,7-benzomorphan derivatives as antagonists of the NMDA receptor-channel complex. J. Med. Chem. 1997, 40, 2922–2930. [Google Scholar] [CrossRef] [PubMed]
- Kokic, M.; Honer, M.; Kessler, L.J.; Grauert, M.; Schubiger, P.A.; Ametamey, S.M. Synthesis and in vitro and in vivo evaluation of [11C]methyl-BIII277CL for imaging the PCP-binding site of the NMDA receptor by PET. J. Recept. Signal Transduc. Res. 2002, 22, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Kurosaki, F.; Haradahira, T.; Yamamoto, F.; Maeda, J.; Okauchi, T.; Suzuki, K.; Suhara, T.; Maeda, M. Synthesis of 11C-labelled bis(phenylalkyl)amines and their in vitro and in vivo binding properties in rodent and monkey brains. Biol. Pharm. Bull. 2004, 27, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haradahira, T.; Maeda, J.; Okauchi, T.; Zhang, M.-R.; Hojo, J.; Kida, T.; Arai, T.; Yamamoto, F.; Sasaki, S.; Maeda, M.; et al. Synthesis, in vitro and in vivo pharmacology of a C-11 labeled analog of CP-101,606, (±)threo-1-(4-hydroxyphenyl)-2-[4-hydroxy-4-(p-[11C]methoxyphenyl)piperidino]-1-propanol, as a PET tracer for NR2B subunit-containing NMDA receptors. Nucl. Med. Biol. 2002, 29, 517–525. [Google Scholar] [CrossRef]
- Rigby, M.; Bourdellès, B.L.; Heavens, R.P.; Kelly, S.; Smith, D.; Butler, A.; Hammans, R.; Hills, R.; Xuereb, J.H.; Hill, R.G.; et al. The messenger RNAs for the N-methyl-D-aspartate receptor subunits show region-specific expression of different subunit composition in the human brain. Neuroscience 1996, 73, 429–447. [Google Scholar] [CrossRef]
- Årstad, E.; Platzer, S.; Berthele, A.; Pilowsky, L.S.; Luthra, S.K.; Wester, H.-J. Towards NR2B receptor selective imaging agents for PET-synthesis and evaluation of N-[11C]-(2-methoxy)benzyl (E)-styrene-, 2-naphthyl- and 4-trifluoromethoxyphenylamidine. Bioorg. Med. Chem. 2006, 14, 6307–6313. [Google Scholar] [CrossRef]
- Tewes, B.; Frehland, B.; Schepmann, D.; Schmidtke, K.U.; Winckler, T.; Wünsch, B. Design, Synthesis, and Biological Evaluation of 3-Benzazepin-1-ols as NR2B-Selective NMDA Receptor Antagonists. ChemMedChem 2010, 5, 687–695. [Google Scholar] [CrossRef]
- Krämer, S.D.; Betzel, T.; Mu, L.; Haider, A.; Adrienne, M.H.; Boninsegni, A.K.; Keller, C.; Szermerski, M.; Schibli, R.; Wünsch, B.; et al. Evaluation of 11C-Me-NB1 as a potential PET radioligand for measuring GluN2B-containing NMDA receptors, drug occupancy, and receptor cross talk. J. Nucl. Med. 2018, 59, 698–703. [Google Scholar] [CrossRef] [Green Version]
- Haider, A.; Herde, A.M.; Krämer, S.D.; Varisco, J.; Keller, C.; Frauenknecht, K.; Auberson, Y.P.; Temme, L.; Robaa, D.; Sippl, W.; et al. Preclinical Evaluation of Benzazepine-Based PET Radioligands (R)- and (S)-11C-Me-NB1 Reveals Distinct Enantiomeric Binding Patterns and a Tightrope Walk Between GluN2B- and σ1-Receptor-Targeted PET Imaging. J. Nucl. Med. 2019, 60, 1167–1173. [Google Scholar] [CrossRef] [Green Version]
- Haider, A.; Iten, I.; Ahmed, H.; Müller Herder, A.; Gruber, S.; Krämer, S.D.; Keller, C.; Schibli, R.; Wünsch, B.; Mu, L.; et al. Identification and preclinical evaluation of a radiofluorinated benzazepine derivative for imaging the GluN2B subunit of the ionotropic NMDA receptor. J. Nucl. Med. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, H.; Haider, A.; Varisco, J.; Stanković, M.; Wallimann, R.; Gruber, S.; Iten, I.; Häne, S.; Müller Herde, A.; Keller, C.; et al. Structure-affinity relationships of 2,3,4,5-tetrahydro-1H-3-benzazepine and 6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-amine analogues and the discovery of a radiofluorinated 2,3,4,5-tetrahydro-1H-3-benzazepine congener for imaging GluN2B subunit-containing N-methyl-D-aspartate receptors. J. Med. Chem. 2019, 62, 9450–9470. [Google Scholar]
- Ahmed, H.; Wallimann, R.; Haider, A.; Hosseini, V.; Gruber, S.; Robledo, M.; Nguyen, T.; Herde, A.M.; Iten, I.; Keller, C.; et al. Preclinical development of 18F-OF-NB1 for imaging GluN2B-containing N-methyl-D-aspartate receptors and its utility as a biomarker for amyotrophic lateral sclerosis. J. Nucl. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.E.; Bruderer, H.; Rieder, J.; Brossi, A. Metabolic studies of Versidyne, a new analgesic, in the rabbit and in man. Biochem. Pharmacol. 1964, 13, 777–778. [Google Scholar] [CrossRef]
- Jakobsson, J.E.; Gourni, E.; Khanapur, S.; Brito, B.; Riss, P.J. Synthesis and characterization in rodent brain of the subtype-selective NR2B NMDA receptor ligand [11C]Ro04-5595 as a potential radiotracer for positron emission tomography. ACS Omega 2019, 4, 9925–9931. [Google Scholar] [CrossRef]
- Wilson, A.A.; Garcia, A.; Jin, L.; Houle, S. Radiotracer synthesis from [11C]iodomethane: A remarkably simple captive solvent method. Nucl. Med. Biol. 2000, 27, 529–532. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Liow, J.-S.; Morse, C.; Telu, S.; Davies, R.; Frankland, M.; Zoghbi, S.; Cheng, K.; Hall, M.; Innis, R.B.; et al. Evaluation of 11C-NR2B-SMe and its enantiomers as PET radioligands for imaging the NR2B subunit within the NMDA receptor complex in rats. J. Nucl. Med. 2020, 61, 1212–1220. [Google Scholar] [CrossRef]
- Thum, S.; Schepmann, D.; Ayet, E.; Pujol, M.; Nieto, F.R.; Ametamey, S.M.; Wünsch, B. Tetrahydro-3-benzazepines with fluorinated side chains as NMDA and sigma1 receptor antagonists: Synthesis, receptor affinity, selectivity and antiallodynic activity. Eur. J. Med. Chem. 2019, 177, 47–62. [Google Scholar] [CrossRef]
- Thum, S.; Schepmann, D.; Reinoso, R.F.; Alvarez, I.; Ametamey, S.M.; Wünsch, B. Synthesis and pharmacological evaluation of fluorinated benzo[7]annulen-7-amines as GluN2B-selective NMDA receptor antagonists. J. Label. Compd. Radiopharm. 2019, 62, 354–379. [Google Scholar] [CrossRef]
- Fuchigami, T.; Yamaguchi, H.; Ogawa, M.; Biao, L.; Nakayama, M.; Haratake, M.; Magata, Y. Synthesis and biological evaluation of radio-iodinated benzimidazoles as SPECT imaging agents for NR2B subtype of NMDA receptor. Bioorg. Med. Chem. 2010, 18, 7497–7506. [Google Scholar] [CrossRef] [Green Version]
