
Designing Nanoparticles and Nanoalloys for Gas-Phase Catalysis with Controlled Surface Reactivity Using Colloidal Synthesis and Atomic Layer Deposition

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Colloidal Synthesis in Gas-Phase Catalysis
2.1. Principles of Colloidal Synthesis for Supported Catalyst Preparation
2.2. Monometallic Supported Catalysts by Colloidal Synthesis
2.2.1. Oxidation Reactions
2.2.2. Hydrogenation Reactions
2.3. Bimetallic Supported Catalysts by Colloidal Synthesis
2.3.1. Oxidation Reactions
2.3.2. Hydrogenation Reactions
2.3.3. Dehydrogenation Reactions
2.3.4. Reforming Reactions
2.4. Gas-Phase Catalysis by Embedded NPs
2.4.1. Oxidation Reactions
2.4.2. Hydrogenation Reactions
2.4.3. Reforming Reactions
2.4.4. Water-Gas Shift
3. ALD in Gas-Phase Catalysis
3.1. Principles of ALD for Supported Catalyst Preparation
3.2. Monometallic Supported Catalysts by ALD
3.2.1. Oxidation Reactions
3.2.2. Hydrogenation Reactions
3.2.3. Reforming Reactions
3.3. Bimetallic Supported Catalysts by ALD
- the NP size can be tailored by adjusting the total number of ALD cycles;
- the relative number of ALD cycles of each element determines the composition;
- the order of deposition determines the NP architecture.
3.3.1. Dehydrogenation Reactions
3.3.2. Reforming Reactions
3.4. Oxide-Coated Catalysts by ALD
3.4.1. Oxidation Reactions
3.4.2. Hydrogenation Reactions
3.4.3. Reforming Reactions
3.5. Area-Selective ALD for Next-Level Catalyst Design
4. Conclusions
5. Outlook: Colloidal Synthesis and ALD Applied to Heterogeneous Catalysis
Author Contributions
Funding
Conflicts of Interest
Acronyms
| 3DOM | three-dimensionally ordered macroporous |
| ALD | atomic layer deposition |
| BDE | 1,3-butadiene |
| BTB | borane tert-butylamine complex |
| CNT | carbon nanotube |
| Cp | cyclopentadienyl |
| CVD | chemical vapor deposition |
| DFT | density functional theory |
| DME | dimethyl ether |
| DP | deposition-precipitation |
| DRM | dry reforming of methane |
| FTO | Fischer Tropsch to olefins |
| FTS | Fischer Tropsch synthesis |
| GC | graphitic carbon |
| GHSV | gas hourly space velocity |
| GISAXS | grazing incidence small-angle X-ray scattering |
| H2-TPR | hydrogen temperature-programmed reduction |
| HAADF | high-angle annular dark-field |
| H-Al2O3 | hydrophobic Al2O3 |
| h-BN | hexagonal boron nitride |
| HDP | homogeneous deposition-precipitation |
| IR | infrared |
| IWI | incipient wetness impregnation |
| ME | modulation-excitation |
| MeOH | methanol |
| MLD | molecular layer deposition |
| MOF | metal-organic framework |
| MSI | metal-support interactions |
| m-SiO2 | mesoporous SiO2 |
| NAP-XPS | near-ambient pressure X-ray photoelectron spectroscopy |
| NEXAFS-TEY | near edge X-ray absorption fine structure spectroscopy with total electron yield |
| NMR | nuclear magnetic resonance |
| NP | nanoparticle |
| OAm | oleylamine |
| P | pressure |
| PAMAM | polyamidoamine |
| pair distribution function | |
| PDH | propane dehydrogenation |
| PODH | propane oxidative dehydrogenation |
| PROX | preferential oxidation of CO in H2 |
| PVA | polyvinyl alcohol |
| RCT | raspberry colloid-template |
| SRM | steam reforming of methane |
| SSITKA | steady-state isotope transient kinetic analysis |
| STD | syngas-to-dimethyl |
| STEM-EDX | scanning transmission electron microscopy energy dispersive X-ray |
| T | temperature |
| TAP | temporal analysis of products |
| TEM | transmission electron microscopy |
| TGA | thermogravimetric analysis |
| thd | 2,2,6,6-tetramethylheptane-3,5-dione |
| TMA | trimethylaluminum |
| TOF | turnover frequency |
| TOS | time-on-stream |
| TTAB | trimethyl(tetradecyl)ammonium bromide |
| UHV | ultra-high vacuum |
| V | volume |
| WHSV | weight hourly space velocity |
| WI | wet impregnation |
| XAS | X-ray absorption spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
Glossary
| activation (catalyst) | The procedure whereby a catalyst is put into a state (activated state) which can catalyze the reaction. Depending on the reaction at hand, this can be, for example, reduction (active phase = metal) or sulfidation (active phase = sulfide). |
| active site | An ensemble of atoms on the catalyst which directly catalyzes a reaction. |
| activity (catalyst) | Catalytic activity refers to the reaction rate, i.e., the speed at which a reaction takes place. This is typically quantified by turnover frequency (TOF). |
| calcination | The process of heating a sample, in this context a catalyst, to high temperature under an oxygen or air environment. |
| cavitation bubble | A small bubble, typically with sizes on the order of µm–mm, that can collapse explosively and generate a localized increase in pressure (e.g., shock waves) and temperature. In the case of sonication (ultrasound) of a liquid, these bubbles originate from rapid compressions and expansions of the liquid medium. |
| chemical reduction (colloidal synthesis) | A colloidal synthesis protocol whereby reduction to zerovalent state of the metal is achieved-in part-through the addition of a reducing agent. |
| coking | The process whereby cokes are formed, i.e., carbonaceous species, as undesired by-products of a reaction. As cokes occupy active sites, this leads to catalyst poisoning. |
| colloid | A type of mixture whereby solid or liquid particles, in the range of 1–1000 nm, are dispersed in a liquid phase and surrounded by a protective layer which prevents their agglomeration. |
| conformal | With even thickness over the exposed surface. |
| conversion (of a reagent) | In reaction engineering, the conversion of a reagent is defined as the ratio of [the number of moles of a certain reagent that have reacted] over [the initial number of moles of reagent]. |
| core-shell | An architecture consisting of a core material (A) surrounded by a concentric shell of a second material (B). Typically denoted as A@B. |
| crystal facet | A planar side of a geometrical shape. |
| deactivation (catalyst) | The process whereby a catalyst loses its activity over time. Typical deactivation phenomena include coking and metal sintering. |
| embedded nanoparticles | Nanoparticles which are located within the support material, rather than on the support’s surface. |
| in situ reduction (colloidal synthesis) | Colloidal deposition protocol whereby the colloidal synthesis takes place in the presence of the support. |
| incipient wetness impregnation | Catalyst synthesis protocol whereby the metal precursor is first dissolved in a solvent, after which this dissolved precursor is added to the support. Herein, the volume of solution used is the same as the support pore volume. |
