
One Pot Synthesis of Micromolar BACE-1 Inhibitors Based on the Dihydropyrimidinone Scaffold and Their Thia and Imino Analogues


 ,
,  ,
,  ,
,
Abstract
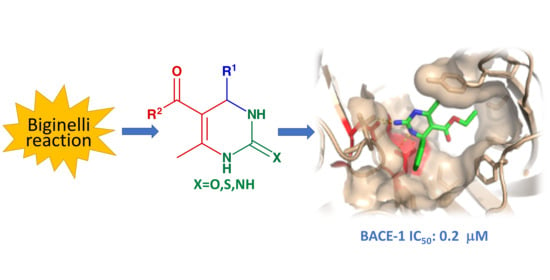
:
1. Introduction
2. Results and Discussion
3. Docking Analysis
4. Materials and Methods
4.1. General Experimental Information
4.2. Synthesis and Characterization
4.2.1. General Method for the Synthesis of 3,4-Diihydropyrimidin-2(1H)-Ones 1a–16a and 3,4-Dihydropyrimidin-2(1H)-Thiones 1b–3b; 9b,10b; 13b
4.2.2. General Method for the Synthesis 3,4-Diihydropyrimidin-2(1H)-Imines 1c–3c, 9c, and 10c
4.2.3. Synthesis of 5-Carboxy-4-Phenyl-6-Methyl-3,4-Dihydropyrimidine-2(1H)-Imine 16c
- (a)
- Synthesis of 5-benzyloxycarbonyl-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-imine17c. From benzaldehyde, benzylacetoacetate, guanidine hydrochloride under the same conditions as above [38]; 0.151 g, 47%; white solid, m.p. 168–170 °C; IR: 3359, 3500–2500 (broad), 1701, 1629, cm−1; 1H-NMR (500 MHz, CDCl3) (Figure S14): δ 2.20 (s, 3H, C(6)CH3), 4.92, 4.98 (AB system, J = 12.6 Hz, PhCH2), 5.22 (s, 1H, H-4), 6.21 (bs, 2H, H-1 and C=NH), 7.09-7.29 (m, 10H, 2xPhH) 7.33 (s, 1H, H-3) ppm; 13C-NMR (125.68 MHz, CDCl3) (Figure S15): δ 24.2, 53.0, 64.3, 96.9, 126.4, 127.4, 127.7, 127.8, 128.6, 137.6 146.8, 156.0, 162.4, 166.2 ppm; HRMS-ESI, m/z (Figure S16): Found: 322.1544 [M+H]+; Calcd for [C19H20N3O2]+ 322.1550.
- (b)
- Hydrogenolysis of 17c. A MeOH solution of the benzylester 17c (0.100 g, 3.1 mmol) was added of 20 mg 10% Pd/C. The mixture was stirred overnight under a H2 atmosphere, then the solvent was removed in vacuo, to give the corresponding carboxylic acid 18c (0.109 g) in a quantitative yield, m.p. 175–176 °C. IR: 3700–2300 (broad), 1700–1600 (multiple bands) cm−1; 1H-NMR (500 MHz, MeOD) (Figure S17): δ 2.22 (s, 3H, C(6)CH3), 5.50 (s, 1H, H-4), 6.16 (br, 2H, H-3 and C=NH), 7.15–7.30 (m, 5H, PhH), 7.39 (s, 1H, H-1) ppm; 13C-NMR (125.68 MHz, MeOD) (Figure S18): δ 21.5, 55.2, 109.0, 126.4, 127.8, 128.5, 145.6, 153.3, 160.0, 175.0 ppm; HRMS-ESI, m/z: Found: 232.1082 [M+H]+; Calcd for [C12H14N3O2]+ 232.1081; Anal. Calcd. for C12H13N3O2 C, 62.33; H, 5.67; N, 18.17; Found C, 62.37; H, 5.80; N, 18.08.
