
LRRK2 Regulates CPT1A to Promote β-Oxidation in HepG2 Cells


Abstract
:1. Introduction
2. Results
2.1. LRRK2 Was Down-Regulated in the Liver of High-Fat Diet Induced NAFLD Mice
2.2. Palmitic Acid Reduced the Expression of LRRK2 in HepG2 Cells
2.3. Overexpression of LRRK2 Promoted Catabolism of Free Fatty Acid in PA-Treated HepG2 Cells
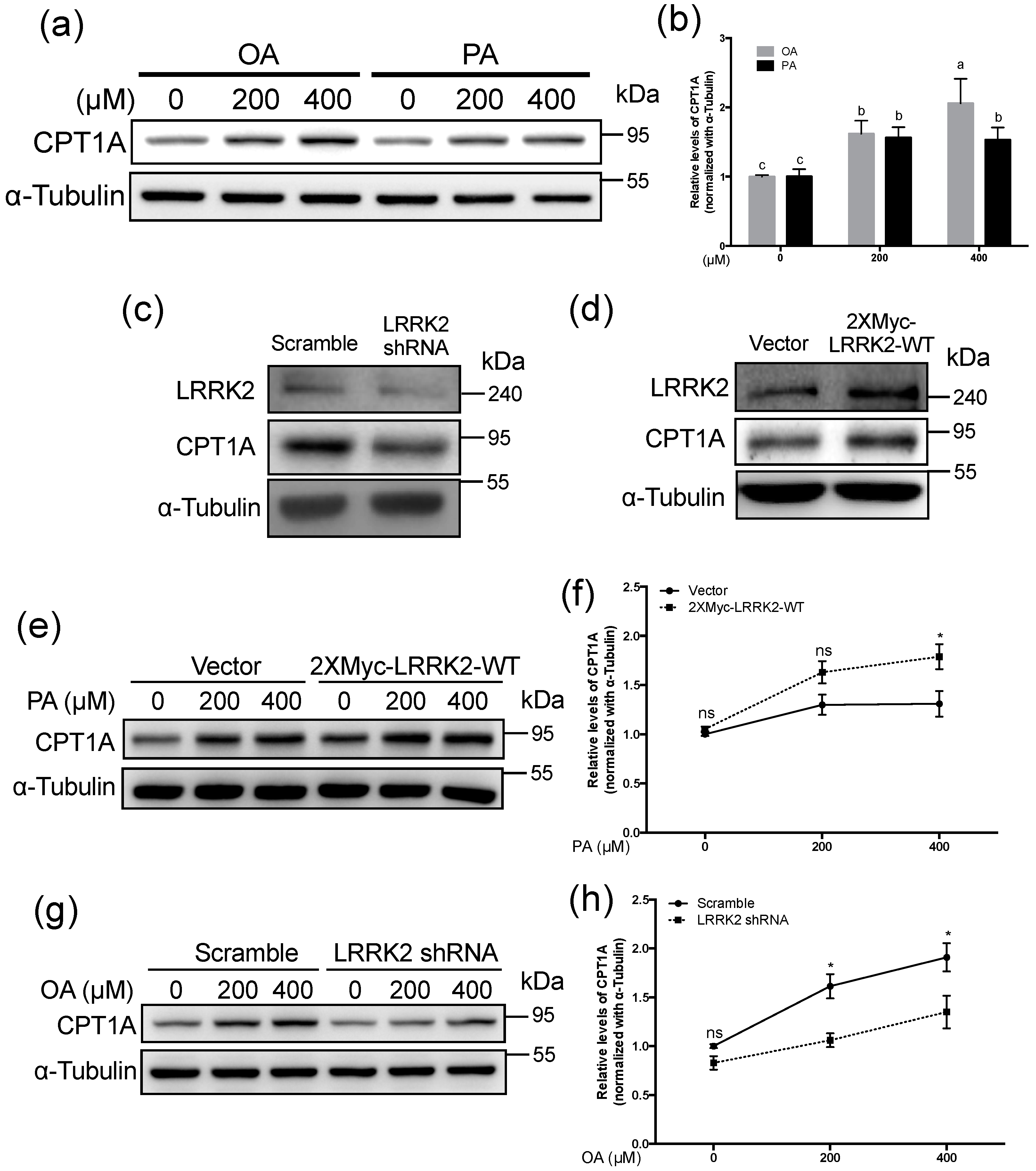
2.4. LRRK2 Positively Regulated CPT1A in HepG2 Cells
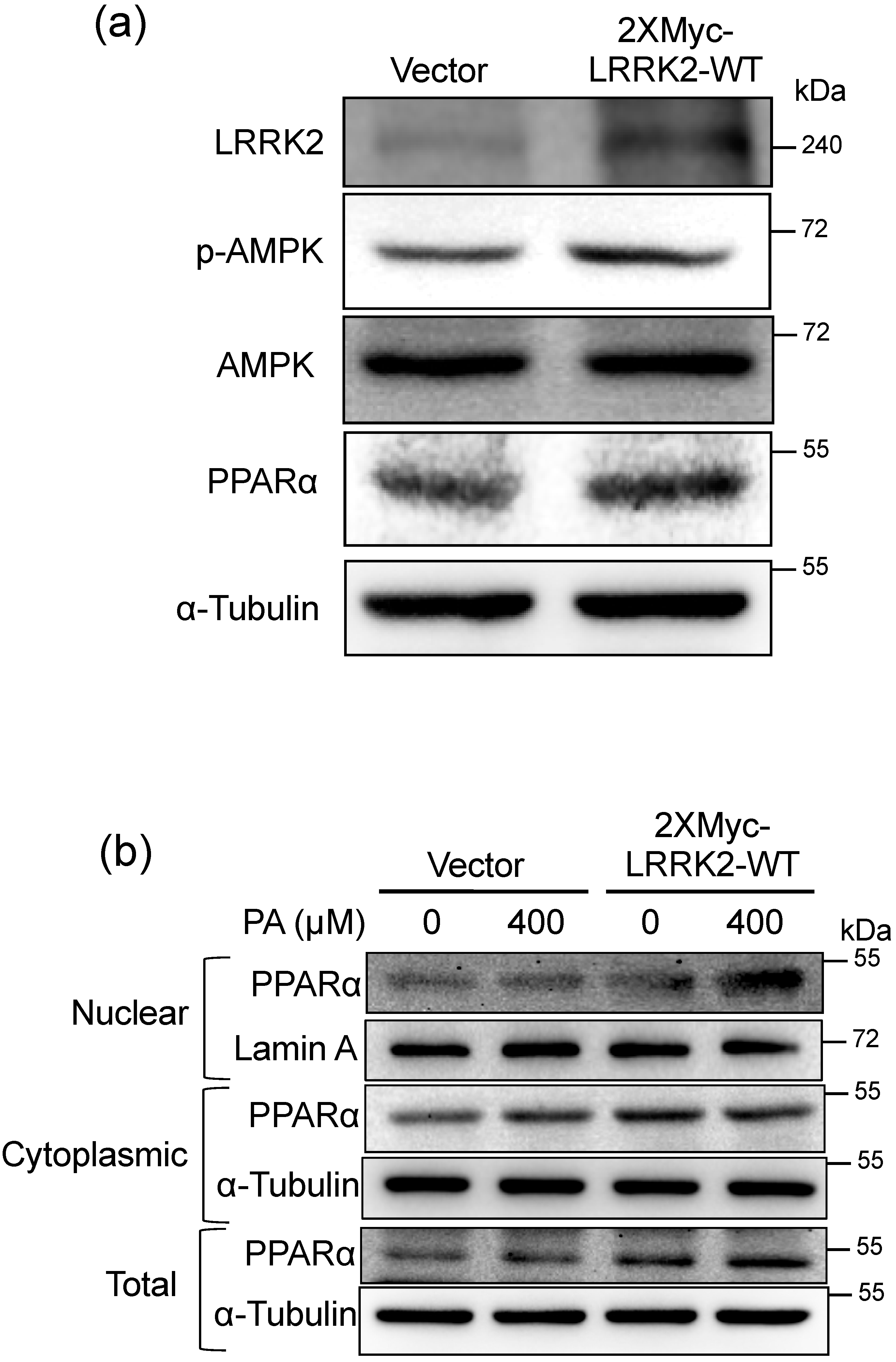
2.5. LRRK2 Activated AMPK and PPARα in HepG2 Cells
2.6. LRRK2 Suppressed the Levels of TNFα in HepG2 Cells after PA Treatment
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Histological Analyses
4.3. RNA Extraction and Real-Time PCR Analyses
4.4. Protein Extraction and Western Blot
4.5. Cell Culture and Fatty Acid Treatment
4.6. Knockdown of LRRK2 in HepG2 Cells
4.7. Overexpression of LRRK2
4.8. Measurement of TG and NEFA
4.9. Measurement the Activity of Fatty Acid Oxidation
4.10. Extraction of Cytoplasmic and Nuclear Proteins
4.11. ELISA Analysis of TNFα and Interleukin 8 Concentrations
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1. AMPK | AMP-activated protein kinase |
| 2. CPT1A | Carnitine palmitoyltransferase 1A |
| 3. H&E | Hematoxylin and eosin |
| 4. IHC | Immunohistochemistry |
| 5. IL-8 | Interleukin-8 |
| 6. LRRK2 | Leucine-Rich Repeat Kinase 2 |
| 7. NAFLD | Nonalcoholic fatty liver disease |
| 8. NASH | Nonalcoholic steatohepatitis |
| 9. NEFA | Non-esterified fatty acid |
| 10. NF-κB | Nuclear factor kappa B |
| 11. OA | Oleic acid |
| 12. PA | Palmitic acid |
| 13. PPARα | Peroxisome proliferator-activated receptor α |
| 14. shRNA | Short hairpin RNA |
| 15. TG | Triglyceride |
| 16. TNFα | Tumor necrosis factor α |
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Kabbany, M.N.; Conjeevaram Selvakumar, P.K.; Watt, K.; Lopez, R.; Akras, Z.; Zein, N.; Carey, W.; Alkhouri, N. Prevalence of Nonalcoholic Steatohepatitis-Associated Cirrhosis in the United States: An Analysis of National Health and Nutrition Examination Survey Data. Am. J. Gastroenterol. 2017, 112, 581–587. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Bril, F.; Barb, D.; Portillo-Sanchez, P.; Biernacki, D.; Lomonaco, R.; Suman, A.; Weber, M.H.; Budd, J.T.; Lupi, M.E.; Cusi, K. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipose tissue, obesity and non-alcoholic fatty liver disease. Minerva. Endocrinol. 2017, 42, 92–108. [Google Scholar]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Ubhayasekera, S.J.; Staaf, J.; Forslund, A.; Bergsten, P.; Bergquist, J. Free fatty acid determination in plasma by GC-MS after conversion to Weinreb amides. Anal. Bioanal. Chem. 2013, 405, 1929–1935. [Google Scholar] [CrossRef]