| Janus-like architecture | Nanoparticle architectural pattern that exhibits two distinguishable subunits. For instance, in a bimetallic CoFe particle, one subunit can exhibit low Co concentration, whereas the other subunit exhibits much higher Co concentration. |
| mesoporous | Containing pores with pore diameters between 2 and 50 nm. |
| metal precursor | A compound that contains a metal in its ionic state and serves as the metal source for catalyst synthesis. |
| microporous | Containing pores with pore diameters smaller than 2 nm. |
| monodisperse distribution | Displaying a narrow distribution. Synonym for uniform. |
| multimetallic | In this paper, multimetallic refers to any composition that contains more than 3 metals. |
| nanoparticle | A particle in the nanoscale regime, i.e., 1–100 nm. |
| nucleation | The initial process whereby a novel phase starts growing within or onto a pre-existing phase. |
| organometallic salt | A substance which is partially ionic (in the case of a metal precursor, this is a metal ion) and partially organic. An example is Ni(acac)2, made up of Ni2+ with surrounding (organic) acetylacetonate (acac) groups. |
| performance (catalyst) | A catalyst’s performance refers to its behavior during reaction. Commonly quantified via the terms reagent conversion, product selectivity, product yield, activity and stability. |
| poisoning (catalyst) | The act of blocking active sites of a catalyst with unwanted material, e.g., cokes, thus inhibiting contact of these active centers with reagents, which is detrimental to the catalytic activity. |
| promoter (catalyst) | A substance added to a catalyst to improve its performance. |
| protective agent | A chemical agent used in colloidal synthesis. It has a dual purpose: controlling the nanoparticle growth (capping agent) and preventing coalescence of colloidal nanoparticles (stabilizing agent). |
| rational/knowledge-driven catalyst design | The design of novel and improved catalysts in an efficient and cost-effective way. The creation of monodisperse materials helps to achieve this goal as follows. Materials with well-defined properties (e.g., nanoparticle size) allow investigating the effect of this property on the catalytic performance for a given reaction. As such, the optimal catalyst property (e.g., size which gives maximum activity) can be identified, which gives guidelines (knowledge) for the design of a new and improved catalyst. |
| reactant (ALD) | One of the two main components, besides a metal precursor, making up an ALD cycle. Reactants typically are gases, e.g., H2O, NH3, H2S, O3 etc., which, unlike the metal precursor, do not contain any metals. |
| reducing agent | A chemical added during colloidal synthesis (in chemical reduction protocols) which helps implement the reduction of the metal ions to their zerovalent state |
| selectivity (towards a product) | In reaction engineering, the conversion of a reagent is defined as the ratio of [the number of moles of the product that have formed] per [mole of reagent that has reacted]. |
| self-limiting surface reaction | A surface reaction which stops when the surface is depleted of functional groups. |
| single crystal | A solid wherein an orderly three-dimensional arrangement of atoms is repeated throughout the entire material. |
| sintering | The thermally-induced process whereby small particles coalesce to form bigger particles. When this occurs for the nanoparticles in a catalyst, this leads to lower surface area of the active phase. |
| stability (catalyst) | A term denoting to what degree a catalyst retains its (initial) performance after a certain reaction time. Typically measured by monitoring the performance over an extended reaction time and observing the changes in activity/selectivity/conversion/etc. relative to initial values. |
| structure-sensitive reaction | A reaction of which the reaction kinetics are dependent on the surface structure of the catalyst. |
| support | A solid material, onto which the active nanoparticles are fixed, supplying mechanical strength and a large area to disperse the active phase. Sometimes also called substrate, though this latter term is more used in the context of planar support materials. |
| thermal decomposition (of an organometallic metal precursor) | A type of colloidal synthesis protocol whereby the metal precursor is organometallic in nature, e.g., an organometallic salt, and, through exposure to high temperatures, is decomposed and (thermally) reduced to its zerovalent state. |
| turnover frequency | A measure used to quantify the number of reagent conversions an active site performs per second. |
| wet impregnation | Catalyst synthesis protocol whereby the metal precursor is first dissolved in a solvent, after which this dissolved precursor is added to the support. Herein, the volume of solution used is much larger than the support pore volume. |
| yield (of a product) | In reaction engineering, the yield of a product is defined as the ratio of [the number of moles of the product that have formed] over [the initial number of moles of reagent]. Mathematically, this follows from multiplying the reagent conversion with the product selectivity. |
References
- Armor, J.N. A history of industrial catalysis. Catal. Today 2011, 163, 3–9. [Google Scholar] [CrossRef]
- White, R.J.; Luque, R.; Budarin, V.L.; Clark, J.H.; Macquarrie, D.J. Supported metal nanoparticles on porous materials. Methods and applications. Chem. Soc. Rev. 2009, 38, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Munnik, P.; De Jongh, P.E.; De Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- An, K.; Somorjai, G.A. Nanocatalysis I: Synthesis of Metal and Bimetallic Nanoparticles and Porous Oxides and Their Catalytic Reaction Studies. Catal. Lett. 2015, 145, 233–248. [Google Scholar] [CrossRef]
- Pan, C.-J.; Tsai, M.-C.; Su, W.-N.; Rick, J.; Akalework, N.G.; Agegnehu, A.K.; Cheng, S.-Y.; Hwang, B.-J. Tuning/exploiting Strong Metal-Support Interaction (SMSI) in Heterogeneous Catalysis. J. Taiwan Inst. Chem. Eng. 2017, 74, 154–186. [Google Scholar] [CrossRef]
- Penner, S.; Armbrüster, M. Formation of intermetallic compounds by reactive metal—support interaction: A frequently encountered phenomenon in catalysis. ChemCatChem 2015, 7, 374–392. [Google Scholar] [CrossRef]
- Cargnello, M.; Fornasiero, P.; Gorte, R.J. Opportunities for Tailoring Catalytic Properties Through Metal-Support Interactions. Catal. Lett. 2012, 142, 1043–1048. [Google Scholar] [CrossRef]
- Cao, S.; Tao, F.; Tang, Y.; Li, Y.; Yu, J. Size- and shape-dependent catalytic performances of oxidation and reduction reactions on nanocatalysts. Chem. Soc. Rev. 2016, 45, 4747–4765. [Google Scholar] [CrossRef]