4.3. BACE1 Inhibition Assay
4.4. Docking Studies
4.5. Prediction of ADME Properties
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Biginelli, P. Aldureides of ethylic acetoacetate and ethylic oxaloacetate. Gazz. Chim. Ital. 1893, 23, 360–413. [Google Scholar]
- Kappe, C.O.; Stadler, A. The Biginelli dihydropyrimidinone synthesis. Org. React. 2004, 63, 1–116. [Google Scholar] [CrossRef]
- Nagarajaiah, H.; Mukhopadhyay, A.; Narasimha Moorth, J. Biginelli reaction: An overview. Tetrahedron Lett. 2016, 57, 5135–5148. [Google Scholar] [CrossRef]
- Kaur, R.; Sandeep, C.; Kumar, K.; Gupta, M.K.; Rawal, R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. Eur. J. Med. Chem. 2017, 132, 108–134. [Google Scholar] [CrossRef] [PubMed]
- Neto, B.A.D.; de Fernandes, T.A.; Correia, M.V. Chemistry and Biology of 3,4-Dihydropyrimidin-2(1H)-one (or thione) derivatives obtained by the Biginelli Multicomponent Reaction. Targets Heterocycl. Syst. 2018, 22, 356–376. [Google Scholar] [CrossRef]
- Naidu, B.N.; Sorenson, M.E.; Patel, M.; Ueda, Y.; Banville, J.; Beaulieu, F.; Bollini, S.; Dicker, I.B.; Higley, H.; Lin, Z. Synthesis and evaluation of C2-carbon-linked heterocyclic-5-hydroxy-6-oxo-dihydropyrimidine-4-carboxamides as HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 717–720. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Fauber, B.P.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Ge, H.; Giannetti, A.M.; Hunsaker, T.; Labadie, S.; Liu, Y. Identification of substituted 2-thio-6-oxo-1,6-dihydropyrimidines as inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2013, 23, 3186–3194. [Google Scholar] [CrossRef]
- Atwal, K.S.; Swanson, B.N.; Unger, S.E.; Floyd, D.M.; Moreland, S.; Hedberg, A.; O’Reilly, B.C. Dihydropyrimidine calcium channel blockers. 3. 3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-pyrimidinecarboxylic acid esters as orally effective antihypertensive agents. J. Med. Chem. 1991, 34, 806–811. [Google Scholar] [CrossRef]
- Mayer, U.; Kapoor, T.M.; Haggarty, S.J.; King, R.W.; Schreiber, S.L.; Mitchison, T.J. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999, 286, 971–974. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.S.; Chen, T.B.; O’Malley, S.S.; Pettibone, D.J.; DiSalvo, J.; Francis, B.; Bock, M.G.; Freidinger, R.; Nagarathnam, D.; Miao, S.W.; et al. In vitro studies on L-771,688 (SNAP 6383), a new potent and selective α1A-adrenoceptor antagonist. Eur. J. Pharmacol. 2000, 409, 301–312. [Google Scholar] [CrossRef]
- Lauro, G.; Strocchia, M.; Terracciano, S.; Bruno, I.; Fischer, K.; Pergola, C.; Werz, O.; Riccio, R.; Bifulco, G. Exploration of the dihydropyrimidine scaffold for the development of new potential anti-inflammatory agents blocking prostaglandin E2 synthase-1 enzyme (mPGES-1). Eur. J. Med. Chem. 2014, 80, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O. Biologically active dihydropyrimidones of the Biginelli type—A literature survey. Eur. J. Med. Chem. 2000, 35, 1043–1052. [Google Scholar] [CrossRef]
- Safari, S.; Ghavimi, R.; Razzaghi-Asl, N.; Sepehri, S. Synthesis, biological evaluation and molecular docking study of dihydropyrimidine derivatives as potential anticancer agents. J. Heterocycl. Chem. 2020, 57, 1023–1033. [Google Scholar] [CrossRef]