- de Almeida, I.T.; Cortez-Pinto, H.; Fidalgo, G.; Rodrigues, D.; Camilo, M.E. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin. Nutr. 2002, 21, 219–223. [Google Scholar] [CrossRef]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xue, R.; Zhang, Z.; Yang, X.; Shi, H. Palmitic and linoleic acids induce ER stress and apoptosis in hepatoma cells. Lipids Health. Dis. 2012, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar] [CrossRef]
- Miura, K.; Yang, L.; van Rooijen, N.; Brenner, D.A.; Ohnishi, H.; Seki, E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013, 57, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Pimenta, A.S.; Gaidhu, M.P.; Habib, S.; So, M.; Fediuc, S.; Mirpourian, M.; Musheev, M.; Curi, R.; Ceddia, R.B. Prolonged exposure to palmitate impairs fatty acid oxidation despite activation of AMP-activated protein kinase in skeletal muscle cells. J. Cell. Physiol. 2008, 217, 478–485. [Google Scholar] [CrossRef]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vazquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef]
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Park, J.S.; Roh, Y.S. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch. Pharm. Res. 2019, 42, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Attia, R.R.; Connaughton, S.; Niesen, M.I.; Ness, G.C.; Elam, M.B.; Hori, R.T.; Cook, G.A.; Park, E.A. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol. Cell. Endocrinol. 2010, 325, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Tobita, H.; Sato, S.; Yazaki, T.; Mishiro, T.; Ishimura, N.; Ishihara, S.; Kinoshita, Y. Alogliptin alleviates hepatic steatosis in a mouse model of nonalcoholic fatty liver disease by promoting CPT1a expression via Thr172 phosphorylation of AMPKalpha in the liver. Mol. Med. Rep. 2018, 17, 6840–6846. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.A.; Pinkosky, S.L.; Filippov, S.; Hanselman, J.C.; Cramer, C.T.; Newton, R.S. AMP-activated protein kinase: An emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. J. Lipid Res. 2012, 53, 2490–2514. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Kim, M.; Park, H.S.; Kim, H.S.; Jeon, M.J.; Oh, K.S.; Koh, E.H.; Won, J.C.; Kim, M.S.; Oh, G.T.; et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem. Biophys. Res. Commun. 