- Fuchigami, T.; Fujimoto, N.; Haradahira, T.; Nojiri, Y.; Okauchi, T.; Maeda, J.; Suhara, T.; Yamamoto, F.; Nakayama, M.; Maeda, M.; et al. Synthesis and characterization of 11C-labeled benzyl amidine derivatives as PET radioligands for GluN2B subunit of the NMDA receptors. J. Label. Compd. Radiopharm. 2018, 61, 1095–1105. [Google Scholar] [CrossRef]
- Fu, H.; Tang, W.; Chen, Z.; Belov, V.V.; Zhang, G.; Shao, T.; Zhang, X.; Yu, Q.; Rong, J.; Deng, X.; et al. Synthesis and preliminary evaluations of a triazole-cored antagonist as a PET imaging probe ([18F]N2B-0518) for GluN2B subunit in the brain. ACS Chem. Neurosci. 2019, 10, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Christiaans, J.A.M.; Klein, P.J.; Metaxas, A.; Kooijman, E.J.M.; Schuit, R.C.; Leysen, J.E.; Lammertsma, A.A.; van Berckel, B.N.M.; Windhorst, A.D. Synthesis and preclinical evaluation of carbon-11 labelled N-((5-(4-fluoro-2-[11C]methoxyphenyl)pyridin-3-yl)methyl)cyclopentanamine as a PET tracer for NR2B subunit-containing NMDA receptors. Nucl. Med. Biol. 2014, 41, 670–680. [Google Scholar] [CrossRef] [PubMed]
- van der Aart, J.; Yaqub, M.; Kooijman, E.J.M.; Bakker, J.; Langermans, J.A.M.; Schuit, R.C.; Hofman, M.B.M.; Christiaans, J.A.M.; Lammertsma, A.A.; Windhorst, A.D.; et al. Evaluation of the novel PET Tracer [11C]HACH242 for imaging the GluN2B NMDA receptor in non-human primates. Mol. Imaging Biol. 2019, 21, 676–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannhardt, G.; Gruchalla, M.; Kohl, B.K.; Parsons, C.G. A Novel Series of 2-Carboxytetrahydro quinolines Provides New Insights into the Eastern Region of Glycine Site NMDA Antagonists. Arch. Pharm. Pharm. Med. Chem. 2000, 333, 267–274. [Google Scholar] [CrossRef]
- Waterhouse, R.N.; Sultana, A.; Laruelle, M. In vivo evaluation of [11C]-3-[2-[(3-methoxyphenylamino) carbonyl]ethenyl]-4,6-dichloroindole-2-carboxylic acid ([11C]3MPICA) as a PET radiotracer for the glycine site of the NMDA ion channel. Nucl. Med. Biol. 2002, 29, 791–794. [Google Scholar] [CrossRef]
- Haradahira, T.; Zhang, M.-R.; Maeda, J.; Okauchi, T.; Kawabe, K.; Kida, T.; Suzuki, K.; Suhara, T. A strategy for increasing the brain uptake of a radioligand in animals: Use of a drug that inhibits plasma protein binding. Nucl. Med. Biol. 2000, 27, 357–360. [Google Scholar] [CrossRef]
- Haradahira, T.; Okauchi, T.; Maeda, J.; Zhang, M.R.; Kida, T.; Kawabe, K.; Mishina, M.; Watanabe, Y.; Suzuki, K.; Suhara, T. A positron-emitter labeled glycineB site antagonist, [11C]L-703,717, preferentially binds to a cerebellar NMDA receptor subtype consisting of GluR epsilon3 subunit in vivo, but not in vitro. Synapse 2002, 43, 131–133. [Google Scholar] [CrossRef]
- Sun, J.-Y.; Kumata, K.; Chen, Z.; Zhang, Y.-D.; Chen, J.-H.; Hatori, A.; Fu, H.-L.; Rong, J.; Deng, X.-Y.; Yamasaki, T.; et al. Synthesis and preliminary evaluation of novel 11C-labeled GluN2B-selective NMDA receptor negative allosteric modulators. Acta Pharmacol. Sin. 2020. [Google Scholar] [CrossRef]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef]
- Henley, J.; Wilkinson, K. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, L.; Beaver, K.; Jane, D.; Taylor, P.; Sunter, D.; Roberts, P. Characterization of the pharmacology and regional distribution of (S)-[3H]-5-fluorowillardiine binding in rat brain. Br. J. Pharmacol. 1995, 116, 2033–2039. [Google Scholar] [CrossRef] [Green Version]
- Michalak, A.; Butterworth, R.F. Selective loss of binding sites for the glutamate receptor ligands [3H]kainate and (S)-[3H]5-fluorowillardiine in the brains of rats with acute liver failure. Hepatology 1997, 25, 631–635. [Google Scholar] [CrossRef]
- Körösi, J.; Láng, T. Heterocyclische Verbindungen, I. Synthese des ersten 5H-2,3-Benzodiazepins, des 5-Äthyl-1-(3,4-dimethoxyphenyl)-7,8-dimethoxy-4-methyl-5H-2,3-benzodiazepins. Chem. Ber. 1974, 107, 3883–3893. [Google Scholar] [CrossRef]
- Láng, T.; Körösi, J.; Andrasi, F.; Botka, P.; Hamori, T.; Berzsenyi, P.; Goldschmidt, K.; Zólyomi, G.; Elekes, I.; Láng-Rihmer, Z. 5H-2, 3-benzodiazepine Derivatives and Pharmaceutical Compositions Containing the Same. US Patent 4,614,740, 30 September 1986. [Google Scholar]
- Tarnawa, I.; Farkas, S.; Berzsenyi, P.; Pataki, Á.; Andrási, F. Electrophysiological studies with a 2,3-benzodiazepine muscle relaxant: GYKI 52466. Eur. J. Pharm. 1989, 167, 193–199. [Google Scholar] [CrossRef]
- Donevan, S.D.; Rogawski, M.A. GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron 1993, 10, 51–59. [Google Scholar] [CrossRef]
- Tarnawa, I.; Vizi, S.E. 2,3-Benzodiazepine AMPA antagonists. Restor. Neurol. Neurosci. 1998, 13, 41–57. [Google Scholar]
- Sólyom, S.; Tarnawa, I. Non-competitive AMPA Antagonists of 2,3-Benzodiazepine Type. Curr. Pharm. Des. 2002, 8, 913–939. [Google Scholar] [CrossRef]
- Gao, M.; Kong, D.; Clearfield, A.; Zheng, Q.-H. Synthesis of carbon-11 and fluorine-18 labeled N-acetyl-1-aryl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline derivatives as new potential PET AMPA receptor ligands. Bioorg. Med. Chem. Lett. 2006, 16, 2229–2233. [Google Scholar] [CrossRef]
- Chen, J.; Gan, J.; Sun, J.; Chen, Z.; Fu, H.; Rong, J.; Deng, X.; Shang, J.; Gong, J.; Shao, T.; et al. Radiosynthesis and preliminary evaluation of 11C-labeled 4-cyclopropyl-7-(3-methoxyphenoxy)-3,4-dihydro-2H-benzo[e] [1,2,4] thiadiazine 1,1-dioxide for PET imaging AMPA receptors. Tetrahedron Lett. 2020, 61, 151635. [Google Scholar] [CrossRef]
- Miyazaki, T.; Nakajima, W.; Hatano, M.; Shibata, Y.; Kuroki, Y.; Arisawa, T.; Serizawa, A.; Sano, A.; Kogami, S.; Yamanoue, T.; et al. Visualization of AMPA receptors in living human brain with positron emission tomography. Nat. Med. 2020, 26, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Nadler, J.V.; Evenson, D.A.; Cuthbertson, G.J. Comparative toxicity of kainic acid and other acidic amino acids toward rat hippocampal neurons. Neuroscience 1981, 6, 2505–2517. [Google Scholar] [CrossRef]
- Evans, A.J.; Gurung, S.; Henley, J.M.; Nakamura, Y.; Wilkinson, K.A. Exciting times: New advances towards understanding the regulation and roles of kainate receptors. Neurochem. Res. 2019, 44, 572–584. [Google Scholar] [CrossRef] [Green Version]