- Graebin, C.S.; Ribeiro, F.V.; Rogerio, K.R.; Kummerle, A.E. Multicomponent Reactions for the Synthesis of Bioactive Compounds: A Review. Curr. Org. Synth. 2019, 16, 855–899. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, P.; Nardi, M.; Oliverio, M. Similarity and Competition between Biginelli and Hantzsch Reactions: An Opportunity for Modern Medicinal Chemistry. Eur. J. Org. Chem. 2020, 2020, 3954–3964. [Google Scholar] [CrossRef]
- Hsiao, C.-C.; Rombouts, F.; Gijsen, H.J.M. New evolutions in the BACE1 inhibitor field from 2014 to 2018. Bioorg. Med. Chem. Lett. 2019, 29, 761–777. [Google Scholar] [CrossRef]
- Silvestri, R. Boom in the development of non-peptidic β-secretase (BACE1) inhibitors for the treatment of Alzheimer’s disease. Med. Res. Rev. 2009, 29, 295–338. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Cárdenas, E.L.; Osswald, H.L. The design, development, and evaluation of BACE1 inhibitors for the treatment of Alzheimer’s disease. In Topics in Medicinal Chemistry; Wolfe, M.S., Ed.; Springer International Publishing: Berlin, Germany, 2017; Volume 24, pp. 27–86. [Google Scholar]
- Calugia, L.; Lencia, E.; Innocenti, R.; Trabocchi, A. Synthesis of morpholine derivatives using the Castagnoli-Cushman reaction as BACE1 inhibitors: Unexpected binding activity of cyclic thioamides. Bioorg. Med. Chem. Lett. 2020, 30, 127211–127214. [Google Scholar] [CrossRef]
- Pettus, L.H.; Bourbeau, M.P.; Bradley, J.; Bartberger, M.D.; Chen, K.; Hickman, D.; Johnson, M.; Liu, Q.; Manning, J.R.; Nanez, A.; et al. Discovery of AM-6494: A Potent and Orally Efficacious β-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1) Inhibitor with in Vivo Selectivity over BACE2. J. Med. Chem. 2020, 63, 2263–2281. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, A.D.; Kondekar, N.B.; Hung, P.-Y.; Hsieh, C.-E.; Yang, C.-R.; Chen, G.S.; Cher, J.-W. 4-Substituted 2-amino-3,4-dihydroquinazolines with a 3-hairpin turn side chain as novel inhibitors of BACE-1. Bioorg. Chem. 2020, 95, 103135–103149. [Google Scholar] [CrossRef] [PubMed]
- Iraji, A.; Khoshneviszadeh, M.; Firuzi, O.; Khoshneviszadeh, M.; Edraki, N. Novel small molecule therapeutic agents for Alzheimer disease: Focusing on BACE1 and multi-target directed ligands. Bioorg. Chem. 2020, 97, 103649–103678. [Google Scholar] [CrossRef] [PubMed]
- Finder, V.H.J. Alzheimer’s disease: A general introduction and pathomechanism. Alzheimer’s Dis. 2010, 22, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Moussa-Pacha, N.M.; Abdin, S.N.; Omar, H.A.; Alniss, H.; Al Tel, T.H. BACE1 inhibitors: Current Status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Watling, M.; Imbimbo, B.P. Do BACE inhibitor failures in Alzheimer patients challenge the amyloid hypothesis of the disease? Expert Rev. Neurother. 2019, 19, 599–602. [Google Scholar] [CrossRef]
- Maia, M.A.; Sousa, E. BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals 2019, 12, 41. [Google Scholar] [CrossRef] [Green Version]
- Felluga, F.; Benedetti, F.; Berti, F.; Drioli, S.; Regini, G. Efficient Biginelli Synthesis of 2-Aminopyrimidines under Microwave Irradiation. Synlett 2018, 29, 1047–1054. [Google Scholar] [CrossRef]