2006, 340, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Li, K.; Hui, X.; Kong, X.; Sweeney, G.; Wang, Y.; Xu, A.; Teng, M.; Liu, P.; Wu, D. Carnitine palmitoyltransferase 1A prevents fatty acid-induced adipocyte dysfunction through suppression of c-Jun N-terminal kinase. Biochem. J. 2011, 435, 723–732. [Google Scholar] [CrossRef]
- Ai, L.; Xu, Q.; Wu, C.; Wang, X.; Chen, Z.; Su, D.; Jiang, X.; Xu, A.; Lin, Q.; Fan, Z. A20 Attenuates FFAs-induced Lipid Accumulation in Nonalcoholic Steatohepatitis. Int. J. Biol. Sci. 2015, 11, 1436–1446. [Google Scholar] [CrossRef] [Green Version]
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gomez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.; Boldt, K.; et al. Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. USA 2016, 113, 4357–4366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata, I.F.; Wedemeyer, W.J.; Farrer, M.J.; Taylor, J.P.; Gallo, K.A. LRRK2 in Parkinson’s disease: Protein domains and functional insights. Trends Neurosci. 2006, 29, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Seol, W.; Nam, D.; Son, I. Rab GTPases as Physiological Substrates of LRRK2 Kinase. Exp. Neurobiol. 2019, 28, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef]
- Rolli-Derkinderen, M.; Leclair-Visonneau, L.; Bourreille, A.; Coron, E.; Neunlist, M.; Derkinderen, P. Is Parkinson’s disease a chronic low-grade inflammatory bowel disease? J. Neurol. 2020, 267, 2207–2213. [Google Scholar] [CrossRef]
- Zhang, F.R.; Huang, W.; Chen, S.M.; Sun, L.D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.X.; Yang, H.T.; Yang, R.D.; et al. Genomewide association study of leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptista, M.A.; Dave, K.D.; Frasier, M.A.; Sherer, T.B.; Greeley, M.; Beck, M.J.; Varsho, J.S.; Parker, G.A.; Moore, C.; Churchill, M.J.; et al. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS ONE 2013, 8, e80705. [Google Scholar] [CrossRef] [Green Version]
- Boddu, R.; Hull, T.D.; Bolisetty, S.; Hu, X.; Moehle, M.S.; Daher, J.P.; Kamal, A.I.; Joseph, R.; George, J.F.; Agarwal, A.; et al. Leucine-rich repeat kinase 2 deficiency is protective in rhabdomyolysis-induced kidney injury. Hum. Mol. Genet. 