
Mechanistic Insights into the Chaperoning of Human Lysosomal-Galactosidase Activity: Highly Functionalized Aminocyclopentanes and C-5a-Substituted Derivatives of 4-epi-Isofagomine

 , ,
, ,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Synthesis of 4-epi-Isofagomines
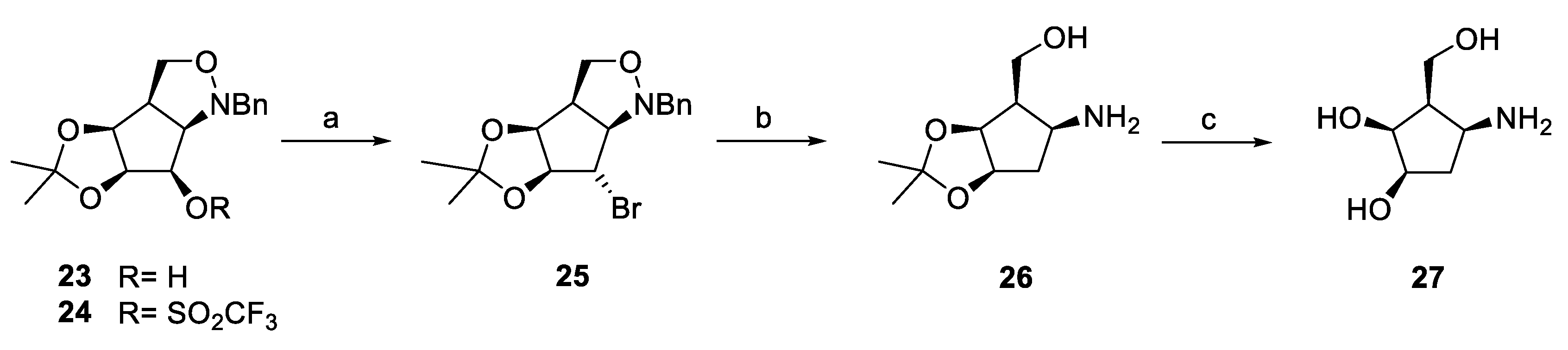
2.2. Synthesis of Cyclopentylamines
2.3. Inhibitory Activities
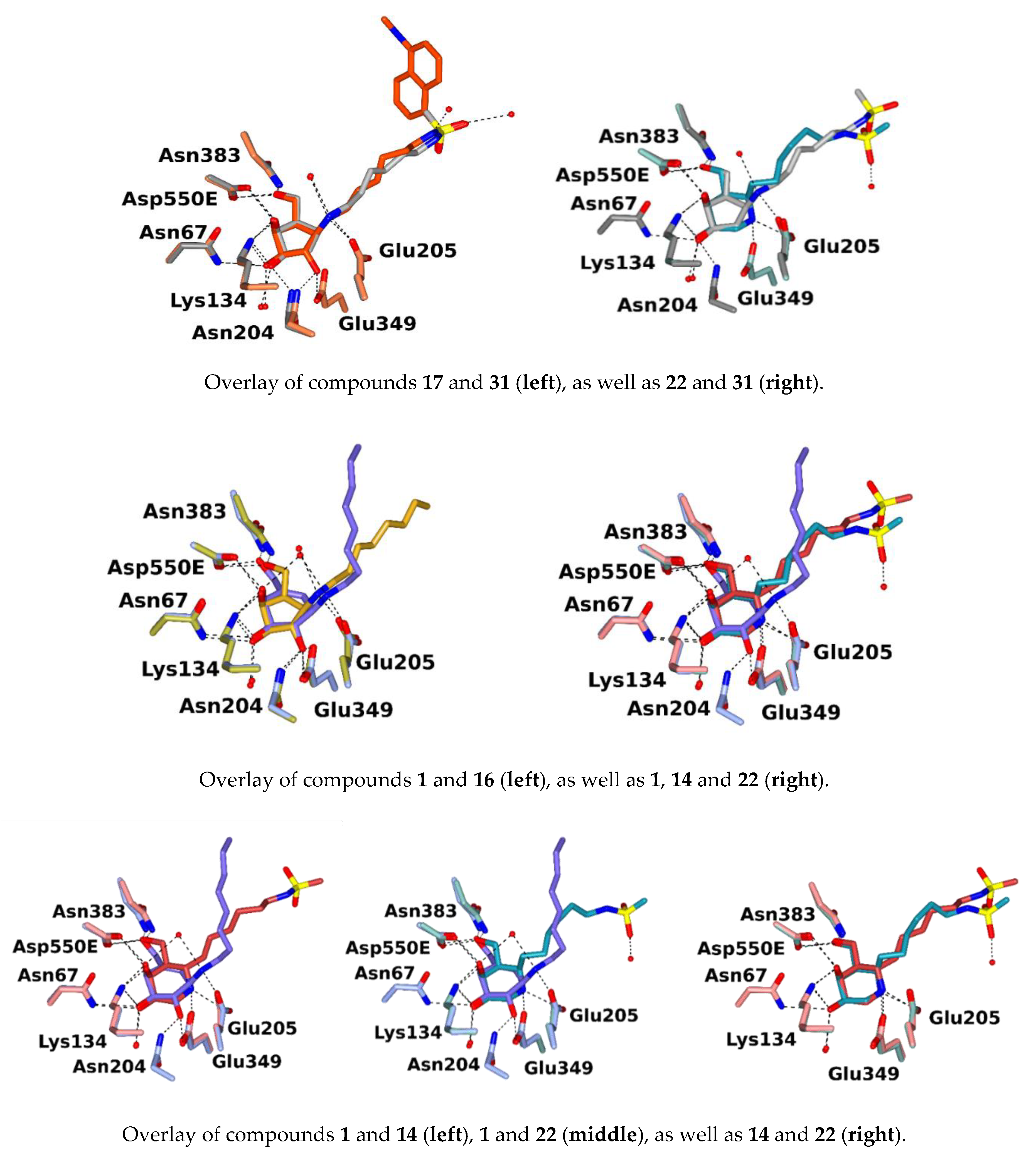
2.4. Inhibitor Binding in Crystal Structures of a Model β-Galactosidase from Cellvibrio Japonicus
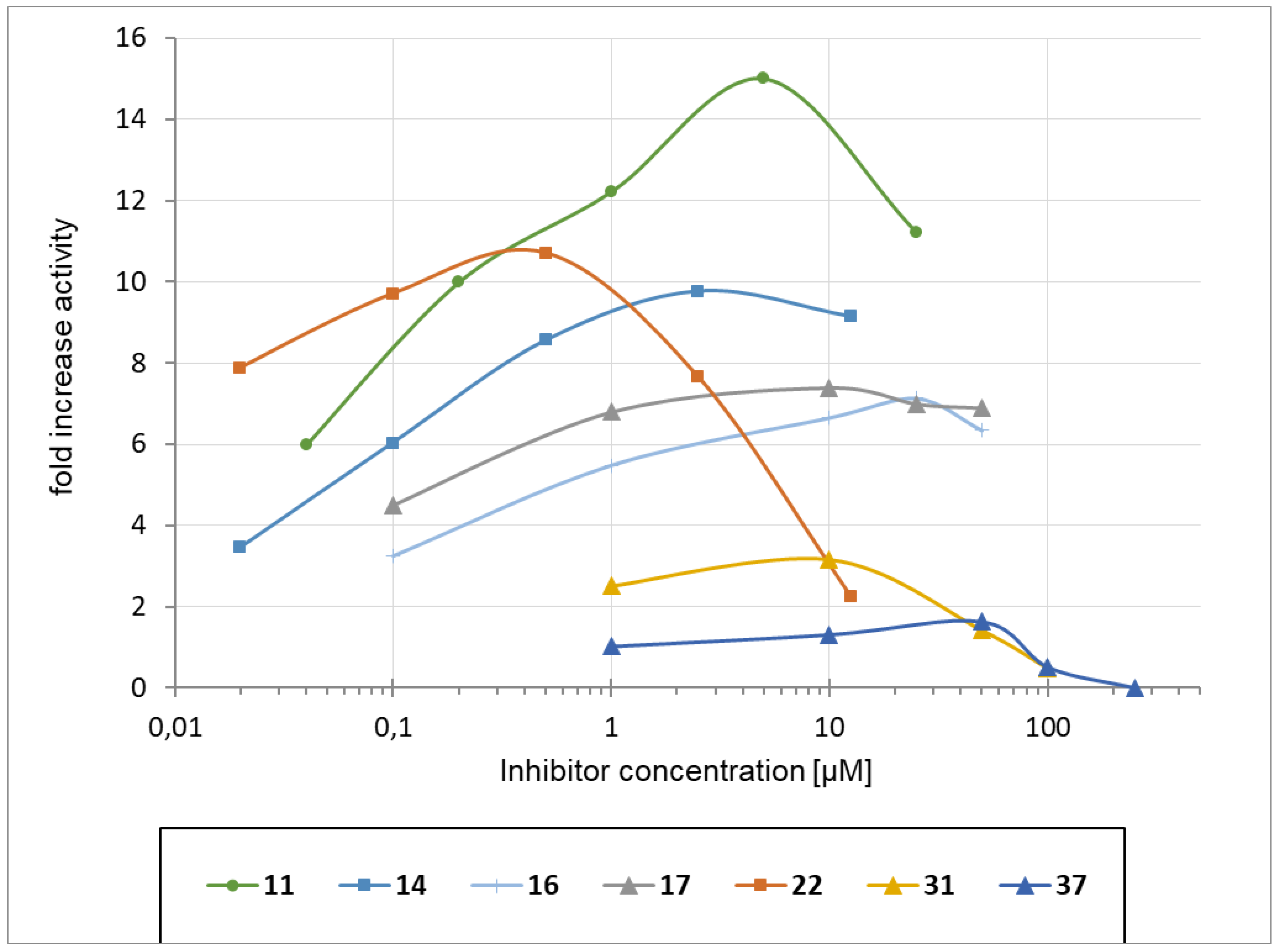
2.5. Chaperoning Activities
3. Discussion
4. Experimental
4.1. Synthetic Section
- 6-[(5aR)-(N-tert-Butyloxycarbonyl-3,4-O-isopropylidene-6-O-methoxymethylene-4-epi-isofagomin-5a-yl)-hex-4-enoic nitrile (19)
- (5aR)-N-tert-Butyloxycarbonyl-5a-C-(6-amino)hexyl-3,4-O-isopropylidene-6-methoxymethylene-4-epi-isofagomine (20)
- (5aR)-N-tert-Butyloxycarbonyl-5a-C-(6-dansylamino)hexyl-3,4-O-isopropylidene-6-O-methoxymethylene-4-epi-isofagomine (21)
- (5aR)-5a-C-(6-Dansylamino)hexyl-4-epi-isofagomine (22)
- (3aR,3bS,6aS,7S,7aR)-1-Benzyl-7-bromo-5,5-dimethylhexahydro-1H[1,3]dioxolo[4′5′:3,4]cyclopenta[1,2-c]isoxazole (25)
- [(3aS,4R,5S,6aR)-5-Amino-2,3-dimethyltetrahydro-4H-cyclopenta[d][1,3]dioxol-4-yl]methanol (26)
- (1R,2S,3R,4S)-4-Amino-3-(hydroxymethyl)cyclopentane-1,2-diol (27)
- 6-[(3aS,4R,5S,6aR)-4-Hydroxymethyl-2,2-dimethyltetrahydro-4H-cyclopenta[d][1,3]dioxol-5-yl)amino]hexanoic nitrile (28)
- (3aS,4R,5S,6aR)-5-(6-aminohexyl)amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1,3]dioxol-4-yl]methanol (29)
- (3aS,4R,5S,6aR)-2,2-Dimethyl-5-[6-(dansylamino)hexyl]aminotetrahydro-4H-cyclopenta[d][1,3]dioxol-4-yl)methanol (30)
- (1R,2S,3R,4S)-3-Hydroxymethyl-4-(6′-dansylaminohexylamino)cyclopentane-1,2-diol (31)
- (3aR,3bS,6aS,7S,7aR)-1-Benzyl-7-fluoro-5,5-dimethylhexahydro-1H-[1,3]dioxolo[4′,5′:3,4]cyclopenta[1,2-c]isoxazole (32)
- [(3aS,4R,5R,6S,6aS)-5-Amino-6-fluoro-2,2-dimethyltetrahydro-4H-cyclopenta[d][1,3]dioxol-4-yl]methanol (33)
- (1S,2S,3S,4R,5R)-4-Amino-3-fluoro-5-hydroxymethylcyclopentane-1,2-diol (34)
- Benzyl 6-(1R,2S,3S,4S,5R)-2-fluoro-3,4-dihydroxy-5-(hydroxymethyl)cyclopentylamino hexylcarbamate (35)
- (1S,2S,3S,4R,5R)-4-(6′-Dansylaminohexyl)amino-3-fluoro-5-hydroxymethylcyclopentane-1,2-diol (37)
4.2. Kinetic Studies
Specific Assay Conditions for Each Enzyme
4.3. CjGH35 Purification and Crystallisation
4.4. Data Collection and Structure Refinement
4.5. Patients and Cell Lines
4.6. Treatment of Cultured Fibroblasts