- Nüchter, M.; Ondruschka, B. Tools for microwave-assisted parallel syntheses and combinatorial chemistry. Mol. Divers. 2003, 7, 253–264. [Google Scholar] [CrossRef]
- Wipf, P.; Cunningham, A. A solid phase protocol of the Biginelli dihydropyrimidine synthesis suitable for combinatorial chemistry. Tetrahedron Lett. 1995, 36, 7819–7822. [Google Scholar] [CrossRef]
- Kappe, C.O. Synthesis and reactions of Biginelli compounds. Part 17. Highly versatile solid phase synthesis of biofunctional 4-aryl-3,4-dihydropyrimidines using resin-bound isothiourea building blocks and multidirectional resin cleavage. Bioorg. Med. Chem. Lett. 2000, 10, 49–51. [Google Scholar] [CrossRef]
- Gomes, C.; Vinagreiro, C.S.; Damas, L.; Aquino, G.; Quaresma, J.; Chaves, C.; Pimenta, J.; Campos, J.; Pereira, M.; Pineiro, M. Advanced Mechanochemistry Device for Sustainable Synthetic Processes. ACS Omega 2020, 5, 10868–10877. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, A.; Bazgir, A.; Teimouri, F. Ammonium chloride-catalyzed one-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones under solvent-free conditions. Tetrahedron Lett. 2003, 44, 857–859. [Google Scholar] [CrossRef]
- Niu, Y.; Gao, H.; Xu, F.; Wang, C.; Liu, P.; Yang, G.; Sun, Q.; Xu, P. Synthesis, in vitro biological evaluation and molecular docking studies of benzimidamides as potential BACE1 inhibitors. Chem. Biol. Drug Des. 2012, 80, 775–780. [Google Scholar] [CrossRef]
- Chiummiento, L.; Funicello, M.; Lupattelli, P.; Tramutola, F.; Berti, F. Synthesis and biological evaluation of novel small non-peptidic HIV-1 PIs: The benzothiophene ring as an effective moiety. Bioorg. Med. Chem. Lett. 2012, 22, 2948–2950. [Google Scholar] [CrossRef]
- Vanden Eynde, J.J.; Hecq, N.; Kataeva, O.; Kappe, C.O. Microwave-mediated regioselective synthesis of novel pyrimido[1,2-a]pyrimidines under solvent-free conditions. Tetrahedron 2001, 57, 1785–1791. [Google Scholar] [CrossRef]
- Ahmad, M.J.; Hassan, S.F.; Nisa, R.U.; Ayub, K.; Nadeem, M.S.; Nazir, S.; Ansari, F.L.; Qureshi, N.A.; Rashid, U. Synthesis, in vitro potential and computational studies on 2-amino-1, 4-dihydropyrimidines as multitarget antibacterial ligands. Med. Chem. Res. 2016, 25, 1877–1894. [Google Scholar] [CrossRef]
- Ahmeda, B.; Khan, R.A.; Habibullah; Keshari, M. An improved synthesis of Biginelli-type compounds via phase-transfer catalysis. Tetrahedron Lett. 2009, 50, 2889–2892. [Google Scholar] [CrossRef]
- Oehlrich, D.; Prokopcova, H.; Gijsen, J.M. The evolution of amidine-based brain penetrant BACE1 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2033–2045. [Google Scholar] [CrossRef] [Green Version]
- Rankovic, Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, E.W.; Conway, A.K.; Kennis, L.; Bischoff, F.; Mercken, M.H.; De Winter, H.L.; Reynolds, C.H.; Tounge, B.A.; Luo, C.; Scott, M.K.; et al. 2-Amino-3,4-dihydroquinazolines as Inhibitors of BACE-1 (-Site APP Cleaving Enzyme): Use of Structure Based Design to Convert a Micromolar Hit into a Nanomolar Lead. J. Med. Chem. 2007, 50, 4261–4264. [Google Scholar] [CrossRef]