- Van Santen, R.A. Complementary structure sensitive and insensitive catalytic relationships. Acc. Chem. Res. 2009, 42, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Den Breejen, J.; Radstake, P.; Bezemer, G.; Bitter, J.; Frøseth, V.; Holmen, A.; De Jong, K.d. On the origin of the cobalt particle size effects in Fischer-Tropsch catalysis. J. Am. Chem. Soc. 2009, 131, 7197–7203. [Google Scholar] [CrossRef] [PubMed]
- Somorjai, G.A.; Park, J.Y. Colloid Science of Metal Nanoparticle Catalysts in 2D and 3D Structures. Challenges of Nucleation, Growth, Composition, Particle Shape, Size Control and Their Influence on Activity and Selectivity. Top. Catal. 2008, 49, 126–135. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Park, J.Y. Molecular Factors of Catalytic Selectivity. Angew. Chem. Int. Ed. 2008, 47, 9212–9228. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Somorjai, G.A. Size and shape control of metal nanoparticles for reaction selectivity in catalysis. ChemCatChem 2012, 4, 1512–1524. [Google Scholar] [CrossRef]
- Goodman, D.W.; Kelley, R.D.; Madey, T.E.; Yates, J.T. Kinetics of the hydrogenation of CO over a single crystal nickel catalyst. J. Catal. 1980, 63, 226–234. [Google Scholar] [CrossRef]
- Oh, S.H.; Fisher, G.B.; Carpenter, J.E.; Goodman, D.W. Comparative kinetic studies of CO–O2 and CO–NO reactions over single crystal and supported rhodium catalysts. J. Catal. 1986, 100, 360–376. [Google Scholar] [CrossRef]
- Oosterbeek, H. Bridging the pressure and material gap in heterogeneous catalysis: Cobalt Fischer-Tropsch catalysts from surface science to industrial application. Phys. Chem. Chem. Phys. 2007, 9, 3570–3576. [Google Scholar] [CrossRef]
- Imbihl, R.; Behm, R.J.; Schlögl, R. Bridging the pressure and material gap in heterogeneous catalysis. Phys. Chem. Chem. Phys. 2007, 9, 3459. [Google Scholar] [CrossRef]
- Marceau, E.; Carrier, X.; Che, M.; Clause, O.; Marcilly, C. Ion exchange and impregnation. In Handbook of Heterogeneous Catalysis, 2nd ed.; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2008; Volume 1, pp. 467–484. [Google Scholar] [CrossRef]
- Behrens, M. Coprecipitation: An excellent tool for the synthesis of supported metal catalysts—From the understanding of the well known recipes to new materials. Catal. Today 2015, 246, 46–54. [Google Scholar] [CrossRef]
- Geus, J.W.; Van Dillen, A.J. Preparation of supported catalysts by deposition–precipitation. In Handbook of Heterogeneous Catalysis, 2nd ed.; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2008; Volume 1, pp. 428–467. [Google Scholar] [CrossRef]
- Jia, C.-J.; Schüth, F. Colloidal metal nanoparticles as a component of designed catalyst. Phys. Chem. Chem. Phys. 2011, 13, 2457–2487. [Google Scholar] [CrossRef]
- Tao, A.R.; Habas, S.; Yang, P. Shape Control of Colloidal Metal Nanocrystals. Small 2008, 4, 310–325. [Google Scholar] [CrossRef]
- Collins, G.; Holmes, J.D. Engineering Metallic Nanoparticles for Enhancing and Probing Catalytic Reactions. Adv. Mater. 2016, 28, 5689–5695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Tang, Z. Noble metal nanoparticle@metal oxide core/yolk-shell nanostructures as catalysts: Recent progress and perspective. Nanoscale 2014, 6, 3995–4011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lee, I.; Joo, J.B.; Zaera, F.; Yin, Y. Core-Shell Nanostructured Catalysts. Acc. Chem. Res. 2013, 46, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Albiter, M.A.; Zhang, Q.; Ge, J.; Yin, Y.; Zaera, F. New nanostructured heterogeneous catalysts with increased selectivity and stability. Phys. Chem. Chem. Phys. 2011, 13, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Li, X.; Zeng, L.; Gong, J. Recent Advances on the Design of Group VIII Base-Metal Catalysts with Encapsulated Structures. ACS Catal. 2015, 5, 4959–4977. [Google Scholar] [CrossRef]
- Lu, J.; Elam, J.W.; Stair, P.C. Synthesis and stabilization of supported metal catalysts by atomic layer deposition. Acc. Chem. Res. 2013, 46, 1806–1815. [Google Scholar] [CrossRef]
- Detavernier, C.; Dendooven, J.; Pulinthanathu Sree, S.; Ludwig, K.F.; Martens, J.A. Tailoring nanoporous materials by atomic layer deposition. Chem. Soc. Rev. 2011, 40, 5242–5253. [Google Scholar] [CrossRef]
- Singh, J.A.; Yang, N.; Bent, S.F. Nanoengineering heterogeneous catalysts by atomic layer deposition. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 41–62. [Google Scholar] [CrossRef]
- O’Neill, B.J.; Jackson, D.H.K.; Lee, J.; Canlas, C.; Stair, P.C.; Marshall, C.L.; Elam, J.W.; Kuech, T.F.; Dumesic, J.A.; Huber, G.W. Catalyst Design with Atomic Layer Deposition. ACS Catal. 2015, 5, 1804–1825. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Elam, J.W.; Stair, P.C. Atomic layer deposition—Sequential self-limiting surface reactions for advanced catalyst “bottom-up” synthesis. Surf. Sci. Rep. 2016, 71, 410–472. [Google Scholar] [CrossRef] [Green Version]
- Mackus, A.J.M.; Merkx, M.J.M.; Kessels, W.M.M. From the Bottom-Up: Toward Area-Selective Atomic Layer Deposition with High Selectivity. Chem. Mater. 2019, 31, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaltonen, T.; Ritala, M.; Sajavaara, T.; Keinonen, J.; Leskelä, M. Atomic Layer Deposition of Platinum Thin Films. Chem. Mater. 2003, 15, 1924–1928. [Google Scholar] [CrossRef]
- Aaltonen, T.; Alén, P.; Ritala, M.; Leskelä, M. Ruthenium Thin Films Grown by Atomic Layer Deposition. Chem. Vap. Depos. 2003, 9, 45–49. [Google Scholar] [CrossRef]
- Aaltonen, T.; Rahtu, A.; Ritala, M.; Leskelä, M. Reaction Mechanism Studies on Atomic Layer Deposition of Ruthenium and Platinum. Electrochem. Solid-State Lett. 2003, 6, C130. [Google Scholar] [CrossRef]
- Cao, K.; Cai, J.; Shan, B.; Chen, R. Surface functionalization on nanoparticles via atomic layer deposition. Sci. Bull. 2020, 65, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Lu, J. Atomic-scale engineering of metal-oxide interfaces for advanced catalysis using atomic layer deposition. Catal. Sci. Technol. 2020, 10, 2695–2710. [Google Scholar] [CrossRef]
- Cao, K.; Cai, J.; Liu, X.; Chen, R. Review Article: Catalysts design and synthesis via selective atomic layer deposition. J. Vac. Sci. Technol. A 2018, 36, 010801. [Google Scholar] [CrossRef] [Green Version]
- Bönnemann, H.; Richards, R.M. Nanoscopic metal particles—synthetic methods and potential applications. Eur. J. Inorg. Chem. 2001, 2001, 2455–2480. [Google Scholar] [CrossRef]
- Lu, Z.; Yin, Y. Colloidal nanoparticle clusters: Functional materials by design. Chem. Soc. Rev. 2012, 41, 6874–6887. [Google Scholar] [CrossRef]