- Straub, C.; Hunt, D.L.; Yamasaki, M.; Kim, K.S.; Watanabe, M.; Castillo, P.E.; Tomita, S. Distinct functions of kainate receptors in the brain are determined by the auxiliary subunit Neto1. Nat. Neurosci. 2011, 14, 866–873. [Google Scholar] [CrossRef]
- Behuet, S.; Cremer, J.N.; Cremer, M.; Palomero-Gallagher, N.; Zilles, K.; Amunts, K. Developmental changes of glutamate and GABA receptor densities in Wistar rats. Front. Neuroanat. 2019, 13, 100. [Google Scholar] [CrossRef] [PubMed]
- Cremer, J.N.; Amunts, K.; Graw, J.; Piel, M.; Rösch, F.; Zilles, K. Neurotransmitter receptor density changes in Pitx3ak mice—A model relevant to Parkinson’s disease. Neuroscience 2015, 285, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Künig, G.; Hartmann, J.; Krause, F.; Deckert, J.; Heinsen, H.; Ransmayr, G.; Beckmann, H.; Riederer, P. Regional differences in the interaction of the excitotoxins domoate and L-beta-oxalyl-amino-alanine with [3H]kainate binding sites in human hippocampus. Neurosci. Lett. 1995, 187, 107–110. [Google Scholar] [CrossRef]
- Flajolet, M.; Rakhilin, S.; Wang, H.; Starkova, N.; Nuangchamnong, N.; Nairn, A.C.; Greengard, P. Protein phosphatase 2C binds selectively to and dephosphorylates metabotropic glutamate receptor 3. Proc. Nat. Acad. Sci. USA 2003, 100, 16006–16011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.-B.; Castillo, P.E. Role of glutamate autoreceptors at hippocampal Mossy fiber synapses. Neuron 2008, 60, 1082–1094. [Google Scholar] [CrossRef] [Green Version]
- Matta, J.A.; Ashby, M.C.; Sanz-Clemente, A.; Roche, K.W.; Isaac, J.T.R. mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 2011, 70, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Pöschel, B.; Wroblewska, B.; Heinemann, U.; Manahan-Vaughan, D. The metabotropic glutamate receptor mGluR3 is critically required for hippocampal long-term depression and modulates long-term potentiation in the dentate gyrus of freely moving rats. Cereb. Cortex 2005, 15, 1414–1423. [Google Scholar] [CrossRef] [Green Version]
- Xi, D.; Li, Y.-C.; Snyder, M.A.; Gao, R.Y.; Adelman, A.E.; Zhang, W.; Shumsky, J.S.; Gao, W.-J. Group II Metabotropic Glutamate Receptor Agonist Ameliorates MK801-Induced Dysfunction of NMDA Receptors via the Akt/GSK-3β Pathway in Adult Rat Prefrontal Cortex. Neuropsychopharmacology 2011, 63, 1260–1274. [Google Scholar] [CrossRef] [Green Version]
- Li, M.-L.; Yang, S.-S.; Xing, B.; Ferguson, B.R.; Gulchina, Y.; Li, Y.-C.; Li, F.; Hu, X.-Q.; Gao, W.-J. LY395756, an mGluR2 agonist and mGluR3 antagonist, enhances NMDA receptor expression and function in the normal adult rat prefrontal cortex, but fails to improve working memory and reverse MK801-induced working memory impairment. Exp. Neurol. 2015, 273, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Dhull, D.K.; Mishra, P.S. Therapeutic potential of mGluR5 targeting in Alzheimer’s disease. Front. Neurosci. 2015, 9, 215. [Google Scholar]
- Hamilton, A.; Esseltine, J.L.; DeVries, R.A.; Cregan, S.P.; Ferguson, S.S.G. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 2014, 7, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahlquist, A. Etretinate pharmacokinetics in chronic renal failure. A preliminary study in psoriasis patients. Dermatologica 1987, 175, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.M.; Vieira, L.B.; Pires, R.G.; Olmo, R.P.; Ferguson, S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol Res. 2017, 115, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.R.; Seo, S.S.; Barnes, S.A.; Louros, S.R.; Muscas, M.; Dando, O.; Kirby, C.; Wyllie, D.J.A.; Hardingham, G.E.; Kind, P.C.; et al. Cell-Type-Specific Translation Profiling Reveals a Novel Strategy for Treating Fragile X Syndrome. Neuron 2017, 95, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, R.; Wong, R.K.S.; Merlin, L.R. Glutamate receptors in epilepsy: Group I mGluR-mediated epileptogenesis. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Ebrahimi-Ghiri, M.; Khakpai, F.; Zarrindast, M.R. Combined treatment of scopolamine and group III mGluR antagonist, CPPG, exerts antidepressant activity without affecting anxiety-related behaviors. Physiol. Behav. 2020, 224, 113034. [Google Scholar] [CrossRef]
- Yu, F.; Zhong, P.; Liu, X.; Sun, D.; Gao, H.Q.; Liu, Q.S. Metabotropic glutamate receptor I (mGluR1) antagonism impairs cocaine-induced conditioned place preference via inhibition of protein synthesis. Neuropsychopharmacology 2013, 38, 1308–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Narendran, R.; Bischoff, F.; Guo, N.; Zhu, Z.; Bae, S.-A.; Lesage, A.S.; Laruelle, M. A positron emission tomography radioligand for the in vivo labeling of metabotropic glutamate 1 receptor: (3-ethyl-2-[11C]methyl-6-quinolinyl)(cis- 4-methoxycyclohexyl)methanone. J. Med. Chem. 2005, 48, 5096–5099. [Google Scholar] [CrossRef] [PubMed]
- Konno, F.; Odawara, C.; Yamasaki, T.; Kawamura, K.; Hatori, A.; Yui, J.; Wakizaka, H.; Nengaki, N.; Takei, M.; Zhang, M.R. Radiosynthesis and evaluation of [11C]YM-202074 as a PET ligand for imaging the metabotropic glutamate receptor type 1. Nucl. Med. Biol. 2010, 37, 615–624. [Google Scholar]
- Sasikumar, T.K.; Qiang, L.; Burnett, D.A.; Greenlee, W.J.; Li, C.; Heimark, L.; Pramanik, B.; Grilli, M.; Bertorelli, R.; Lozza, G.; et al. Tricyclic thienopyridine-pyrimidones/thienopyrimidine- pyrimidones as orally efficacious mGluR1 antagonists for neuropathic pain. Bioorg. Med. Chem. Lett. 2009, 19, 3199–3203. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, J.; Majo, V.J.; Milak, M.S.; Kassir, S.A.; Palner, M.; Savenkova, L.; Mali, P.; Arango, V.; Mann, J.J.; Parsey, R.V.; et al. Synthesis, in vitro and in vivo evaluation of [11C]MMTP: A potential PET ligand for mGluR1 receptors. Bioorg. Med. Chem. Lett. 2010, 20, 3499–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostetler, E.D.; Eng, W.; Joshi, A.D.; Sanabria-Bohórquez, S.; Kawamoto, H.; Ito, S.; O’Malley, S.; Krause, S.; Ryan, C.; Patel, S.; et al. Synthesis, characterization, and monkey PET studies of [1⁸F]MK-1312, a PET tracer for quantification of mGluR1 receptor occupancy by MK-5435. Synapse 2011, 65, 125–135. [Google Scholar] [CrossRef]