2015, 24, 4078–4093. [Google Scholar] [CrossRef]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Arshad, M.; Wang, W.; Zhao, D.; Xu, L.; Zhou, L. LRRK2 mediated Rab8a phosphorylation promotes lipid storage. Lipids Health. Dis. 2018, 17, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, 825–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Greggio, E.; Zambrano, I.; Kaganovich, A.; Beilina, A.; Taymans, J.M.; Daniels, V.; Lewis, P.; Jain, S.; Ding, J.; Syed, A.; et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 2008, 283, 16906–16914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Pan, P.; Jing, J.; Hu, X.; Chen, L.; Qiu, X.; Ma, R.; Jueraitetibaike, K.; Huang, X.; Yao, B. Rosiglitazone ameliorates palmitic acid-induced cytotoxicity in TM4 Sertoli cells. Reprod. Biol. Endocrinol. 2018, 16, 98. [Google Scholar] [CrossRef] [PubMed]
- Cleophas, M.C.P.; Crisan, T.O.; Kluck, V.; Hoogerbrugge, N.; Netea-Maier, R.T.; Dinarello, C.A.; Netea, M.G.; Joosten, L.A.B. Romidepsin suppresses monosodium urate crystal-induced cytokine production through upregulation of suppressor of cytokine signaling 1 expression. Arthritis Res. 2019, 21, 50. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Suaga, P.; Luzon-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef] [Green Version]
- Malandrino, M.I.; Fucho, R.; Weber, M.; Calderon-Dominguez, M.; Mir, J.F.; Valcarcel, L.; Escote, X.; Gomez-Serrano, M.; Peral, B.; Salvado, L.; et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am. J. Physiol. Endocrinol. Metab. 2015, 308, 756–769. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Xiao, K.; Zhang, Z.; Ma, L. Altered Expression of EPO Might Underlie Hepatic Hemangiomas in LRRK2 Knockout Mice. Biomed Res. Int. 2016, 2016, 7681259. [Google Scholar] [CrossRef] [Green Version]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Miklossy, J.; Arai, T.; Guo, J.P.; Klegeris, A.; Yu, S.; McGeer, E.G.; McGeer, P.L. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006, 65, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moravcova, A.; Cervinkova, Z.; Kucera, O.; Mezera, V.; Rychtrmoc, D.; Lotkova, H. The effect of oleic and palmitic acid on induction of steatosis and cytotoxicity on rat hepatocytes in primary culture. Physiol. Res. 2015, 64, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Mei, S.; Ni, H.M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.X. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J. Pharm. Exp. 2011, 339, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Gerhart-Hines, Z.; Dominy, J.E.; Lee, Y.; Kim, S.; Tabata, M.; Xiang, Y.K.; Puigserver, P. Oleic acid stimulates complete oxidation of fatty acids through protein kinase A-dependent activation of SIRT1-PGC1alpha complex. J. Biol. Chem. 2013, 288, 7117–7126. [Google Scholar] [CrossRef] [Green Version]