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Compain, P.; Martin, O.R. (Eds.) Iminosugars—From Synthesis to Therapeutic Applications; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Wennekes, T.; van den Berg, R.J.B.H.N.; Boot, R.G.; van der Marel, G.A.; Overkleeft, H.S.; Aerts, J.M.F.G. Glycosphingolipids—Nature, Function, and Pharmacological Modulation. Angew. Chem. Int. Ed. 2009, 48, 8848–8869. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, R.H. Enzyme replacement therapy for lysosomal storage diseases. Pediatrics 2011, 6, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Condori, J.; Acosta, W.; Ayala, J.; Katta, V.; Flory, A.; Martin, R.; Radin, J.; Cramer, C.J.; Radin, D.N. Enzyme replacement for GM1-gangliosidosis: Uptake, lysosomal activation, and cellular disease correction using a novel β-galactosidase: RTB lectin fusion. Mol. Genet. Metab. 2016, 117, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.C. Hematopoietic stem cell transplantation for lysosomal storage diseases. Pediatr. Endocrinol. Rev. 2013, 11, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Cell Press 2017, 25, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A. Gene therapy for lysosomal storage disorders: A good start. Hum. Mol. Genet. 2016, 25, R65–R75. [Google Scholar] [CrossRef] [Green Version]
- Yew, N.S.; Cheng, S.H. Gene therapy for lysosomal storage disorders. Pediatr. Endocrinol. Rev. 2013, 11, 99–109. [Google Scholar] [CrossRef]
- Macauley, S.L. Combination Therapies for Lysosomal Storage Diseases: A Complex Answer to a Simple Problem. Pediatr. Endocrinol. Rev. 2016, 13, 639–648. [Google Scholar]
- Platt, F.M.; Jeyakumar, M.; Andersson, U.; Priestman, D.A.; Dwek, R.A.; Butters, T.D.; Cox, T.M.; Lachmann, R.; Hollak, C.; Aerts, J.M.F.G.; et al. Inhibition of Substrate Synthesis as a Strategy for Glycolipid Storage Disease Therapy. J. Inherit. Metab. Dis. 2001, 24, 275–290. [Google Scholar] [CrossRef]
- Lachmann, R.H.; Platt, F.M. Substrate Reduction Therapy for Glycosphingolipid Storage Disorders. Expert Opin. Investig. Drugs 2001, 10, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Santos, J.I.; Matos, L.; Alves, S. Genetic Substrate Reduction Therapy: A Promising Approach for Lysosomal Storage Disorders. Diseases 2016, 4, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.-Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Morello, J.-P.; Petäjä-Repo, U.E.; Bichet, D.G.; Bouvier, M. Pharmacological chaperones: A new twist on receptor folding. Trends Pharm. Sci. 2000, 21, 466–469. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ogawa, S.; Sakakibara, Y. Chaperone Therapy for Neuronopathic Lysosomal Diseases: Competitive Inhibitors as Chemical Chaperones for Enhancement of Mutant Enzyme Activities. Persp. Med. Chem. 2009, 3, 7–19. [Google Scholar] [CrossRef]