- Huang, Y.; Narendran, R.; Bischoff, F.; Guo, N.; Bae, S.A.; Hwang, D.R.; Lesage, A.S.; Laruelle, M. Synthesis and characterization of two PET radioligands for the metabotropic glutamate 1 (mGlu1) receptor. Synapse 2012, 66, 1002–1014. [Google Scholar] [CrossRef]
- Lee, B.; Kim, Y.K.; Lee, J.Y.; Kim, Y.J.; Lee, Y.-S.; Lee, D.S.; Chung, J.-K.; Jeong, J.M. Preclinical anaylses of [18F]cEFQ as a PET tracer for imaging metabotropic glutamate receptor type 1 (mGluR1). J. Cereb. Blood Flow Metab. 2017, 37, 2283–2293. [Google Scholar] [CrossRef] [Green Version]
- Satoh, A.; Nagatomi, Y.; Hirata, Y.; Ito, S.; Suzuki, G.; Kimura, T.; Maehara, S.; Hikichi, H.; Satow, A.; Hata, M.; et al. Discovery and in vitro and in vivo profiles of 4-fluoro- N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide as novel class of an orally active metabotropic glutamate receptor 1 (mGluR1) antagonist. Bioorg. Med. Chem. Lett. 2009, 19, 5464–5468. [Google Scholar] [CrossRef]
- Yamasaki, T.; Fujinaga, M.; Yui, J.; Wakizaka, H.; Ohya, T.; Nengaki, N.; Ogawa, M.; Ikoma, Y.; Hatori, A.; Xie, L.; et al. Improved Visualization and Specific Binding for Metabotropic Glutamate Receptor Subtype 1 (mGluR1) Using [11C]ITMM with Ultra-High Specific Activity in Small-Animal PET. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef]
- Yui, J.; Xie, L.; Fujinaga, M.; Yamasaki, T.; Hatori, A.; Kumata, K.; Nengaki, N.; Zhang, M.R. Monitoring neuroprotective effects using positron emission tomography with [11C]ITMM, a radiotracer for metabotropic glutamate 1 receptor. Stroke 2013, 44, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Fujinaga, M.; Yamasaki, T.; Kawamura, K.; Kumata, K.; Hatori, A.; Yui, J.; Yanamoto, K.; Yoshida, Y.; Ogawa, M.; Nengaki, N.; et al. Synthesis and evaluation of 6-[1-(2-[18F]fluoro-3-pyridyl)-5-methyl-1H-1,2,3-triazol-4-yl]quinoline for positron emission tomography imaging of the metabotropic glutamate receptor type 1 in brain. Bioorg. Med. Chem. 2011, 19, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, M.; Maeda, J.; Yui, J.; Hatori, A.; Yamasaki, T.; Kawamura, K.; Kumata, K.; Yoshida, Y.; Nagai, Y.; Higuchi, M.; et al. Characterization of 1-(2-[18F]fluoro-3-pyridyl)- 4-(2-isopropyl-1-oxo- isoindoline-5-yl)-5-methyl-1H-1,2,3-triazole, a PET ligand for imaging the metabotropic glutamate receptor type 1 in rat and monkey brains. J. Neurochem. 2012, 121, 115–124. [Google Scholar] [CrossRef]
- Yamasaki, T.; Fujinaga, M.; Maeda, J.; Kawamura, K.; Yui, J.; Hatori, A.; Yoshida, Y.; Nagai, Y.; Tokunaga, M.; Higuchi, M.; et al. Imaging for metabotropic glutamate receptor subtype 1 in rat and monkey brains using PET with [18F]FITM. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Miura, Y.; Ishikawa, K.; Ishii, K.; Ishiwata, K. Decreased metabotropic glutamate receptor type 1 availability in a patient with spinocerebellar ataxia type 6: A (11)C-ITMM PET study. J. Neurol. Sci. 2015, 355, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Miura, Y.; Toyohara, J.; Ishii, K.; Ishiwata, K. Comparison of imaging using 11C-ITMM and 18F-FDG for the detection of cerebellar ataxia. J. Neurol. Sci. 2017, 375, 97–102. [Google Scholar] [CrossRef]
- Ishibashi, K.; Miura, Y.; Toyohara, J.; Ishiwata, K.; Ishii, K. Unchanged type 1 metabotropic glutamate receptor availability in patients with Alzheimer’s disease: A study using 11C-ITMM positron emission tomography. NeuroImage Clin. 2019, 22, 101783. [Google Scholar] [CrossRef]
- Sakata, M.; Toyohara, J.; Ishibashi, K.; Wagatsuma, K.; Ishii, K.; Zhang, M.R.; Ishiwata, K. Age and gender effects of 11C-ITMM binding to metabotropic glutamate receptor type 1 in healthy human participants. Neurobiol. Aging 2017, 55, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Maeda, J.; Fujinaga, M.; Nagai, Y.; Hatori, A.; Yui, J.; Xie, L.; Nengaki, N.; Zhang, M.R. PET brain kinetics studies of 11C-ITMM and 11C-ITDM, radioprobes for metabotropic glutamate receptor type 1, in a nonhuman primate. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 260–269. [Google Scholar] [PubMed]
- Bertoglio, D.; Verhaeghe, J.; Korat, Š.; Miranda, A.; Wyffels, L.; Stroobants, S.; Mrzljak, L.; Dominguez, C.; Liu, L.; Skinbjerg, M.; et al. In vitro and In vivo Assessment of Suitable Reference Region and Kinetic Modelling for the mGluR1 Radioligand [11C]ITDM in Mice. Mol. Imaging Biol. 2020, 22, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Bertoglio, D.; Verhaeghe, J.; Korat, Š.; Miranda, A.; Cybulska, K.; Wyffels, L.; Stroobants, S.; Mrzljak, L.; Dominguez, C.; Skinbjerg, M.; et al. Elevated Type 1 Metabotropic Glutamate Receptor Availability in a Mouse Model of Huntington’s Disease: A Longitudinal PET Study. Mol. Neurobiol. 2020, 57, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, T.; Fujinaga, M.; Kawamura, K.; Furutsuka, K.; Nengaki, N.; Shimoda, Y.; Shiomi, S.; Takei, M.; Hashimoto, H.; Yui, J.; et al. Dynamic Changes in Striatal mGluR1 but Not mGluR5 during Pathological Progression of Parkinson’s Disease in Human Alpha-Synuclein A53T Transgenic Rats: A Multi-PET Imaging Study. J. Neurosci. 2016, 36, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, T.; Fujinaga, M.; Mori, W.; Zhang, Y.; Wakizaka, H.; Nengaki, N.; Xie, L.; Hatori, A.; Zhang, M.R. In Vivo Monitoring for Regional Changes of Metabotropic Glutamate Receptor Subtype 1 (mGluR1) in Pilocarpine-Induced Epileptic Rat Brain by Small-Animal PET. Sci. Rep. 2017, 7, 14945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Krause, S.M.; Hamill, T.; Chaudhary, A.; Burns, D.H.; Gibson, R.A. In vitro characterization of [3H]MethoxyPyEP, an mGluR5 selective radioligand. Life Sci. 2003, 73, 371–379. [Google Scholar] [CrossRef]