- Nemecz, M.; Constantin, A.; Dumitrescu, M.; Alexandru, N.; Filippi, A.; Tanko, G.; Georgescu, A. The Distinct Effects of Palmitic and Oleic Acid on Pancreatic Beta Cell Function: The Elucidation of Associated Mechanisms and Effector Molecules. Front. Pharm. 2018, 9, 1554. [Google Scholar] [CrossRef] [Green Version]
- Papazyan, R.; Sun, Z.; Kim, Y.H.; Titchenell, P.M.; Hill, D.A.; Lu, W.; Damle, M.; Wan, M.; Zhang, Y.; Briggs, E.R.; et al. Physiological Suppression of Lipotoxic Liver Damage by Complementary Actions of HDAC3 and SCAP/SREBP. Cell Metab. 2016, 24, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef] [Green Version]
- Schilling, J.D.; Machkovech, H.M.; He, L.; Sidhu, R.; Fujiwara, H.; Weber, K.; Ory, D.S.; Schaffer, J.E. Palmitate and lipopolysaccharide trigger synergistic ceramide production in primary macrophages. J. Biol. Chem. 2013, 288, 2923–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Zhang, X.; Lu, Z.; Perry, D.M.; Li, Y.; Russo, S.B.; Cowart, L.A.; Hannun, Y.A.; Huang, Y. Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am. J. Physiol. Endocrinol. Metab. 2013, 305, 853–867. [Google Scholar] [CrossRef]
- Rico, J.E.; Mathews, A.T.; Lovett, J.; Haughey, N.J.; McFadden, J.W. Palmitic acid feeding increases ceramide supply in association with increased milk yield, circulating nonesterified fatty acids, and adipose tissue responsiveness to a glucose challenge. J. Dairy Sci. 2016, 99, 8817–8830. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulou, M.; Gordillo, R.; Koliaki, C.; Gancheva, S.; Jelenik, T.; De Filippo, E.; Herder, C.; Markgraf, D.; Jankowiak, F.; Esposito, I.; et al. Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes Care 2018, 41, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Li, C.; Liu, Q.; Wang, A.; Lei, M. Inhibiting Ceramide Synthesis Attenuates Hepatic Steatosis and Fibrosis in Rats With Non-alcoholic Fatty Liver Disease. Front. Endocrinol. (Lausanne) 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Di Benedetto, G.; Kaganovich, A.; Ding, J.; Mercatelli, D.; Morari, M.; Cookson, M.R.; Bubacco, L.; Greggio, E. Leucine-rich repeat kinase 2 controls protein kinase A activation state through phosphodiesterase 4. J. Neuroinflamm. 2018, 15, 297. [Google Scholar] [CrossRef]