- Pareek, V.; Bhargava, A.; Gupta, R.; Jain, N.; Panwar, J. Synthesis and applications of noble metal nanoparticles: A review. Adv. Sci. Eng. Med. 2017, 9, 527–544. [Google Scholar] [CrossRef]
- Amendola, V.; Meneghetti, M. Laser ablation synthesis in solution and size manipulation of noble metal nanoparticles. Phys. Chem. Chem. Phys. 2009, 11, 3805–3821. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Zhang, W.; Hu, L.; Ding, W.; Wu, F.; Li, J. Etching synthesis of iron oxide nanoparticles for adsorption of arsenic from water. RSC Adv. 2016, 6, 15900–15910. [Google Scholar] [CrossRef]
- Xu, C.; De, S.; Balu, A.M.; Ojeda, M.; Luque, R. Mechanochemical synthesis of advanced nanomaterials for catalytic applications. Chem. Commun. 2015, 51, 6698–6713. [Google Scholar] [CrossRef] [PubMed]
- Roucoux, A.; Schulz, J.; Patin, H. Reduced transition metal colloids: A novel family of reusable catalysts? Chem. Rev. 2002, 102, 3757–3778. [Google Scholar] [CrossRef]
- Fiévet, F.; Ammar-Merah, S.; Brayner, R.; Chau, F.; Giraud, M.; Mammeri, F.; Peron, J.; Piquemal, J.Y.; Sicard, L.; Viau, G. The polyol process: A unique method for easy access to metal nanoparticles with tailored sizes, shapes and compositions. Chem. Soc. Rev. 2018, 47, 5187–5233. [Google Scholar] [CrossRef]
- Dong, H.; Chen, Y.C.; Feldmann, C. Polyol synthesis of nanoparticles: Status and options regarding metals, oxides, chalcogenides, and non-metal elements. Green Chem. 2015, 17, 4107–4132. [Google Scholar] [CrossRef]
- An, K.; Alayoglu, S.; Ewers, T.; Somorjai, G.A. Colloid chemistry of nanocatalysts: A molecular view. J. Colloid Interface Sci. 2012, 373, 1–13. [Google Scholar] [CrossRef]
- Kuhn, J.N.; Tsung, C.-K.; Huang, W.; Somorjai, G.A. Effect of organic capping layers over monodisperse platinum nanoparticles upon activity for ethylene hydrogenation and carbon monoxide oxidation. J. Catal. 2009, 265, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.-H.; Meijboom, R. Catalytic evaluation of dendrimer-templated Pd nanoparticles in the reduction of 4-nitrophenol using Langmuir-Hinshelwood kinetics. Appl. Surf. Sci. 2014, 320, 400–413. [Google Scholar] [CrossRef]
- Hinterwirth, H.; Kappel, S.; Waitz, T.; Prohaska, T.; Lindner, W.; Lämmerhofer, M. Quantifying Thiol Ligand Density of Self-Assembled Monolayers on Gold Nanoparticles by Inductively Coupled Plasma-Mass Spectrometry. ACS Nano 2013, 7, 1129–1136. [Google Scholar] [CrossRef]
- Serrano-Maldonado, A.; Martin, E.; Guerrero-Ríos, I. Pyridine-Stabilized Rhodium Nanoparticles in Ionic Liquids as Selective Hydrogenation and Transfer Hydrogenation Catalysts. Eur. J. Inorg. Chem. 2019, 2019, 2863–2870. [Google Scholar] [CrossRef]
- Gyergyek, S.; Makovec, D.; Drofenik, M. Colloidal stability of oleic- and ricinoleic-acid-coated magnetic nanoparticles in organic solvents. J. Colloid Interface Sci. 2011, 354, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Chawla, P.; Jain, S.; Sharma, S.N. Tapping the potential of trioctylphosphine (TOP) in the realization of highly luminescent blue-emitting colloidal indium phosphide (InP) quantum dots. Phys. E Low Dimens. Syst. Nanostruct. 2017, 90, 175–182. [Google Scholar] [CrossRef]
- Eriksson, S.; Nylén, U.; Rojas, S.; Boutonnet, M. Preparation of catalysts from microemulsions and their applications in heterogeneous catalysis. Appl. Catal. A Gen. 2004, 265, 207–219. [Google Scholar] [CrossRef]
- Martínez, A.; Prieto, G. The key role of support surface tuning during the preparation of catalysts from reverse micellar-synthesized metal nanoparticles. Catal. Commun. 2007, 8, 1479–1486. [Google Scholar] [CrossRef]
- Park, J.; Joo, J.; Kwon, S.G.; Jang, Y.; Hyeon, T. Synthesis of Monodisperse Spherical Nanocrystals. Angew. Chem. Int. Ed. 2007, 46, 4630–4660. [Google Scholar] [CrossRef]
- Thanh, N.T.K.; Maclean, N.; Mahiddine, S. Mechanisms of Nucleation and Growth of Nanoparticles in Solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef]
- Polte, J. Fundamental growth principles of colloidal metal nanoparticles—A new perspective. CrystEngComm 2015, 17, 6809–6830. [Google Scholar] [CrossRef] [Green Version]
- Murray, C.B.; Sun, S.; Gaschler, W.; Doyle, H.; Betley, T.A.; Kagan, C.R. Colloidal synthesis of nanocrystals and nanocrystal superlattices. IBM J. Res. Dev. 2001, 45, 47–56. [Google Scholar] [CrossRef]
- Mori, K.; Kumami, A.; Tomonari, M.; Yamashita, H. A pH-induced size controlled deposition of colloidal Ag nanoparticles on alumina support for catalytic application. J. Phys. Chem. C 2009, 113, 16850–16854. [Google Scholar] [CrossRef]
- Pol, V.G.; Srivastava, D.N.; Palchik, O.; Palchik, V.; Slifkin, M.A.; Weiss, A.M.; Gedanken, A. Sonochemical Deposition of Silver Nanoparticles on Silica Spheres. Langmuir 2002, 18, 3352–3357. [Google Scholar] [CrossRef]
- Doktycz, S.J.; Suslick, K.S. Interparticle collisions driven by ultrasound. Science 1990, 247, 1067–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cullen, D.A.; Zhai, P.; Ding, K. Adsorption of Colloidal Metal Nanoparticles via Solvent Engineering. ACS Catal. 2020, 10, 2378–2383. [Google Scholar] [CrossRef]
- Tovstun, S.A.; Razumov, V.F. Theory of size-selective precipitation. J. Nanopart. Res. 2016, 19, 8. [Google Scholar] [CrossRef]
- Quang, D.V.; Lee, J.E.; Kim, J.-K.; Kim, Y.N.; Shao, G.N.; Kim, H.T. A gentle method to graft thiol-functional groups onto silica gel for adsorption of silver ions and immobilization of silver nanoparticles. Powder Technol. 2013, 235, 221–227. [Google Scholar] [CrossRef]
- Maria Claesson, E.; Philipse, A.P. Thiol-functionalized silica colloids, grains, and membranes for irreversible adsorption of metal(oxide) nanoparticles. Colloids Surf. Physicochem. Eng. Aspects 2007, 297, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Rioux, R.M.; Song, H.; Hoefelmeyer, J.D.; Yang, P.; Somorjai, G.A. High-Surface-Area Catalyst Design: Synthesis, Characterization, and Reaction Studies of Platinum Nanoparticles in Mesoporous SBA-15 Silica. J. Phys. Chem. B 2005, 109, 2192–2202. [Google Scholar] [CrossRef]
- Haneveld, J.; Tas, N.R.; Brunets, N.; Jansen, H.V.; Elwenspoek, M. Capillary filling of sub-10nm nanochannels. J. Appl. Phys. 2008, 104, 014309. [Google Scholar] [CrossRef]
- Blavo, S.O.; Qayyum, E.; Baldyga, L.M.; Castillo, V.A.; Sanchez, M.D.; Warrington, K.; Barakat, M.A.; Kuhn, J.N. Verification of Organic Capping Agent Removal from Supported Colloidal Synthesized Pt Nanoparticle Catalysts. Top. Catal. 2013, 56, 1835–1842. [Google Scholar] [CrossRef]
- Huang, W.X.; Hua, Q.; Cao, T. Influence and Removal of Capping Ligands on Catalytic Colloidal Nanoparticles. Catal. Lett. 2014, 144, 1355–1369. [Google Scholar] [CrossRef]