- Parenti, G.; Pignata, C.; Vajro, P.; Salerno, M. New strategies for the treatment of lysosomal storage diseases. Int. J. Mol. Med. 2013, 31, 11–20. [Google Scholar] [CrossRef]
- Suzuki, Y. Emerging novel concept of chaperone therapies for protein misfolding diseases. Proc. Jap. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 145–162. [Google Scholar] [CrossRef] [Green Version]
- Parenti, G.; Moracci, M.; Fecarotta, S.; Andria, G. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med. Chem. 2014, 6, 1031–1045. [Google Scholar] [CrossRef]
- Small, S.A. Pharmacological chaperones in the age of proteomic pathology. Proc. Natl. Acad. Sci. USA 2014, 111, 12274–12275. [Google Scholar] [CrossRef] [Green Version]
- Covertino, M.; Das, J.; Dokholyan, N.V. Pharmacological Chaperones: Design and Development of New Therapeutic Strategies for the Treatment of Conformational Diseases. Chem. Biol. 2016, 11, 1471–1489. [Google Scholar] [CrossRef]
- Sanchez-Fernandez, E.M.; Garcia Fernandez, J.M.; Ortiz Mellet, C. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: Lessons from Gaucher, GM1-gangliosidosis and Fabry diseases. Chem. Commun. 2016, 5497–5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Wrodnigg, T.M.; Stütz, A.E. The Two Faces of Iminoalditols: Powerful Inhibitors Trigger Glycosidase Activation. Curr. Enzym. Inhib. 2012, 8, 47–99. [Google Scholar] [CrossRef]
- Shin, M.H.; Lim, H.-S. Screening methods for identifying pharmacological chaperones. Mol. Biosyst. 2017, 13, 638–647. [Google Scholar] [CrossRef]
- Arenz, C. Recent advances and novel treatments for sphingolipidoses. Future Med. Chem. 2017, 9, 1685–1698. [Google Scholar] [CrossRef]
- Benito, J.M.; García Fernández, J.M.; Ortiz Mellet, C. Pharmacological chaperone therapy for Gaucher disease: A patent review. Expert Opin. Pat. 2011, 21, 885–903. [Google Scholar] [CrossRef] [Green Version]
- Sawkar, A.R.; Adamski-Werner, S.L.; Cheng, W.-C.; Wong, C.-H.; Beutler, E.; Zimmer, K.-P.; Kelly, J.W. Gaucher Disease-Associated Glucocerebrosidases Show Mutation-Dependent Chemical Chaperoning Profiles. Chem. Biol. 2005, 12, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Wennekes, T.; van den Berg, R.J.B.H.N.; Donker, W.; van der Marel, G.A.; Strijland, A.; Aerts, M.F.G.; Overkleeft, H.S. Development of Adamantan-1-yl-methoxy-Functionalized 1-Deoxynojirimycin Derivatives as Selective Inhibitors of Glucosylceramide Metabolism in Man. J. Org. Chem. 2007, 72, 1088–1097. [Google Scholar] [CrossRef]