- Russo, I.; Berti, G.; Plotegher, N.; Bernardo, G.; Filograna, R.; Bubacco, L.; Greggio, E. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-kappaB p50 signaling in cultured microglia cells. J. Neuroinflamm. 2015, 12, 230. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.J.; Shen, T.L.; Chen, Y.S.; Mersmann, H.J.; Liu, B.H.; Ding, S.T. Adiponectin and adiponectin receptor 1 overexpression enhance inflammatory bowel disease. J. Biomed. Sci. 2018, 25, 24. [Google Scholar] [CrossRef]
- Cui, A.; Hu, Z.; Han, Y.; Yang, Y.; Li, Y. Optimized Analysis of In Vivo and In Vitro Hepatic Steatosis. J. Vis. Exp. 2017, 121, 55178. [Google Scholar]
- Robinet, P.; Wang, Z.; Hazen, S.L.; Smith, J.D. A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J. Lipid Res. 2010, 51, 3364–3369. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer Sequence | Reference Sequence |
|---|---|---|
| Mouse Fasn | F: GGAGGTGGTGATAGCCGGTAT R: TGGGTAATCCATAGAGCCCAG | NM_007988.3 |
| Mouse Srebp1c | F: GGAGCCATGGATTGCACATT R: GGCCCGGGAAGTCACTGT | NM_001358314.1 |
| Mouse Acc | F: TAATGGGCTGCTTCTGTGACTC R: CTCAATATCGCCATCAGTCTTG | NM_133360.2 |
| Mouse Pparg | F: TTGCTGTGGGGATGTCTCAC R: AACAGCTTCTCCTTCTCGGC | NM_001127330.2 |
| Mouse Tnfa | F: CCACGTCGTAGCAAACCAC R: TTGTCCCTTGAAGAGAACCTG | NM_013693.3 |
| Mouse Il1b | F: GCAGTGGTTCGAGGCCTAAT R: GCTGCTTCAGACACTTGCAC | NM_008361.4 |
| Mouse Lrrk2 | F: ATGGAGTTGGCCTCCAAAGG R: GATCCCGTAGTCCGCAATCT | NM_025730.3 |
| Mouse Ppia | F: AGGATTCATGTGCCAGGGTG R: GATGCCAGGACCTGTATGCT | NM_008907.2 |
| Human TNFA | F: AGCCTCTTCTCCTTCCTGAT R: AAGATGATCTGACTGCCTGG | NM_000594.4 |
| Human IL8 | F: CCAGGAAGAAACCACCGGA R: GAAATCAGGAAGGCTGCCAAG | NM_000584.4 |
| Human ACTB | F: GAAGATCAAGATCATTGCTCCTC R: CTAAGTCATAGTCCGCCTAGAAG | NM_001101.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-W.; Peng, Y.-J.; Lin, Y.-Y.; Mersmann, H.J.; Ding, S.-T. LRRK2 Regulates CPT1A to Promote β-Oxidation in HepG2 Cells. Molecules 2020, 25, 4122. https://doi.org/10.3390/molecules25184122
Lin C-W, Peng Y-J, Lin Y-Y, Mersmann HJ, Ding S-T. LRRK2 Regulates CPT1A to Promote β-Oxidation in HepG2 Cells. Molecules. 2020; 25(18):4122. https://doi.org/10.3390/molecules25184122
Chicago/Turabian StyleLin, Chiao-Wei, Yu-Ju Peng, Yuan-Yu Lin, Harry John Mersmann, and Shih-Torng Ding. 2020. "LRRK2 Regulates CPT1A to Promote β-Oxidation in HepG2 Cells" Molecules 25, no. 18: 4122. https://doi.org/10.3390/molecules25184122