- Severance, A.J.; Parsey, R.V.; Kumar, J.S.; Underwood, M.D.; Arango, V.; Majo, V.J.; Prabhakaran, J.; Simpson, N.R.; Van Heertum, R.L.; Mann, J.J. In vitro and in vivo evaluation of [11C]MPEPy as a potential PET ligand for mGlu5 receptors. Nucl. Med. Biol. 2006, 33, 1021–1027. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Kessler, L.; Honer, M.; Auberson, Y.; Gasparini, F.; Schubiger, P.A. Synthesis and evaluation of [11C]M-FPEP as a PET ligand for imaging the metabotropic glutamate receptor subtype 5 (mGluR5). J. Label. Compd. Radiopharm. 2003, 46, S188. [Google Scholar]
- Yu, M.; Tueckmantel, W.; Wang, X.; Zhu, A.; Kozikowski, A.P.; Brownell, A.L. Methoxyphenylethynyl, methoxypyridylethynyl and phenylethynyl derivatives of pyridine: Synthesis, radiolabeling and evaluation of new PET ligands for metabotropic glutamate subtype 5 receptors. Nucl. Med. Biol. 2005, 32, 631–640. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Kessler, L.J.; Honer, M.; Wyss, M.T.; Buck, A.; Hintermann, S.; Auberson, Y.P.; Gasparini, F.; Schubiger, P.A. Radiosynthesis and preclinical evaluation of 11C-ABP688 as a probe for imaging the metabotropic glutamate receptor subtype 5. J. Nucl. Med. 2006, 47, 698–705. [Google Scholar]
- Wyckhuys, T.; Verhaeghe, J.; Wyffels, L.; Langlois, X.; Schmidt, M.; Stroobants, S.; Staelens, S. N-acetylcysteine- and MK-801-induced changes in glutamate levels do not affect in vivo binding of metabotropic glutamate 5 receptor radioligand 11C-ABP688 in rat brain. J. Nucl. Med. 2013, 54, 1954–1961. [Google Scholar] [CrossRef] [Green Version]
- Miyake, N.; Skinbjerg, M.; Easwaramoorthy, B.; Kumar, D.; Girgis, R.R.; Xu, X.; Slifstein, M.; Abi-Dargham, A. Imaging changes in glutamate transmission in vivo with the metabotropic glutamate receptor 5 tracer [11C] ABP688 and N-acetylcysteine challenge. Biol. Psychiatry 2011, 69, 822–824. [Google Scholar] [CrossRef]
- Kosten, L.; Verhaeghe, J.; Wyffels, L.; Stroobants, S.; Staelens, S. Acute Ketamine Infusion in Rat Does Not Affect In Vivo [11C]ABP688 Binding to Metabotropic Glutamate Receptor Subtype 5. Mol. Imaging 2018, 17, 1536012118788636. [Google Scholar] [CrossRef] [Green Version]
- Müller Herde, A.; Boss, S.D.; He, Y.; Schibli, R.; Mu, L.; Ametamey, S.M. Ketamine and Ceftriaxone-Induced Alterations in Glutamate Levels Do Not Impact the Specific Binding of Metabotropic Glutamate Receptor Subtype 5 Radioligand [18F]PSS232 in the Rat Brain. Pharmaceuticals (Basel Switz.) 2018, 11, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandiego, C.M.; Nabulsi, N.; Lin, S.F.; Labaree, D.; Najafzadeh, S.; Huang, Y.; Cosgrove, K.; Carson, R.E. Studies of the metabotropic glutamate receptor 5 radioligand [11C]ABP688 with N-acetylcysteine challenge in rhesus monkeys. Synapse 2013, 67, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosten, L.; Deleye, S.; Stroobants, S.; Wyffels, L.; Mingote, S.; Rayport, S.; Staelens, S. Molecular Imaging of mGluR5 Availability with [11C]ABP68 in Glutaminase Heterozygous Mice. Cell. Mol. Neurobiol. 2019, 39, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, E.R.; Parent, M.J.; Leuzy, A.; Aliaga, A.; Aliaga, A.; Moquin, L.; Schirrmacher, E.S.; Soucy, J.P.; Skelin, I.; Gratton, A.; et al. Imaging in vivo glutamate fluctuations with [11C]ABP688: A GLT-1 challenge with ceftriaxone. J. Cereb. Blood Flow Metab. 2015, 35, 1169–1174. [Google Scholar] [CrossRef] [Green Version]
- Mathews, W.B.; Kuwabara, H.; Stansfield, K.; Valentine, H.; Alexander, M.; Kumar, A.; Hilton, J.; Dannals, R.F.; Wong, D.F.; Gasparini, F. Dose-dependent, saturable occupancy of the metabotropic glutamate subtype 5 receptor by fenobam as measured with [11C]ABP688 PET imaging. Synapse 2014, 68, 565–573. [Google Scholar] [CrossRef]
- Varnäs, K.; Juréus, A.; Finnema, S.J.; Johnström, P.; Raboisson, P.; Amini, N.; Takano, A.; Stepanov, V.; Halldin, C.; Farde, L. The metabotropic glutamate receptor 5 radioligand [11C]AZD9272 identifies unique binding sites in primate brain. Neuropharmacology 2018, 135, 455–463. [Google Scholar] [CrossRef]
- Fang, X.T.; Eriksson, J.; Antoni, G.; Yngve, U.; Cato, L.; Lannfelt, L.; Sehlin, D.; Syvänen, S. Brain mGluR5 in mice with amyloid beta pathology studied with in vivo [11C]ABP688 PET imaging and ex vivo immunoblotting. Neuropharmacology 2017, 113 Pt A, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Lee, H.J.; Jeong, Y.J.; Oh, S.J.; Kang, K.J.; Han, S.J.; Nam, K.R.; Lee, Y.J.; Lee, K.C.; Ryu, Y.H.; et al. Age dependency of mGluR5 availability in 5xFAD mice measured by PET. Neurobiol. Aging 2019, 84, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, H.J.; Park, I.S.; Park, J.A.; Kwon, Y.J.; Ryu, Y.H.; Kim, C.H.; Kang, J.H.; Hyun, I.Y.; Lee, K.C.; et al. Aβ pathology downregulates brain mGluR5 density in a mouse model of Alzheimer. Neuropharmacology 2018, 133, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Bertoglio, D.; Kosten, L.; Verhaeghe, J.; Thomae, D.; Wyffels, L.; Stroobants, S.; Wityak, J.; Dominguez, C.; Mrzljak, L.; Staelens, S. Longitudinal Characterization of mGluR5 Using 11C-ABP688 PET Imaging in the Q175 Mouse Model of Huntington Disease. J. Nucl. Med. 2018, 59, 1722–1727. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Kim, Y.K.; Oh, S.W.; Im, H.-J.; Hwang, D.W.; Kang, H.; Lee, B.; Lee, Y.-S.; Jeong, J.M.; Kim, E.E.; et al. In Vivo Imaging of mGluR5 Changes during Epileptogenesis Using [11C]ABP688 PET in Pilocarpine-Induced Epilepsy Rat Model. PLoS ONE 2014, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownell, A.-L.; Kuruppu, D.; Kil, K.-E.; Jokivarsi, K.; Poutiainen, P.; Zhu, A.; Maxwell, M. PET imaging studies show enhanced expression of mGluR5 and inflammatory response during progressive degeneration in ALS mouse model expressing SOD1-G93A gene. J. Neuroinflamm. 2015, 12, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller Herde, A.; Schibli, R.; Weber, M.; Ametamey, S.M. Metabotropic glutamate receptor subtype 5 is altered in LPS-induced murine neuroinflammation model and in the brains of AD and ALS patients. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Glorie, D.; Verhaeghe, J.; Miranda, A.; Kertesz, I.; Wyffels, L.; Stroobants, S.; Staelens, S. Progression of obsessive compulsive disorder-like grooming in Sapap3 knockout mice: A longitudinal [11C]ABP688 PET study. Neuropharmacology 2020, 177, 108160. [Google Scholar] [CrossRef]