- Alfonso, P.; Andreu, V.; Pino-Angeles, A.; Moya-Garcia, A.A.; Garcia-Moreno, M.I.; Rodriguez-Rey, J.C.; Sanchez-Jimenez, F.; Pocovi, M.; Ortiz Mellet, C.; Garcia Fernandez, J.M.; et al. Bicyclic Derivatives of L-Idonojirimycin as Pharmacological Chaperones for Neuronopathic Forms of Gaucher Disease. ChemBioChem 2013, 14, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, T.; Dai, S.; Xie, X.; Ma, X.; Zhao, W.; Zhang, W.; Li, J.; Wang, P.G. New Insights into the Pharmacological Chaperone Activity of C2-Substituted Glucoimidazoles for the Treatment of Gaucher Disease. ChemBioChem 2013, 14, 1239–1247. [Google Scholar] [CrossRef]
- Suzuki, Y.; Oshima, A.; Nanba, E. The Metabolic and Molecular Bases of Inherited Disease; Shriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3775–3809. [Google Scholar]
- Brunetti-Pierri, N.; Scaglia, F. GM1 Gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metabol. 2008, 94, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.; Suzuki, O.; Oshima, A.; Yamamoto, Y.; Noguchi, A.; Takimoto, K.; Itoh, M.; Matsuzaki, Y.; Yasuda, Y.; Sakata, Y.; et al. Chemical chaperone therapy for brain pathology in GM1-gangliosidosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15912–15917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higaki, K.; Li, L.; Okuzawa, S.; Takamuram, A.; Yamamoto, K.; Adachi, K.; Paraguison, R.C.; Takai, T.; Ikehata, H.; Tominaga, L.; et al. Chemical Chaperone Therapy: Chaperone Effect on Mutant Enzyme and Cellular Pathophysiology in β-Galactosidase Deficiency. Hum. Mutat. 2011, 32, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Kuno, S.; Higaki, K.; Takahashi, A.; Nanba, E.; Ogawa, S. Potent chemical chaperone compounds for GM1-gangliosidosis: N-substituted (+)-conduramine F-4 derivatives. Med. Chem. Commun. 2015, 6, 306–310. [Google Scholar] [CrossRef]
- Takai, T.; Higaki, K.; Aguilar-Moncayo, M.; Mena-Barragan, T.; Hirano, Y.; Yura, K.; Yu, L.; Ninomiya, H.; Garcia-Moreno, M.I.; Sakakibara, Y.; et al. A Bicyclic 1-Deoxygalactonojirimycin Derivative as a Novel Pharmacological Chaperone for GM1 Gangliosidosis. Mol. Ther. 2013, 21, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Siriwardena, A.; Sonawane, D.P.; Bande, O.P.; Markad, P.R.; Yonekawa, S.; Tropak, M.B.; Ghosh, S.; Chopade, B.A.; Mahuran, D.J.; Dhavale, D.D. Synthesis of 1,5-Dideoxy-1,5-iminoribitol C-Glycosides through a Nitrone—Olefin Cycloaddition Domino Strategy: Identification of Pharmacological Chaperones of Mutant Human Lysosomal β-Galactosidase. J. Org. Chem. 2014, 79, 4398–4404. [Google Scholar] [CrossRef]
- Kasperzyk, J.L.; El-Abbadi, M.M.; Hauser, E.C.; d’Azzo, A.; Platt, F.M.; Seyfried, T.N. N-butyldeoxygalactonojirimycin reduces neonatal brain ganglioside content in a mouse model of GM1 gangliosidosis. J. Neurochem. 2004, 89, 645–653. [Google Scholar] [CrossRef]