- Delgado, J.A.; Claver, C.; Castillon, S.; Curulla-Ferre, D.; Ordomsky, V.V.; Godard, C. Effect of polymeric stabilizers on Fischer-Tropsch synthesis catalyzed by cobalt nanoparticles supported on TiO2. J. Mol. Catal. A-Chem. 2016, 417, 43–52. [Google Scholar] [CrossRef]
- He, B.; Zhao, Q.; Zeng, Z.; Wang, X.; Han, S. Effect of hydrothermal reaction time and calcination temperature on properties of Au@CeO2 core-shell catalyst for CO oxidation at low temperature. J. Mater. Sci. 2015, 50, 6339–6348. [Google Scholar] [CrossRef]
- Tsubota, S.; Nakamura, T.; Tanaka, K.; Haruta, M. Effect of calcination temperature on the catalytic activity of Au colloids mechanically mixed with TiO2 powder for CO oxidation. Catal. Lett. 1998, 56, 131–135. [Google Scholar] [CrossRef]
- Freund, H.-J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chem. Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef]
- Al Soubaihi, R.M.; Saoud, K.M.; Dutta, J. Critical Review of Low-Temperature CO Oxidation and Hysteresis Phenomenon on Heterogeneous Catalysts. Catalysts 2018, 8, 660. [Google Scholar] [CrossRef] [Green Version]
- Xi, K.; Wang, Y.; Jiang, K.; Xie, J.; Zhou, Y.; Lu, H. Support interaction of Pt/CeO2 and Pt/SiC catalysts prepared by nano platinum colloid deposition for CO oxidation. J. Rare Earths 2020, 38, 376–383. [Google Scholar] [CrossRef]
- Ciriminna, R.; Pandarus, V.; Béland, F.; Xu, Y.-J.; Pagliaro, M. Heterogeneously Catalyzed Alcohol Oxidation for the Fine Chemical Industry. Org. Process Res. Dev. 2015, 19, 1554–1558. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, V.P.; Srikanth, A.; Vishwanathan, V.; Chary, K.V.R. Vapor Phase Oxidation of Benzyl Alcohol over Nano Au/SBA-15 Catalysts: Effect of Preparation Methods. Catal. Lett. 2016, 146, 35–46. [Google Scholar] [CrossRef]
- Saadatjou, N.; Jafari, A.; Sahebdelfar, S. Synthesis and Characterization of Ru/Al2O3 Nanocatalyst for Ammonia Synthesis. Iran. J. Chem. Chem. Eng.-Int. Engl. Ed. 2015, 34, 1–9. [Google Scholar]
- Abashar, M.E.E. Ultra-clean hydrogen production by ammonia decomposition. J. King Saud Univ. Eng. Sci. 2018, 30, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, T.V.; Sivadinarayana, C.; Goodman, D.W. Catalytic ammonia decomposition: COx-free hydrogen production for fuel cell applications. Catal. Lett. 2001, 72, 197–201. [Google Scholar] [CrossRef]
- Bell, T.E.; Torrente-Murciano, L. H2 Production via Ammonia Decomposition Using Non-Noble Metal Catalysts: A Review. Top. Catal. 2016, 59, 1438–1457. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wen, J.; Ali, A.M.; Duan, M.; Zhu, W.; Zhang, H.; Chen, C.; Li, Y. Size structure–catalytic performance correlation of supported Ni/MCF-17 catalysts for COx-free hydrogen production. Chem. Commun. 2018, 54, 6364–6367. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, J.; Xu, H.; Li, W. NH3 Decomposition Kinetics on Supported Ru Clusters: Morphology and Particle Size Effect. Catal. Lett. 2007, 119, 311–318. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, H.; Li, W. Kinetic study of NH3 decomposition over Ni nanoparticles: The role of La promoter, structure sensitivity and compensation effect. Appl. Catal. A Gen. 2005, 296, 257–267. [Google Scholar] [CrossRef]
- Mahmoudi, H.; Mahmoudi, M.; Doustdar, O.; Jahangiri, H.; Tsolakis, A.; Gu, S.; LechWyszynski, M. A review of Fischer Tropsch synthesis process, mechanism, surface chemistry and catalyst formulation. Biofuels Eng. 2017, 2. [Google Scholar] [CrossRef]
- Dalai, A.K.; Davis, B.H. Fischer-Tropsch synthesis: A review of water effects on the performances of unsupported and supported Co catalysts. Appl. Catal. A Gen. 2008, 348, 1–15. [Google Scholar] [CrossRef]
- Jahangiri, H.; Bennett, J.; Mahjoubi, P.; Wilson, K.; Gu, S. A review of advanced catalyst development for Fischer-Tropsch synthesis of hydrocarbons from biomass derived syn-gas. Catal. Sci. Technol. 2014, 4, 2210–2229. [Google Scholar] [CrossRef] [Green Version]
- Steynberg, A.P.; Nel, H.G. Clean coal conversion options using Fischer-Tropsch technology. Fuel 2004, 83, 765–770. [Google Scholar] [CrossRef]
- Melaet, G.; Ralston, W.T.; Li, C.-S.; Alayoglu, S.; An, K.; Musselwhite, N.; Kalkan, B.; Somorjai, G.A. Evidence of Highly Active Cobalt Oxide Catalyst for the Fischer-Tropsch Synthesis and CO2 Hydrogenation. J. Am. Chem. Soc. 2014, 136, 2260–2263. [Google Scholar] [CrossRef]
- Delgado, J.A.; Claver, C.; Castillón, S.; Curulla-Ferré, D.; Ordomsky, V.V.; Godard, C. Fischer-Tropsch synthesis catalysed by small TiO2 supported cobalt nanoparticles prepared by sodium borohydride reduction. Appl. Catal. A Gen. 2016, 513, 39–46. [Google Scholar] [CrossRef]
- Glavee, G.N.; Klabunde, K.J.; Sorensen, C.M.; Hadjipanayis, G.C. Borohydride reduction of cobalt ions in water. Chemistry leading to nanoscale metal, boride, or borate particles. Langmuir 1993, 9, 162–169. [Google Scholar] [CrossRef]
- Tan, K.F.; Chang, J.; Borgna, A.; Saeys, M. Effect of boron promotion on the stability of cobalt Fischer-Tropsch catalysts. J. Catal. 2011, 280, 50–59. [Google Scholar] [CrossRef]
- Saeys, M.; Tan, K.F.; Chang, J.; Borgna, A. Improving the Stability of Cobalt Fischer-Tropsch Catalysts by Boron Promotion. Ind. Eng. Chem. Res. 2010, 49, 11098–11100. [Google Scholar] [CrossRef]
- Krans, N.A.; Weber, J.L.; Van den Bosch, W.; Zecevic, J.; De Jongh, P.E.; De Jong, K.P. Influence of Promotion on the Growth of Anchored Colloidal Iron Oxide Nanoparticles during Synthesis Gas Conversion. ACS Catal. 2020, 10, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Torres Galvis, H.M.; Koeken, A.C.J.; Kirilin, A.; Dugulan, A.I.; Ruitenbeek, M.; De Jong, K.P. Size and Promoter Effects on Stability of Carbon-Nanofiber-Supported Iron-Based Fischer-Tropsch Catalysts. ACS Catal. 2016, 6, 4017–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hoof, A.J.F.; Michel-Ligthart, D.A.J.; Friedrich, H.; Hensen, E.J.M. The Influence and Removability of Colloidal Capping Agents on Carbon Monoxide Hydrogenation by Zirconia-Supported Rhodium Nanoparticles. Chemcatchem 2017, 9, 1018–1024. [Google Scholar] [CrossRef]
- Lam, E.; Larmier, K.; Wolf, P.; Tada, S.; Safonova, O.V.; Copéret, C. Isolated Zr Surface Sites on Silica Promote Hydrogenation of CO2 to CH3OH in Supported Cu Catalysts. J. Am. Chem. Soc. 2018, 140, 10530–10535. [Google Scholar] [CrossRef]
- Singh, A.K.; Xu, Q. Synergistic Catalysis over Bimetallic Alloy Nanoparticles. ChemCatChem 2013, 5, 652–676. [Google Scholar] [CrossRef]
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef]
- Jiang, H.-L.; Xu, Q. Recent progress in synergistic catalysis over heterometallic nanoparticles. J. Mater. Chem. 2011, 21, 13705–13725. [Google Scholar] [CrossRef]