- Cai, G.; Wang, M.; Wang, S.; Liu, Y.; Zhao, Y.; Zhu, Y.; Zhao, S.; Zhang, M.; Guo, B.; Yao, H.; et al. Brain mGluR5 in Shank3B-/- Mice Studied With in vivo [18F]FPEB PET Imaging and ex vivo Immunoblotting. Front. Psychiatry 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Wong, D.F.; Brašić, J.R.; Kuwabara, H.; Mathur, A.; Folsom, T.D.; Jacob, S.; Realmuto, G.M.; Pardo, J.V.; Lee, S. Metabotropic glutamate receptor 5 tracer [18F]-FPEB displays increased binding potential in postcentral gyrus and cerebellum of male individuals with autism: A pilot PET study. Cerebellum Ataxias 2018, 5, 3. [Google Scholar] [CrossRef]
- Smart, K.; Cox, S.; Kostikov, A.; Shalai, A.; Scala, S.G.; Tippler, M.; Jaworska, N.; Boivin, M.; Séguin, J.R.; Benkelfat, C.; et al. Effect of (Z)-isomer content on [11C]ABP688 binding potential in humans. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1175–1178. [Google Scholar] [CrossRef]
- Kawamura, K.; Yamasaki, T.; Kumata, K.; Furutsuka, K.; Takei, M.; Wakizaka, H.; Fujinaga, M.; Kariya, K.; Yui, J.; Hatori, A.; et al. Binding potential of (E)-[11C]ABP688 to metabotropic glutamate receptor subtype 5 is decreased by the inclusion of its 11C-labelled Z-isomer. Nucl. Med. Biol. 2014, 41, 17–23. [Google Scholar] [CrossRef]
- Smart, K.; Cox, S.; Nagano-Saito, A.; Rosa-Neto, P.; Leyton, M.; Benkelfat, C. Test-retest variability of [11C]ABP688 estimates of metabotropic glutamate receptor subtype 5 availability in humans. Synapse 2018, 72, e22041. [Google Scholar] [CrossRef]
- DeLorenzo, C.; Gallezot, J.D.; Gardus, J.; Yang, J.; Planeta, B.; Nabulsi, N.; Ogden, R.T.; Labaree, D.C.; Huang, Y.H.; Mann, J.J.; et al. In vivo variation in same-day estimates of metabotropic glutamate receptor subtype 5 binding using [11C]ABP688 and [18F]FPEB. J. Cereb. Blood Flow Metab. 2017, 37, 2716–2727. [Google Scholar] [CrossRef] [Green Version]
- DeLorenzo, C.; Kumar, J.S.; Mann, J.J.; Parsey, R.V. In vivo variation in metabotropic glutamate receptor subtype 5 binding using positron emission tomography and [11C]ABP688. J. Cereb. Blood Flow Metab. 2011, 31, 2169–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, S.C.; Sousek, A.; Hefti, K.; Saberi-Moghadam, S.; Buck, A.; Ametamey, S.M.; Scheidegger, M.; Franken, P.; Henning, A.; Seifritz, E.; et al. Cerebral mGluR5 availability contributes to elevated sleep need and behavioral adjustment after sleep deprivation. eLife 2017, 6, e28751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smart, K.; Cox, S.; Scala, S.G.; Tippler, M.; Jaworska, N.; Boivin, M.; Séguin, J.R.; Benkelfat, C.; Leyton, M. Sex differences in [11C]ABP688 binding: A positron emission tomography study of mGlu5 receptors. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1179–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuBois, J.M.; Rousset, O.G.; Rowley, J.; Porras-Betancourt, M.; Reader, A.J.; Labbe, A.; Massarweh, G.; Soucy, J.P.; Rosa-Neto, P.; Kobayashi, E. Characterization of age/sex and the regional distribution of mGluR5 availability in the healthy human brain measured by high-resolution [11C]ABP688 PET. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Treyer, V.; Gietl, A.F.; Suliman, H.; Gruber, E.; Meyer, R.; Buchmann, A.; Johayem, A.; Unschuld, P.G.; Nitsch, R.M.; Buck, A.; et al. Reduced uptake of [11C]-ABP688, a PET tracer for metabolic glutamate receptor 5 in hippocampus and amygdala in Alzheimer’s dementia. Brain Behav. 2020, 10, e01632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecca, A.P.; McDonald, J.W.; Michalak, H.R.; Godek, T.A.; Harris, J.E.; Pugh, E.A.; Kemp, E.C.; Chen, M.K.; Salardini, A.; Nabulsi, N.B.; et al. PET imaging of mGluR5 in Alzheimer’s disease. Alzheimer Res. Ther. 2020, 12, 15. [Google Scholar] [CrossRef]
- Kang, Y.; Henchcliffe, C.; Verma, A.; Vallabhajosula, S.; He, B.; Kothari, P.J.; Pryor, K.O.; Mozley, P.D. 18F-FPEB PET/CT Shows mGluR5 Upregulation in Parkinson’s Disease. J. Neuroimaging 2019, 29, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Crabbé, M.; Van der Perren, A.; Weerasekera, A.; Himmelreich, U.; Baekelandt, V.; Van Laere, K.; Casteels, C. Altered mGluR5 binding potential and glutamine concentration in the 6-OHDA rat model of acute Parkinson’s disease and levodopa-induced dyskinesia. Neurobiol. Aging 2018, 61, 82–92. [Google Scholar] [CrossRef]
- Lam, J.; DuBois, J.M.; Rowley, J.; González-Otárula, K.A.; Soucy, J.P.; Massarweh, G.; Hall, J.A.; Guiot, M.C.; Rosa-Neto, P.; Kobayashi, E. In vivo metabotropic glutamate receptor type 5 abnormalities localize the epileptogenic zone in mesial temporal lobe epilepsy. Ann. Neurol. 2019, 85, 218–228. [Google Scholar] [CrossRef]
- DuBois, J.M.; Rousset, O.G.; Guiot, M.C.; Hall, J.A.; Reader, A.J.; Soucy, J.P.; Rosa-Neto, P.; Kobayashi, E. Metabotropic Glutamate Receptor Type 5 (mGluR5) Cortical Abnormalities in Focal Cortical Dysplasia Identified In Vivo With [11C]ABP688 Positron-Emission Tomography (PET) Imaging. Cereb. Cortex 2016, 26, 4170–4179. [Google Scholar] [CrossRef] [Green Version]
- DeLorenzo, C.; DellaGioia, N.; Bloch, M.; Sanacora, G.; Nabulsi, N.; Abdallah, C.; Yang, J.; Wen, R.; Mann, J.J.; Krystal, J.H.; et al. In vivo ketamine-induced changes in [11C]ABP688 binding to metabotropic glutamate receptor subtype 5. Biol. Psychiatry 2015, 77, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, S.E.; Gallezot, J.D.; Davis, M.T.; DellaGioia, N.; Matuskey, D.; Nabulsi, N.; Krystal, J.H.; Javitch, J.A.; DeLorenzo, C.; Carson, R.E.; et al. Measuring the effects of ketamine on mGluR5 using [18F]FPEB and PET. J. Cereb. Blood Flow Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Esterlis, I.; DellaGioia, N.; Pietrzak, R.H.; Matuskey, D.; Nabulsi, N.; Abdallah, C.G.; Yang, J.; Pittenger, C.; Sanacora, G.; Krystal, J.H.; et al. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: An [11C]ABP688 and PET imaging study in depression. Mol. Psychiatry 2018, 23, 824–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLorenzo, C.; Sovago, J.; Gardus, J.; Xu, J.; Yang, J.; Behrje, R.; Kumar, J.S.; Devanand, D.P.; Pelton, G.H.; Mathis, C.A.; et al. Characterization of brain mGluR5 binding in a pilot study of late-life major depressive disorder using positron emission tomography and [11C]ABP688. Transl. Psychiatry 2015, 5, e693. [Google Scholar] [CrossRef]