- Hua, K.M.; Tran, P.H.; Le, T.N. An efficient and recyclable L-proline triflate ionic liquid catalyst for one-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones via the multi-component Biginelli reaction. Arkivoc 2019, 6, 406–415. [Google Scholar] [CrossRef]
- Khorshidi, A.; Tabatabaeian, K.; Azizi, H.; Aghaei-Hashjin, M.; Abbaspour-Gilandeh, E. Efficient one-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones catalyzed by a new heterogeneous catalyst based on Co-functionalized Na+-montmorillonite. RSC Adv. 2017, 7, 17732–17740. [Google Scholar] [CrossRef] [Green Version]
- Crespo, A.; El Maatougui, A.; Biagini, P.; Azuaje, J.; Coelho, A.; Brea, J.; Loza, M.I.; Cadavid, M.I.; García-Mera, X.; Gutierrez de Teran, H.; et al. Discovery of 3,4-Dihydropyrimidin-2(1H)-ones as a novel class of potent and selective A2B adenosine receptor antagonists. ACS Med. Chem. Lett. 2013, 4, 1031–1036. [Google Scholar] [CrossRef] [Green Version]
- Pasunooti, K.K.; Chai, H.; Jensen, C.N.; Gorityala, B.K.; Wang, S.; Liu, X.W. A Microwave-assisted, Copper-Catalyzed Three-Component Synthesis of Dihydropyrimidinones under Mild Conditions. Tetrahedron Lett. 2011, 52, 80–84. [Google Scholar] [CrossRef]
- Gadkari, Y.U.; Hatvate, N.T.; Takale, B.S.; Telvekar, V.N. Concentrated solar radiation as a renewable heat source for a preparative-scale and solvent-free Biginelli reaction. New J. Chem. 2020, 44, 8167–8170. [Google Scholar] [CrossRef]
- Fu, L.-H.; Xie, Z.-B.; Lan, J.; Li, H.-X.; Liu, L.-S.; Le, Z.-G. Biginelli reaction of aliphatic aldehydes catalyzed by α-chymotrypsin: One-pot biocatalytic synthesis of dihydropyrimidinones. Heterocycles 2018, 96, 1808–1820. [Google Scholar] [CrossRef] [Green Version]
- Couto, I.; Tellitu, I.; Domínguez, E. Searching for a direct preparation of dihydropyrimidine-5-carboxamides under Biginelli reaction conditions. Arkivoc 2011, 2, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Soumyanarayanan, U.; Bhat, V.G.; Kar, S.S.; Mathew, J.A. Monastrol mimic Biginelli dihydropyrimidinone derivatives: Synthesis, cytotoxicity screening against HepG2 and HeLa cell lines and molecular modeling study. Org. Med. Chem. Lett. 2012, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Roßbach, J.; Baumeister, J.; Harms, K.; Koert, U. Regio- and Diastereoselective Crotylboration of vic-Tricarbonyl Compounds. Eur. J. Org. Chem. 2013, 6, 662–665. [Google Scholar] [CrossRef]
- Desai, B.; Dallinger, D.; Kappe, C.O. Microwave-assisted solution phase synthesis of dihydropyrimidine C5 amides and esters. Tetrahedron 2006, 62, 4651–4664. [Google Scholar] [CrossRef]
- Stachel, S.J.; Coburn, C.A.; Steele, T.G.; Jones, K.G.; Loutzenhiser, E.F.; Gregro, A.R.; Rajapakse, H.A.; Lai, M.-T.; Crouthamel, M.-C.; Xu, M.; et al. Structure-Based Design of Potent and Selective Cell-Permeable Inhibitors of Human β-Secretase (BACE-1). J. Med. Chem. 2004, 47, 6447–6450. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2015, 12, 281–296. [Google Scholar] [CrossRef]
- Maestro. Schrödinger Release, 2016-4; Schrödinger, Inc.: New York, NY, USA, 2016. [Google Scholar]
- Chemicaliase. Available online: https://chemicalize.com/ (accessed on 30 June 2020).