- Rigat, B.A.; Tropak, M.B.; Buttner, J.; Crushell, E.; Benedict, D.; Callahan, J.W.; Martin, D.R.; Mahuran, D.J. Evaluation of N-nonyl-deoxygalactonojirimycin as a pharmacological chaperone for human GM1 gangliosidosis leads to identification of a feline model suitable for testing enzyme enhancement therapy. Mol. Genet. Metab. 2012, 107, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Fantur, K.; Hofer, D.; Schitter, G.; Steiner, A.J.; Pabst, B.M.; Wrodnigg, T.M.; Stütz, A.E.; Paschke, E. DLHex-DGJ, a novel derivative of 1-deoxygalactonojirimycin with pharmacological chaperone activity in human GM1-gangliosidosis fibroblasts. Mol. Genet. Metab. 2010, 100, 262–268. [Google Scholar] [CrossRef]
- Schitter, G.; Steiner, A.J.; Pototschnig, G.; Scheucher, E.; Thonhofer, M.; Tarling, C.A.; Withers, S.G.; Fantur, K.; Paschke, E.; Mahuran, D.J.; et al. Fluorous Iminoalditols: A New Family of Glycosidase Inhibitors and Pharmacological Chaperones. ChemBioChem 2010, 11, 2026–2033. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Otho, U.; Higaki, K.; Mena-Barragan, T.; Aguilar-Moncayo, M.; Ortiz Mellet, C.; Nanba, E.; Garcia Fernandez, J.M.; Suzuki, Y.; Shimizu, T. Structural Basis of Pharmacological Chaperoning for Human β-Galactosidase. J. Biol. Chem. 2014, 289, 14560–14568. [Google Scholar] [CrossRef] [Green Version]
- Front, S.; Gallienne, E.; Charollais-Thoenig, J.; Demotz, S.; Martin, O. N-Alkyl-, 1-C-Alkyl-, and 5-C-Alkyl-1,5-dideoxy-1,5-imino-L-ribitols as Galactosidase Inhibitors. ChemMedChem 2016, 11, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Igarashi, Y.; Ichikawa, M.; Suhara, Y. 1-N-Iminosugars: Potent and Selective Inhibitors of β-Glycosidases. J. Am. Chem. Soc. 1998, 120, 3007–3018. [Google Scholar] [CrossRef]
- Thonhofer, M.; Hojnik, C.; Schalli, M.; Zoidl, M.; Wrodnigg, T.M.; Stütz, A.E.; Withers, S.G. New 4-epi-Isofagomine Derivatives for Glycosidase Research. In Proceedings of the 97th Canadian Chemisty Conference and Exhibition, Vancouver, BC, Canada, 1–6 June 2014. [Google Scholar]
- Thonhofer, M.; Weber, P.; Gonzalez Santana, A.; Christina, T.; Roland, F.; Bettina, M.P.; Eduard, P.; Michael, S.; Arnold, E.S.; Marion, T.; et al. Synthesis of C-5a-substituted derivatives of 4-epi-isofagomine: Notable β-galactosidase inhibitors and activity promotors of GM1-gangliosidosis related human lysosomal β-galactosidase mutant R201C. Carbohydr. Res. 2016, 429, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.R.; Gallienne, E.; Front, S.; Charollais, J.; Demotz, S. 4-epi-Isofagomine derivatives, Eur. Pat. Appl. 2015, 2015, 90–92. [Google Scholar]