- Deschwanden, A.; Karolewicz, B.; Feyissa, A.M.; Treyer, V.; Ametamey, S.M.; Johayem, A.; Burger, C.; Auberson, Y.P.; Sovago, J.; Stockmeier, C.A.; et al. Reduced metabotropic glutamate receptor 5 density in major depression determined by [11C]ABP688 PET and postmortem study. Am. J. Psychiatry 2011, 168, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Joo, Y.H.; Son, Y.D.; Kim, J.H.; Kim, Y.K.; Kim, H.K.; Lee, S.Y.; Ido, T. In vivo metabotropic glutamate receptor 5 availability-associated functional connectivity alterations in drug-naïve young adults with major depression. Eur. Neuropsychopharmacol. 2019, 29, 278–290. [Google Scholar] [CrossRef]
- Davis, M.T.; Hillmer, A.; Holmes, S.E.; Pietrzak, R.H.; DellaGioia, N.; Nabulsi, N.; Matuskey, D.; Angarita, G.A.; Carson, R.E.; Krystal, J.H.; et al. In vivo evidence for dysregulation of mGluR5 as a biomarker of suicidal ideation. Proc. Nat. Acad. Sci. USA 2019, 116, 11490–11495. [Google Scholar] [CrossRef] [Green Version]
- Akkus, F.; Treyer, V.; Ametamey, S.M.; Johayem, A.; Buck, A.; Hasler, G. Metabotropic glutamate receptor 5 neuroimaging in schizophrenia. Schizophr. Res. 2017, 183, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkus, F.; Terbeck, S.; Ametamey, S.M.; Rufer, M.; Treyer, V.; Burger, C.; Johayem, A.; Mancilla, B.G.; Sovago, J.; Buck, A.; et al. Metabotropic glutamate receptor 5 binding in patients with obsessive-compulsive disorder. Int. J. Neuropsychopharmacol. 2014, 17, 1915–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, S.E.; Girgenti, M.J.; Davis, M.T.; Pietrzak, R.H.; DellaGioia, N.; Nabulsi, N.; Matuskey, D.; Southwick, S.; Duman, R.S.; Carson, R.E.; et al. Altered metabotropic glutamate receptor 5 markers in PTSD: In vivo and postmortem evidence. Proc. Nat. Acad. Sci. USA 2017, 114, 8390–8395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkus, F.; Ametamey, S.M.; Treyer, V.; Burger, C.; Johayem, A.; Umbricht, D.; Gomez Mancilla, B.; Sovago, J.; Buck, A.; Hasler, G. Marked global reduction in mGluR5 receptor binding in smokers and ex-smokers determined by [11C]ABP688 positron emission tomography. Proc. Natl. Acad. Sci. USA 2013, 110, 737–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkus, F.; Mihov, Y.; Treyer, V.; Ametamey, S.M.; Johayem, A.; Senn, S.; Rösner, S.; Buck, A.; Hasler, G. Metabotropic glutamate receptor 5 binding in male patients with alcohol use disorder. Transl. Psychiatry 2018, 8, 17. [Google Scholar] [CrossRef]
- Milella, M.S.; Marengo, L.; Larcher, K.; Fotros, A.; Dagher, A.; Rosa-Neto, P.; Benkelfat, C.; Leyton, M. Limbic system mGluR5 availability in cocaine dependent subjects: A high-resolution PET [11C]ABP688 study. NeuroImage 2014, 98, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Fekete, S.; Simko, J.; Gradosova, I.; Malakova, J.; Zivna, H.; Palicka, V.; Zivny, P. The effect of levetiracetam on rat bone mass, structure and metabolism. Epilepsy Res. 2013, 107, 56–60. [Google Scholar] [CrossRef]
- Ceccarini, J.; Leurquin-Sterk, G.; Crunelle, C.L.; de Laat, B.; Bormans, G.; Peuskens, H.; Van Laere, K. Recovery of Decreased Metabotropic Glutamate Receptor 5 Availability in Abstinent Alcohol-Dependent Patients. J. Nucl. Med. 2020, 61, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Cox, S.; Tippler, M.; Jaworska, N.; Smart, K.; Castellanos-Ryan, N.; Durand, F.; Allard, D.; Benkelfat, C.; Parent, S.; Dagher, A.; et al. mGlu5 Receptor availability in youth at risk for addictions: Effects of vulnerability traits and cannabis use. Neuropsychopharmacology 2020. [Google Scholar] [CrossRef]
- Wanger-Baumann, C.A.; Mu, L.; Honer, M.; Belli, S.; Alf, M.F.; Schubiger, P.A.; Krämer, S.D.; Ametamey, S.M. In vitro and in vivo evaluation of [18F]-FDEGPECO as a PET tracer for imaging the metabotropic glutamate receptor subtype 5 (mGluR5). NeuroImage 2011, 56, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Sephton, S.M.; Dennler, P.; Leutwiler, D.S.; Mu, L.; Wanger-Baumann, C.A.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Synthesis, radiolabelling and in vitro and in vivo evaluation of a novel fluorinated ABP688 derivative for the PET imaging of metabotropic glutamate receptor subtype 5. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 14–28. [Google Scholar] [PubMed]
- Warnock, G.; Sommerauer, M.; Mu, L.; Pla Gonzalez, G.; Geistlich, S.; Treyer, V.; Schibli, R.; Buck, A.; Krämer, S.D.; Ametamey, S.M. A first-in-man PET study of [18F]PSS232, a fluorinated ABP688 derivative for imaging metabotropic glutamate receptor subtype 5. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1041–1051. [Google Scholar] [CrossRef]
- Johnson, B.G.; Wright, R.A.; Arnold, M.B.; Wheeler, W.J.; Ornstein, P.L.; Schoepp, D.D. [3H]-LY341495 as a novel antagonist radioligand for group II metabotropic glutamate (mGlu) receptors: Characterization of binding to membranes of mGlu receptor subtype expressing cells. Neuropharmacology 1999, 38, 1519–1529. [Google Scholar] [CrossRef]
- Waterhouse, R.N.; Schmidt, M.E.; Sultana, A.; Schoepp, D.D.; Wheeler, W.J.; Mozley, P.D.; Laruelle, M. Evaluation of [3H]LY341495 for labeling group II metabotropic glutamate receptors in vivo. Nucl. Med. Biol. 2003, 30, 187–190. [Google Scholar] [CrossRef]
- Ma, Y.; Kumata, K.; Yui, J.; Zhang, Y.; Yamasaki, T.; Hatori, A.; Fujinaga, M.; Nengaki, N.; Xie, L.; Wang, H.; et al. Synthesis and evaluation of 1-(cyclopropylmethyl)-4-(4-[11C]methoxyphenyl)- piperidin-1-yl-2-oxo-1,2-dihydropyridine-3-carbonitrile ([11C]CMDC) for PET imaging of metabotropic glutamate receptor 2 in the rat brain. Bioorg. Med. Chem. 2017, 25, 1014–1021. [Google Scholar] [CrossRef]