- Wang, D.; Li, Y. Bimetallic Nanocrystals: Liquid-Phase Synthesis and Catalytic Applications. Adv. Mater. 2011, 23, 1044–1060. [Google Scholar] [CrossRef] [PubMed]
- Destro, P. Colloidal Nanoparticles for Heterogeneous Catalysis; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Destro, P.; Marras, S.; Manna, L.; Colombo, M.; Zanchet, D. AuCu alloy nanoparticles supported on SiO2: Impact of redox pretreatments in the catalyst performance in CO oxidation. Catal. Today 2017, 282, 105–110. [Google Scholar] [CrossRef]
- Destro, P.; Kokumai, T.M.; Scarpellini, A.; Pasquale, L.; Manna, L.; Colombo, M.; Zanchet, D. The Crucial Role of the Support in the Transformations of Bimetallic Nanoparticles and Catalytic Performance. ACS Catal. 2018, 8, 1031–1037. [Google Scholar] [CrossRef]
- Najafishirtari, S.; Brescia, R.; Guardia, P.; Marras, S.; Manna, L.; Colombo, M. Nanoscale Transformations of Alumina-Supported AuCu Ordered Phase Nanocrystals and Their Activity in CO Oxidation. ACS Catal. 2015, 5, 2154–2163. [Google Scholar] [CrossRef]
- Zaytsev, S.Y.; Plyusnin, P.E.; Slavinskaya, E.M.; Shubin, Y.V. Synthesis of bimetallic nanocompositions AuxPd1-x/γ-Al2O3 for catalytic CO oxidation. J. Nanopart. Res. 2017, 19, 367. [Google Scholar] [CrossRef]
- Nagy, G.; Benkó, T.; Borkó, L.; Csay, T.; Horváth, A.; Frey, K.; Beck, A. Bimetallic Au-Ag/SiO2 catalysts: Comparison in glucose, benzyl alcohol and CO oxidation reactions. React. Kinet. Mech. Catal. 2015, 115, 45–65. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Banerjee, S.; Choudhary, V.R. Catalysts for combustion of methane and lower alkanes. Appl. Catal. A Gen. 2002, 234, 1–23. [Google Scholar] [CrossRef]
- Xiong, H.; Wiebenga, M.H.; Carrillo, C.; Gaudet, J.R.; Pham, H.N.; Kunwar, D.; Oh, S.H.; Qi, G.; Kim, C.H.; Datye, A.K. Design considerations for low-temperature hydrocarbon oxidation reactions on Pd based catalysts. Appl. Catal. B Environ. 2018, 236, 436–444. [Google Scholar] [CrossRef]
- Nie, H.; Howe, J.Y.; Lachkov, P.T.; Chin, Y.-H.C. Chemical and Structural Dynamics of Nanostructures in Bimetallic Pt-Pd Catalysts, Their Inhomogeneity, and Their Roles in Methane Oxidation. ACS Catal. 2019, 9, 5445–5461. [Google Scholar] [CrossRef]
- Nassiri, H.; Lee, K.-E.; Hu, Y.; Hayes, R.E.; Scott, R.W.J.; Semagina, N. Platinum Inhibits Low-Temperature Dry Lean Methane Combustion through Palladium Reduction in Pd-Pt/Al2O3: An In Situ X-ray Absorption Study. Chemphyschem 2017, 18, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, K.; Jansson, K.; Järås, S.G. Characterisation and microstructure of Pd and bimetallic Pd-Pt catalysts during methane oxidation. J. Catal. 2007, 245, 401–414. [Google Scholar] [CrossRef]
- Qu, P.; Wang, S.; Hu, W.; Wu, Y.; Chen, J.; Zhang, G.; Shen, P.; Chen, Y.; Zhong, L. A novel strategy to design PtPd bimetallic catalysts for efficient methane combustion. Catal. Commun. 2020, 135, 105900. [Google Scholar] [CrossRef]
- Cano, L.A.; Garcia Blanco, A.A.; Lener, G.; Marchetti, S.G.; Sapag, K. Effect of the support and promoters in Fischer-Tropsch synthesis using supported Fe catalysts. Catal. Today 2017, 282, 204–213. [Google Scholar] [CrossRef]
- Ismail, A.S.M.; Casavola, M.; Liu, B.; Gloter, A.; Van Deelen, T.W.; Versluijs, M.; Meeldijk, J.D.; Stéphan, O.; De Jong, K.P.; De Groot, F.M.F. Atomic-Scale Investigation of the Structural and Electronic Properties of Cobalt-Iron Bimetallic Fischer-Tropsch Catalysts. ACS Catal. 2019, 9, 7998–8011. [Google Scholar] [CrossRef] [Green Version]
- Dad, M.; Lancee, R.J.; Janse van Vuuren, M.; Van de Loosdrecht, J.; Niemantsverdriet, J.W.H.; Fredriksson, H.O.A. SiO2-supported Fe & FeMn colloids—Fischer-Tropsch synthesis on 3D model catalysts. Appl. Catal. A Gen. 2017, 537, 83–92. [Google Scholar] [CrossRef]
- Jiang, N.; Yang, G.; Zhang, X.; Wang, L.; Shi, C.; Tsubaki, N. A novel silicalite-1 zeolite shell encapsulated iron-based catalyst for controlling synthesis of light alkenes from syngas. Catal. Commun. 2011, 12, 951–954. [Google Scholar] [CrossRef]
- Yang, G.; Tan, Y.; Han, Y.; Qiu, J.; Tsubaki, N. Increasing the shell thickness by controlling the core size of zeolite capsule catalyst: Application in iso-paraffin direct synthesis. Catal. Commun. 2008, 9, 2520–2524. [Google Scholar] [CrossRef]
- Zeng, B.; Hou, B.; Jia, L.; Wang, J.; Chen, C.; Li, D.; Sun, Y. The intrinsic effects of shell thickness on the Fischer-Tropsch synthesis over core-shell structured catalysts. Catal. Sci. Technol. 2013, 3, 3250–3255. [Google Scholar] [CrossRef]
- Haghtalab, A.; Mosayebi, A. Co@Ru nanoparticle with core-shell structure supported over γ-Al2O3 for Fischer-Tropsch synthesis. Int. J. Hydrogen Energy 2014, 39, 18882–18893. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl ether (DME) as an alternative fuel. J. Power Sources 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Anggarani, R.; Wibowo, C.S.; Rulianto, D. Application of dimethyl ether as LPG substitution for household stove. Energy Procedia 2014, 47, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Olah, G.A.; Goeppert, A.; Prakash, G.S. Chemical recycling of carbon dioxide to methanol and dimethyl ether: From greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef] [PubMed]
- French, S.; Sokol, A.; Bromley, S.; Catlow, C.; Sherwood, P. Identification and characterization of active sites and their catalytic processes—The Cu/ZnO methanol catalyst. Top. Catal. 2003, 24, 161–172. [Google Scholar] [CrossRef]
- Stiefel, M.; Ahmad, R.; Arnold, U.; Döring, M. Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions. Fuel Process. Technol. 2011, 92, 1466–1474. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, X.; Asami, K.; Asaoka, S.; Fujimoto, K. Synthesis of LPG from synthesis gas. Fuel Process. Technol. 2004, 85, 1139–1150. [Google Scholar] [CrossRef]
- Gentzen, M.; Habicht, W.; Doronkin, D.E.; Grunwaldt, J.D.; Sauer, J.; Behrens, S. Bifunctional hybrid catalysts derived from Cu/Zn-based nanoparticles for single-step dimethyl ether synthesis. Catal. Sci. Technol. 2016, 6, 1054–1063. [Google Scholar] [CrossRef]
- Gentzen, M.; Doronkin, D.E.; Sheppard, T.L.; Grunwaldt, J.D.; Sauer, J.; Behrens, S. Bifunctional catalysts based on colloidal Cu/Zn nanoparticles for the direct conversion of synthesis gas to dimethyl ether and hydrocarbons. Appl. Catal. A Gen. 2018, 557, 99–107. [Google Scholar] [CrossRef]
- Lebarbier, V.M.; Dagle, R.A.; Kovarik, L.; Lizarazo-Adarme, J.A.; King, D.L.; Palo, D.R. Synthesis of methanol and dimethyl ether from syngas over Pd/ZnO/Al2O3 catalysts. Catal. Sci. Technol. 2012, 2, 2116–2127. [Google Scholar] [CrossRef]