- Front, S.; Biela-Banas, A.; Burda, P.; Ballhausen, D.; Higaki, K.; Caciotti, A.; Morrone, A.; Charollais-Thoenig, J.; Gallienne, E.; Demotz, S.; et al. (5aR)-5a-C-Pentyl-4-epi-isofagomine: A powerful inhibitor of lysosomal β-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B. Eur. J. Med. Chem. 2017, 126, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Front, S.; Almeida, S.; Zoete, V.; Charollais-Thoenig, J.; Gallienne, E.; Marmy, C.; Pilloud, V.; Marti, R.; Wood, T.; Martin, O.R.; et al. 4-epi-Isofagomine derivatives as pharmacological chaperones for the treatment of lysosomal diseases linked to β-galactosidase mutations: Improved synthesis and biological investigations. Bioorg. Med. Chem. 2018, 26, 5462–5469. [Google Scholar] [CrossRef]
- Schalli, M.; Tysoe, C.; Fischer, R.; Pabst, B.M.; Thonhofer, M.; Paschke, E.; Rappitsch, T.; Stütz, A.E.; Tschernutter, M.; Windischhofer, W.; et al. N-Substituted 5-amino-1-hydroxymethyl-cyclopentanetriols: A new family of activity promotors for a GM1-gangliosidosis related human lysosomal β-galactosidase mutant. Carbohydr. Res. 2017, 443–444, 15–22. [Google Scholar] [CrossRef]
- Leroy, E.; Reymond, J.L. Anomer-Selective Inhibition of Glycosidases Using Aminocyclopentanols. Org. Lett. 1999, 1, 775–777. [Google Scholar] [CrossRef]
- Greul, J.N.; Kleban, M.; Schneider, B.; Picasso, S.; Jäger, V. Amino(hydroxymethyl)cyclopentanetriols, an Emerging Class of Potent Glycosidase Inhibitors Part II: Synthesis, Evaluation, and Optimization of β-D-Galactopyranoside Analogues. ChemBioChem 2001, 2, 368–370. [Google Scholar] [CrossRef]
- Gartenmann Dickson, L.; Leroy, E.; Reymond, J.-L. Structure–activity relationships in aminocyclopentitol glycosidase inhibitors. Org. Biomol. Chem. 2004, 2, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Schalli, M.; Weber, P.; Tysoe, C.; Pabst, B.M.; Thonhofer, M.; Paschke, E.; Stütz, A.E.; Tschernutter, M.; Windischhofer, W.; Withers, S.G. A new type of pharmacological chaperone for GM1-gangliosidosis related human lysosomal β-galactosidase: N-Substituted 5-amino-1-hydroxymethyl-cyclopentanetriols. Bioorg. Med. Chem. Lett. 2017, 27, 3431–3435. [Google Scholar] [CrossRef] [PubMed]
- Thonhofer, M.; Weber, P.; Gonzalez Santana, A.; Fischer, R.; Pabst, B.M.; Paschke, E.; Schalli, M.; Stütz, A.E.; Tschernutter, M.; Windischhofer, W.; et al. Synthesis of C-5a-chain extended derivatives of 4-epi-isofagomine: Powerful β-galactosidase inhibitors and low concentration activators of GM1-gangliosidosis-related human lysosomal β-galactosidase. Bioorg. Med. Chem. Lett. 2016, 26, 1438–1442. [Google Scholar] [CrossRef]