- Daina, A.; Michielin, A.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dihydropyrimidine | Yield (%) | IC50 (μM) | LogP2 | Dihydropyrimidine | Yield (%) | IC50 (μM) | LogP2 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
 |  | ||||||||||
| 1a | X=O | 82 | 2.8 ± 0.1 | 1.2 | 9a | X=O | 89 | 3.1 ± 0.4 | 2.2 | ||
| 1b | X=S | 85 | 1.5 ± 0.4 | 2.1 | 9b | X=S | 87 | 0.3 ± 0.1 | 3.1 | ||
| 1c | X=NH | 83 | 0.5 ± 0.1 | 1.5 | 9c | X=NH | 88 | 1.5 ± 0.4 | 2.5 | ||
| (−0.8)3 | (0.1)3 | ||||||||||
 |  | ||||||||||
| 2a | X=O | 83 | 0.32 ± 0.05 | 0.9 | 10a | X=O | 87 | 0.50 ± 0.05 | 2.2 | ||
| 2b | X=S | 87 | 0.85 ± 0.03 | 1.8 | 10b | X=S | 88 | 1.1 ± 0.1 | 3.1 | ||
| 2c | X=NH | 80 | 0.52 ± 0.11 | 1.1 | 10c | X=NH | 90 | 0.2 ± 0.1 | 2.5 | ||
| (−1.2)3 | (0.2)3 | ||||||||||
 |  | 11a | X=O | 73 | 71.3 ± 1.2 | 2.1 | |||||
| 3a | X=O | 70 | 1.7 ± 0.4 | 1.4 | |||||||
| 3b | X=S | 68 | 0.24 ± 0.05 | 2.3 | |||||||
| 3c | X=NH | 70 | 0.30 ± 0.15 | 1.6 | |||||||
| (−0.6)3 | |||||||||||
 | 4a | X=O | 81 | 0.6 ± 0.3 | 2.1 |  | 12a | X=O | 85 | 1.35 ± 0.10 | 2.3 |
| 12b | X=S | 73 | 0.7 ± 0.1 | 3.1 | |||||||
 | 5a | X=O | 78 | 0.65 ± 0.03 | 0.8 |  | 13a | X=O | 50 | 2.3 ± 0.2 | 1.3 |
 | 6a | X=O | 58 | 3.2 ± 0.3 | 1.0 |  | 14a | X=O | 47 | 1.6 ± 0.1 | −0.3 |
 | 7a | X=O | 61 | 2.2 ± 0.2 | 1.2 |  | 15a | X=O | 60 | 1.6 ± 0.1 | −0.1 |
 | 8a | X=O | 50 | 1.2 ± 0.4 | 1.3 |  | 16a | X=O | 454 | 34.0 ± 8.0 | −2.93 |
| 16b | X=S | 804 | 1.2 ± 0.3 | −1.93 | |||||||
| 16c | X=NH | 475 | 0.7 ± 0.2 | −0.91 | |||||||
| Inhibitor | Experimental IC50 1 (nM) | Rel ΔEb 2 (Kcal mol−1) |
|---|---|---|
| (R)-10c | 200 | 0 |
| (S)-10c | 5.2 | |
| (R)-9c | 150 | 5.6 |
| (R)-1c | 500 | 2.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bais, J.; Benedetti, F.; Berti, F.; Cerminara, I.; Drioli, S.; Funicello, M.; Regini, G.; Vidali, M.; Felluga, F. One Pot Synthesis of Micromolar BACE-1 Inhibitors Based on the Dihydropyrimidinone Scaffold and Their Thia and Imino Analogues. Molecules 2020, 25, 4152. https://doi.org/10.3390/molecules25184152
Bais J, Benedetti F, Berti F, Cerminara I, Drioli S, Funicello M, Regini G, Vidali M, Felluga F. One Pot Synthesis of Micromolar BACE-1 Inhibitors Based on the Dihydropyrimidinone Scaffold and Their Thia and Imino Analogues. Molecules. 2020; 25(18):4152. https://doi.org/10.3390/molecules25184152
Chicago/Turabian StyleBais, Jessica, Fabio Benedetti, Federico Berti, Iole Cerminara, Sara Drioli, Maria Funicello, Giorgia Regini, Mattia Vidali, and Fulvia Felluga. 2020. "One Pot Synthesis of Micromolar BACE-1 Inhibitors Based on the Dihydropyrimidinone Scaffold and Their Thia and Imino Analogues" Molecules 25, no. 18: 4152. https://doi.org/10.3390/molecules25184152