- Bahruji, H.; Bowker, M.; Jones, W.; Hayward, J.; Esquius, J.R.; Morgan, D.J.; Hutchings, G.J. PdZn catalysts for CO2 hydrogenation to methanol using chemical vapour impregnation (CVI). Faraday Discuss. 2017, 197, 309–324. [Google Scholar] [CrossRef]
- Gentzen, M.; Doronkin, D.E.; Sheppard, T.L.; Zimina, A.; Li, H.; Jelic, J.; Studt, F.; Grunwaldt, J.-D.; Sauer, J.; Behrens, S. Supported Intermetallic PdZn Nanoparticles as Bifunctional Catalysts for the Direct Synthesis of Dimethyl Ether from CO-Rich Synthesis Gas. Angew. Chem. Int. Ed. 2019, 58, 15655–15659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh Kumar, B.; Saravanan, S. Use of higher alcohol biofuels in diesel engines: A review. Renew. Sust. Energ. Rev. 2016, 60, 84–115. [Google Scholar] [CrossRef]
- Lu, Y.; Cao, B.; Yu, F.; Liu, J.; Bao, Z.; Gao, J. High Selectivity Higher Alcohols Synthesis from Syngas over Three-Dimensionally Ordered Macroporous Cu-Fe Catalysts. ChemCatChem 2014, 6, 473–478. [Google Scholar] [CrossRef]
- Gao, W.; Zhao, Y.; Liu, J.; Huang, Q.; He, S.; Li, C.; Zhao, J.; Wei, M. Catalytic conversion of syngas to mixed alcohols over CuFe-based catalysts derived from layered double hydroxides. Catal. Sci. Technol. 2013, 3, 1324–1332. [Google Scholar] [CrossRef]
- Xiao, K.; Bao, Z.; Qi, X.; Wang, X.; Zhong, L.; Fang, K.; Lin, M.; Sun, Y. Structural evolution of CuFe bimetallic nanoparticles for higher alcohol synthesis. J. Mol. Catal. A Chem. 2013, 378, 319–325. [Google Scholar] [CrossRef]
- Xiao, K.; Qi, X.; Bao, Z.; Wang, X.; Zhong, L.; Fang, K.; Lin, M.; Sun, Y. CuFe, CuCo and CuNi nanoparticles as catalysts for higher alcohol synthesis from syngas: A comparative study. Catal. Sci. Technol. 2013, 3, 1591–1602. [Google Scholar] [CrossRef]
- He, S.; Wang, W.; Shen, Z.; Li, G.; Kang, J.; Liu, Z.; Wang, G.-C.; Zhang, Q.; Wang, Y. Carbon nanotube-supported bimetallic Cu-Fe catalysts for syngas conversion to higher alcohols. Mol. Catal. 2019, 479, 110610. [Google Scholar] [CrossRef]
- Aitbekova, A.; Goodman, E.D.; Wu, L.; Boubnov, A.; Hoffman, A.S.; Genc, A.; Cheng, H.; Casalena, L.; Bare, S.R.; Cargnello, M. Engineering of Ruthenium-Iron Oxide Colloidal Heterostructures: Improved Yields in CO2 Hydrogenation to Hydrocarbons. Angew. Chem. Int. Ed. 2019, 58, 17451–17457. [Google Scholar] [CrossRef]
- McCue, A.J.; Anderson, J.A. Recent advances in selective acetylene hydrogenation using palladium containing catalysts. Front. Chem. Sci. Eng. 2015, 9, 142–153. [Google Scholar] [CrossRef]
- Bruno, J.E.; Dwarica, N.S.; Whittaker, T.N.; Hand, E.R.; Guzman, C.S.; Dasgupta, A.; Chen, Z.; Rioux, R.M.; Chandler, B.D. Supported Ni-Au Colloid Precursors for Active, Selective, and Stable Alkyne Partial Hydrogenation Catalysts. ACS Catal. 2020, 10, 2565–2580. [Google Scholar] [CrossRef]
- Nawaz, Z. Light alkane dehydrogenation to light olefin technologies: A comprehensive review. Rev. Chem. Eng. 2015, 31, 413–436. [Google Scholar] [CrossRef]
- Iglesias-Juez, A.; Beale, A.M.; Maaijen, K.; Weng, T.C.; Glatzel, P.; Weckhuysen, B.M. A combined in situ time-resolved UV-Vis, Raman and high-energy resolution X-ray absorption spectroscopy study on the deactivation behavior of Pt and PtSn propane dehydrogenation catalysts under industrial reaction conditions. J. Catal. 2010, 276, 268–279. [Google Scholar] [CrossRef]
- Bariås, O.A.; Holmen, A.; Blekkan, E.A. Propane dehydrogenation over supported Pt and Pt-Sn catalysts: Catalyst preparation, characterization, and activity measurements. J. Catal. 1996, 158, 1–12. [Google Scholar] [CrossRef]
- Yu, C.; Ge, Q.; Xu, H.; Li, W. Effects of Ce addition on the Pt-Sn/γ-Al2O3 catalyst for propane dehydrogenation to propylene. Appl. Catal. A Gen. 2006, 315, 58–67. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Qiu, A.; Wang, Y.; Xu, Y.; Wu, P. Propane dehydrogenation on PtSn/ZSM-5 catalyst: Effect of tin as a promoter. Catal. Commun. 2006, 7, 860–866. [Google Scholar] [CrossRef]
- Lobera, M.; Tellez, C.; Herguido, J.; Menéndez, M. Transient kinetic modelling of propane dehydrogenation over a Pt-Sn-K/Al2O3 catalyst. Appl. Catal. A Gen. 2008, 349, 156–164. [Google Scholar] [CrossRef]
- Vu, B.K.; Song, M.B.; Ahn, I.Y.; Suh, Y.-W.; Suh, D.J.; Kim, W.-I.; Koh, H.-L.; Choi, Y.G.; Shin, E.W. Pt-Sn alloy phases and coke mobility over Pt-Sn/Al2O3 and Pt-Sn/ZnAl2O4 catalysts for propane dehydrogenation. Appl. Catal. A Gen. 2011, 400, 25–33. [Google Scholar] [CrossRef]
- Zhu, H.; Anjum, D.H.; Wang, Q.; Abou-Hamad, E.; Emsley, L.; Dong, H.; Laveille, P.; Li, L.; Samal, A.K.; Basset, J.-M. Sn surface-enriched Pt-Sn bimetallic nanoparticles as a selective and stable catalyst for propane dehydrogenation. J. Catal. 2014, 320, 52–62. [Google Scholar] [CrossRef]
- Kaylor, N.; Davis, R.J. Propane dehydrogenation over supported Pt-Sn nanoparticles. J. Catal. 2018, 367, 181–193. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Marin, G.B. Enhanced Carbon-Resistant Dry Reforming Fe-Ni Catalyst: Role of Fe. ACS Catal. 2015, 5, 3028–3039. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Sabbe, M.; Poelman, H.; Detavernier, C.; Marin, G.B. Controlling the stability of a Fe-Ni reforming catalyst: Structural organization of the active components. Appl. Catal. B Environ. 2017, 209, 405–416. [Google Scholar] [CrossRef]
- De Lima, S.M.; Assaf, J.M. Ni-Fe Catalysts Based on Perovskite-type Oxides for Dry Reforming of Methane to Syngas. Catal. Lett. 2006, 108, 63–70. [Google Scholar] [CrossRef]
- Medeiros, R.L.B.A.; Macedo, H.P.; Melo, V.R.M.; Oliveira, Â.A.S.; Barros, J.M.F.; Melo, M.A.F.; Melo, D.M.A. Ni supported on Fe-doped MgAl2O4 for dry reforming of methane: Use of factorial design to optimize H2 yield. Int. J. Hydrogen Energy 2016, 41, 14047–14057. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Batchu, R.; Galvita, V.V.; Poelman, H.; Marin, G.B. Carbon gasification from Fe-Ni catalysts after methane dry reforming. Appl. Catal. B Environ. 2016, 185, 42–55. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Dharanipragada, N.V.R.A.; Longo, A.; Meledina, M.; Van Tendeloo, G.; Detavernier, C.; Marin, G.B. Fe-Containing Magnesium Aluminate Support for Stability and Carbon Control during Methane Reforming. ACS Catal. 2018, 8, 5983–5995. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Margossian, T.; Hosseini, D.; Foppa, L.; Armutlulu, A.; Van Beek, W.; Comas-Vives, A.; Copéret, C.; Müller, C. Cooperativity and Dynamics Increase the Performance of NiFe Dry Reforming Catalysts. J. Am. Chem. Soc. 2017, 139, 1937–1949. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Poelman, H.; Marin, G.B.; Galvita, V.V. Chapter 6—How Does the Surface Structure of Ni-Fe Nanoalloys Control Carbon Formation During Methane Steam/Dry Reforming. In Advanced Nanomaterials for Catalysis and Energy; Sadykov, V.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 177–225. [Google Scholar] [CrossRef]