- Lavreysen, H.; Langlois, X.; Ahnaou, A.; Drinkenburg, W.; te Riele, P.; Biesmans, I.; Van der Linden, I.; Peeters, L.; Megens, A.; Wintmolders, C.; et al. Pharmacological Characterization of JNJ-40068782, a New Potent, Selective, and Systemically Active Positive Allosteric Modulator of the mGlu2 Receptor and Its Radioligand [3H]JNJ-40068782. J. Pharm. Exp. Ther. 2013, 346, 514–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrés, J.I.; Alcázar, J.; Cid, J.M.; De Angelis, M.; Iturrino, L.; Langlois, X.; Lavreysen, H.; Trabanco, A.A.; Celen, S.; Bormans, G. Synthesis, evaluation, and radiolabeling of new potent positive allosteric modulators of the metabotropic glutamate receptor 2 as potential tracers for positron emission tomography imaging. J. Med. Chem. 2012, 55, 8685–8699. [Google Scholar] [CrossRef] [PubMed]
- Leurquin-Sterk, G.; Celen, S.; Van Laere, K.; Koole, M.; Bormans, G.; Langlois, X.; Van Hecken, A.; Te Riele, P.; Alcázar, J.; Verbruggen, A.; et al. What We Observe In Vivo Is Not Always What We See In Vitro: Development and Validation of 11C-JNJ-42491293, A Novel Radioligand for mGluR2. J. Nucl. Med. 2017, 58, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Kumata, K.; Yamasaki, T.; Cheng, R.; Hatori, A.; Ma, L.; Zhang, Y.; Xie, L.; Wang, L.; Kang, H.J.; et al. Synthesis and Preliminary Studies of a Novel Negative Allosteric Modulator, 7-((2,5-Dioxopyrrolidin-1-yl)methyl)-4-(2-fluoro-4-[11C]methoxyphenyl) quinoline-2-carboxamide, for Imaging of Metabotropic Glutamate Receptor 2. ACS Chem. Neurosci. 2017, 8, 1937–1948. [Google Scholar] [CrossRef] [Green Version]
- Kumata, K.; Hatori, A.; Yamasaki, T.; Zhang, Y.; Mori, W.; Fujinaga, M.; Xie, L.; Nengaki, N.; Zhang, M.R. Synthesis and evaluation of 4-(2-fluoro-4-[11C]methoxyphenyl)-5-((2-methylpyridin-4-yl)methoxy) picolinamide for PET imaging of the metabotropic glutamate receptor 2 in the rat brain. Bioorg. Med. Chem. 2019, 27, 483–491. [Google Scholar] [CrossRef]
- Neale, J.H.; Bzdega, T.; Wroblewska, B. N-Acetylaspartylglutamate. J. Neurochem. 2000, 75, 443–452. [Google Scholar] [CrossRef]
- Fricker, A.C.; Mok, M.H.; de la Flor, R.; Shah, A.J.; Woolley, M.; Dawson, L.A.; Kew, J.N. Effects of N-acetylaspartylglutamate (NAAG) at group II mGluRs and NMDAR. Neuropharmacology 2009, 56, 1060–1067. [Google Scholar] [CrossRef]
- Di Menna, L.; Joffe, M.E.; Iacovelli, L.; Orlando, R.; Lindsley, C.W.; Mairesse, J.; Gressèns, P.; Cannella, M.; Caraci, F.; Copani, A.; et al. Functional partnership between mGlu3 and mGlu5 metabotropic glutamate receptors in the central nervous system. Neuropharmacology 2018, 128, 301–313. [Google Scholar] [CrossRef]
- Tu, J.C.; Xiao, B.; Naisbitt, S.; Yuan, J.P.; Petralia, R.S.; Brakeman, P.; Doan, A.; Aakalu, V.K.; Lanahan, A.A.; Sheng, M.; et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 1999, 23, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Corti, C.; Aldegheri, L.; Somogyi, P.; Ferraguti, F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience 2002, 110, 403–420. [Google Scholar] [CrossRef]
- Charvin, D.; Di Paolo, T.; Bezard, E.; Gregoire, L.; Takano, A.; Duvey, G.; Pioli, E.; Halldin, C.; Medori, R.; Conquet, F. An mGlu4-Positive Allosteric Modulator Alleviates Parkinsonism in Primates. Mov. Disord. 2018, 33, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Kil, K.E.; Poutiainen, P.; Zhang, Z.; Zhu, A.; Choi, J.K.; Jokivarsi, K.; Brownell, A.L. Radiosynthesis and evaluation of an 18F-labeled positron emission tomography (PET) radioligand for metabotropic glutamate receptor subtype 4 (mGlu4). J. Med. Chem. 2014, 57, 9130–9138. [Google Scholar] [CrossRef] [Green Version]
- Kil, K.E.; Poutiainen, P.; Zhang, Z.; Zhu, A.; Kuruppu, D.; Prabhakar, S.; Choi, J.K.; Tannous, B.A.; Brownell, A.L. Synthesis and evaluation of N-(methylthiophenyl)picolinamide derivatives as PET radioligands for metabotropic glutamate receptor subtype 4. Bioorg. Med. Chem. Lett. 2016, 26, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Takano, A.; Nag, S.; Jia, Z.; Jahan, M.; Forsberg, A.; Arakawa, R.; Grybäck, P.; Duvey, G.; Halldin, C.; Charvin, D. Characterization of [11C]PXT012253 as a PET Radioligand for mGlu4 Allosteric Modulators in Nonhuman Primates. Mol. Imaging Biol. 2019, 21, 500–508. [Google Scholar] [CrossRef]
- Fujinaga, M.; Yamasaki, T.; Nengaki, N.; Ogawa, M.; Kumata, K.; Shimoda, Y.; Yui, J.; Xie, L.; Zhang, Y.; Kawamura, K.; et al. Radiosynthesis and evaluation of 5-methyl-N- (4-[11C]methylpyrimidin-2-yl)-4-(1H-pyrazol-4-yl)thiazol-2-amine ([11C]ADX88178) as a novel radioligand for imaging of metabotropic glutamate receptor subtype 4 (mGluR4). Bioorg. Med. Chem. Lett. 2016, 26, 370–374. [Google Scholar] [CrossRef]
- Laurie, D.J.; Schoeffter, P.; Wiederhold, K.H.; Sommer, B. Cloning, distribution and functional expression of the human mGlu6 metabotropic glutamate receptor. Neuropharmacology 1997, 36, 145–152. [Google Scholar] [CrossRef]
- Ohishi, H.; Akazawa, C.; Shigemoto, R.; Nakanishi, S.; Mizuno, N. Distributions of the mRNAs for L-2-amino-4-phosphonobutyrate-sensitive metabotropic glutamate receptors, mGluR4 and mGluR7, in the rat brain. J. Comp. Neurol. 1995, 360, 555–570. [Google Scholar] [CrossRef]
- Palazzo, E.; Marabese, I.; Luongo, L.; Guida, F.; de Novellis, V.; Maione, S. Nociception modulation by supraspinal group III metabotropic glutamate receptors. J. Neurochem. 2017, 141, 507–519. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Marton, J.; Ametamey, S.M.; Cumming, P. A Review of Molecular Imaging of Glutamate Receptors. Molecules 2020, 25, 4749. https://doi.org/10.3390/molecules25204749
Kim J-H, Marton J, Ametamey SM, Cumming P. A Review of Molecular Imaging of Glutamate Receptors. Molecules. 2020; 25(20):4749. https://doi.org/10.3390/molecules25204749
Chicago/Turabian StyleKim, Jong-Hoon, János Marton, Simon Mensah Ametamey, and Paul Cumming. 2020. "A Review of Molecular Imaging of Glutamate Receptors" Molecules 25, no. 20: 4749. https://doi.org/10.3390/molecules25204749