- Weber, P.; Nasseri, S.A.; Pabst, B.M.; Torvisco, A.; Müller, P.; Paschke, E.; Tschernutter, M.; Windischhofer, W.; Withers, S.G.; Wrodnigg, T.M.; et al. Potent GH20 N-Acetyl-β-D-hexosaminidase Inhibitors: N-Substituted 3-acetamido-4-amino-5-hydroxymethyl-cyclopentanediols. Molecules 2018, 23, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The Carbohydrate-active enzymes database in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- CAZypedia Consortium. Ten years of CAZypedia: A living encyclopedia of carbohydrate-active enzymes. Glycobiology 2018, 28, 3–8. [Google Scholar] [CrossRef]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Cryst. 2011, D67, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Hermetter, A.; Scholze, H.; Stütz, A.E.; Withers, S.G.; Wrodnigg, T.M. Powerful probes for glycosidases: Novel fluorescently tagged glycosidase inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 1339–1342. [Google Scholar] [CrossRef]
- Wrodnigg, T.M.; Diness, F.; Gruber, C.; Häusler, H.; Lundt, I.; Rupitz, K.; Steiner, A.J.; Stütz, A.E.; Tarling, C.A.; Withers, S.G.; et al. Probing the aglycon binding site of a β-glucosidase: A collection of C-1-modified 2,5-dideoxy-2,5-imino-D-mannitol derivatives and their structure-activity relationships as competitive inhibitors. Bioorg. Med. Chem. 2004, 12, 3485–3495. [Google Scholar] [CrossRef]
- Greimel, P.; Häusler, H.; Lundt, I.; Rupitz, K.; Stütz, A.E.; Tarling, C.A.; Withers, S.G.; Wrodnigg, T.M. Fluorescent glycosidase inhibiting 1,4-dideoxy-1,5-iminoalditols. Bioorg. Med. Chem. Lett. 2006, 16, 2067–2070. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; Garcia-Moreno, M.I.; Stütz, A.E.; Garcia Fernandez, J.M.; Wrodnigg, T.M.; Ortiz Mellet, C. Fluorescent-tagged sp2-iminosugars with potent β-glucosidase inhibitory activity. Bioorg. Med. Chem. 2010, 18, 7439–7445. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Krivickas, S.J.; Tamanini, E.; Todd, M.H.; Watkinson, M. Effective Methods for the Biotinylation of Azamacrocycles. J. Org. Chem. 2007, 72, 8280–8289. [Google Scholar] [CrossRef]
- Prade, H.; Mackenzie, L.F.; Withers, S.G. Enzymatic synthesis of disaccharides using Agrobacterium sp. β-glucosidase. Carbohydr. Res. 1998, 305, 371–381. [Google Scholar] [CrossRef]
- Kempton, J.B.; Withers, S.G. Mechanism of Agrobacterium β-Glucosidase: Kinetic Studies. Biochemistry 1992, 31, 9961–9969. [Google Scholar] [CrossRef]
- Chen, H.-M.; Withers, S.G. Facile Synthesis of 2,4-Dinitrophenyl α-D-Glycopyranosides as Chromogenic Substrates for α-Glycosidases. ChemBioChem 2007, 8, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Larsbrink, J.; Thompson, A.J.; Lundqvist, M.; Gardner, J.G.; Davies, G.J.; Brumer, H. A complex gene locus enables xyloglucan utilization in the model saprophyte Cellvibrio japonicus. Mol. Microbiol. 2014, 94, 418–433. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Cryst. Sect. D 2018, 74, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. XDS. Acta Cryst. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Cryst. Sect. D 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. Sect. D 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Potterton, L.; Agirre, J.; Ballard, C.; Cowtan, K.; Dodson, E.; Evans, P.R.; Jenkins, H.T.; Keegan, R.; Krissinel, E.; Stevenson, K.; et al. CCP4i2: The new graphical user interface to theCCP4 program suite. Acta Cryst. Sect. D 2018, 74, 68–84. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compound 17 are available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme (GH Family) | Compounds | ||||||
|---|---|---|---|---|---|---|---|
 22 |  14 [46,47] |  16 |  17 [55] |  31 |  37 |  1 | |
| Abg (β-Glc/Gal) (GH1) | 0.0003 | 0.0175 | 0.0010 | 0.0012 | 0.0035 | 0.077 | 7.7 |
| E. coli (β-Gal) (GH2) | 0.0004 | 0.0021 | 0.0132 | 0.0101 | 0.0041 | 5.2 | 2.8 |
| Bovine liver (β-Gal) (GH35) | 0.0005 | 0.0014 | 0.1167 | 0.0053 | 0.102 | 0.137 | 0.87 [16] |
| Fabrazyme (α-Gal) (GH27) | n.i. | n.i. | n.i. | 238.1 | n.i. | n.i. | n.i. |
| S. cer. (α-Glc) (GH13) | n.i. | n.i. | n.i. | n.i. | n.i. | 22.5 | n.i. |
| GCase (β-Glc) (GH30) | 0.003 | 1.2 | 0.0639 | 0.0193 | 22.6 | 3.0 | 83 |
| β-Gal human lys. (GH35); IC50 | 0.094 | 0.38 | 0.42 | 0.40 | 82.5 | 34.9 | 0.51 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, P.; Thonhofer, M.; Averill, S.; Davies, G.J.; Santana, A.G.; Müller, P.; Nasseri, S.A.; Offen, W.A.; Pabst, B.M.; Paschke, E.; et al. Mechanistic Insights into the Chaperoning of Human Lysosomal-Galactosidase Activity: Highly Functionalized Aminocyclopentanes and C-5a-Substituted Derivatives of 4-epi-Isofagomine. Molecules 2020, 25, 4025. https://doi.org/10.3390/molecules25174025
Weber P, Thonhofer M, Averill S, Davies GJ, Santana AG, Müller P, Nasseri SA, Offen WA, Pabst BM, Paschke E, et al. Mechanistic Insights into the Chaperoning of Human Lysosomal-Galactosidase Activity: Highly Functionalized Aminocyclopentanes and C-5a-Substituted Derivatives of 4-epi-Isofagomine. Molecules. 2020; 25(17):4025. https://doi.org/10.3390/molecules25174025
Chicago/Turabian StyleWeber, Patrick, Martin Thonhofer, Summer Averill, Gideon J. Davies, Andres Gonzalez Santana, Philipp Müller, Seyed A. Nasseri, Wendy A. Offen, Bettina M. Pabst, Eduard Paschke, and et al. 2020. "Mechanistic Insights into the Chaperoning of Human Lysosomal-Galactosidase Activity: Highly Functionalized Aminocyclopentanes and C-5a-Substituted Derivatives of 4-epi-Isofagomine" Molecules 25, no. 17: 4025. https://doi.org/10.3390/molecules25174025