- Margossian, T.; Larmier, K.; Kim, S.M.; Krumeich, F.; Müller, C.; Copéret, C. Supported Bimetallic NiFe Nanoparticles through Colloid Synthesis for Improved Dry Reforming Performance. ACS Catal. 2017, 7, 6942–6948. [Google Scholar] [CrossRef]
- Li, Z.W.; Wang, Z.G.; Kawi, S. Sintering and Coke Resistant Core/Yolk Shell Catalyst for Hydrocarbon Reforming. Chemcatchem 2019, 11, 202–224. [Google Scholar] [CrossRef]
- Park, J.C.; Song, H. Metal@Silica Yolk-Shell Nanostructures as Versatile Bifunctional Nanocatalysts. Nano Res. 2011, 4, 33–49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, T.; Ge, J.; Yin, Y. Permeable Silica Shell through Surface-Protected Etching. Nano Lett. 2008, 8, 2867–2871. [Google Scholar] [CrossRef]
- Zhang, Q.; Lee, I.; Ge, J.; Zaera, F.; Yin, Y. Surface-Protected Etching of Mesoporous Oxide Shells for the Stabilization of Metal Nanocatalysts. Adv. Funct. Mater. 2010, 20, 2201–2214. [Google Scholar] [CrossRef]
- Galeano, C.; Güttel, R.; Paul, M.; Arnal, P.; Lu, A.-H.; Schüth, F. Yolk-Shell Gold Nanoparticles as Model Materials for Support-Effect Studies in Heterogeneous Catalysis: Au, @C and Au, @ZrO2 for CO Oxidation as an Example. Chem. Eur. 2011, 17, 8434–8439. [Google Scholar] [CrossRef] [PubMed]
- Prieto, G.; Tüysüz, H.; Duyckaerts, N.; Knossalla, J.; Wang, G.-H.; Schüth, F. Hollow Nano- and Microstructures as Catalysts. Chem. Rev. 2016, 116, 14056–14119. [Google Scholar] [CrossRef] [PubMed]
- Bakhmutsky, K.; Wieder, N.L.; Cargnello, M.; Galloway, B.; Fornasiero, P.; Gorte, R.J. A Versatile Route to Core-Shell Catalysts: Synthesis of Dispersible M@Oxide (M = Pd, Pt; Oxide = TiO2, ZrO2) Nanostructures by Self-Assembly. Chemsuschem 2012, 5, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Seo, C.Y.; Chen, X.; Sun, K.; Allard, L.F.; Fisher, G.B.; Schwank, J.W. Palladium redispersion at high temperature within the Pd@SiO2 core@shell structure. Catal. Commun. 2018, 108, 73–76. [Google Scholar] [CrossRef]
- Shirman, E.; Shirman, T.; Shneidman, A.V.; Grinthal, A.; Phillips, K.R.; Whelan, H.; Bulger, E.; Abramovitch, M.; Patil, J.; Nevarez, R.; et al. Modular Design of Advanced Catalytic Materials Using Hybrid Organic-Inorganic Raspberry Particles. Adv. Funct. Mater. 2018, 28, 1704559. [Google Scholar] [CrossRef]
- Shirman, T.; Lattimer, J.; Luneau, M.; Shirman, E.; Reece, C.; Aizenberg, M.; Madix, R.J.; Aizenberg, J.; Friend, C.M. New Architectures for Designed Catalysts: Selective Oxidation using AgAu Nanoparticles on Colloid-Templated Silica. Chem.-Eur. J. 2018, 24, 1833–1837. [Google Scholar] [CrossRef] [Green Version]
- Luneau, M.; Shirman, T.; Filie, A.; Timoshenko, J.; Chen, W.; Trimpalis, A.; Flytzani-Stephanopoulos, M.; Kaxiras, E.; Frenkel, A.I.; Aizenberg, J.; et al. Dilute Pd/Au Alloy Nanoparticles Embedded in Colloid-Templated Porous SiO2: Stable Au-Based Oxidation Catalysts. Chem. Mater. 2019, 31, 5759–5768. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.; Jung, W. Sintering Resistance of Pt@SiO2 Core-Shell Catalyst. Chemcatchem 2019, 11, 4653–4659. [Google Scholar] [CrossRef]
- Habibi, A.H.; Hayes, R.E.; Semagina, N. Evaluation of hydrothermal stability of encapsulated PdPt@SiO2 catalyst for lean CH4 combustion. Appl. Catal. A Gen. 2018, 556, 129–136. [Google Scholar] [CrossRef]
- Pei, W.; Liu, Y.; Deng, J.; Zhang, K.; Hou, Z.; Zhao, X.; Dai, H. Partially embedding Pt nanoparticles in the skeleton of 3DOM Mn2O3: An effective strategy for enhancing catalytic stability in toluene combustion. Appl. Catal. B Environ. 2019, 256, 117814. [Google Scholar] [CrossRef]
- Sadakane, M.; Ueda, W. Three-Dimensionally Ordered Macroporous (3DOM) Perovskite Mixed Metal Oxides. In Perovskites and Related Mixed Oxides; Granger, P., Parvulescu, V.I., Prellier, W., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2015; pp. 113–142. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; Van Dillen, A.J.; De Jong, K.P. Cobalt Particle Size Effects in the Fischer-Tropsch Reaction Studied with Carbon Nanofiber Supported Catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wielers, A.F.H.; Kock, A.J.H.M.; Hop, C.E.C.A.; Geus, J.W.; Van Der Kraan, A.M. The reduction behavior of silica-supported and alumina-supported iron catalysts: A Mössbauer and infrared spectroscopic study. J. Catal. 1989, 117, 1–18. [Google Scholar] [CrossRef]
- Ni, Z.; Qin, H.; Kang, S.; Bai, J.; Wang, Z.; Li, Y.; Zheng, Z.; Li, X. Effect of graphitic carbon modification on the catalytic performance of Fe@SiO2-GC catalysts for forming lower olefins via Fischer-Tropsch synthesis. J. Colloid Interface Sci. 2018, 516, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, M.W.; Zhu, C.; Mu, X.L.; Zhang, K.; Zhong, L.S.; Fang, K.G.; Wu, M.H. Fabrication of Highly Stable SiO2 Encapsulated Multiple CuFe Nanoparticles for Higher Alcohols Synthesis via CO Hydrogenation. Catal. Lett. 2018, 148, 1080–1092. [Google Scholar] [CrossRef]
- Ilsemann, J.; Strass-Eifert, A.; Friedland, J.; Kiewidt, L.; Thoming, J.; Baumer, M.; Guttel, R. Cobalt@Silica Core-Shell Catalysts for Hydrogenation of CO/CO2 Mixtures to Methane. Chemcatchem 2019, 11, 4884–4893. [Google Scholar] [CrossRef] [Green Version]
- Luneau, M.; Shirman, T.; Foucher, A.C.; Duanmu, K.; Verbart, D.M.A.; Sautet, P.; Stach, E.A.; Aizenberg, J.; Madix, R.J.; Friend, C.M. Achieving High Selectivity for Alkyne Hydrogenation at High Conversions with Compositionally Optimized PdAu Nanoparticle Catalysts in Raspberry Colloid-Templated SiO2. ACS Catal. 2020, 10, 441–450. [Google Scholar] [CrossRef]
- Zhang, Y.; Diao, W.; Williams, C.T.; Monnier, J.R. Selective hydrogenation of acetylene in excess ethylene using Ag- and Au–Pd/SiO2 bimetallic catalysts prepared by electroless deposition. Appl. Catal. A Gen. 2014, 469, 419–426. [Google Scholar] [CrossRef]
- Li, M.; Shen, J. Microcalorimetric and infrared spectroscopic studies of CO and C2H4 adsorption on Pd/SiO2 and Pd-Ag/SiO2 catalysts. Mater. Chem. Phys. 2001, 68, 204–209. [Google Scholar] [CrossRef]
- Teschner, D.; Borsodi, J.; Wootsch, A.; Révay, Z.; Hävecker, M.; Knop-Gericke, A.; Jackson, S.D.; Schlögl, R. The roles of subsurface carbon and hydrogen in palladium-catalyzed alkyne hydrogenation. Science 2008, 320, 86–89. [Google Scholar] [CrossRef]
- Zhao, S.; Li, Y.; Liu, D.; Liu, J.; Liu, Y.-M.; Zakharov, D.N.; Wu, Q.; Orlov, A.; Gewirth, A.A.; Stach, E.A. Multimodal study of the speciations and activities of supported Pd catalysts during the hydrogenation of ethylene. J. Phys. Chem. C 2017, 121, 18962–18972. [Google Scholar] [CrossRef]
- Yang, H.; Gao, P.; Zhang, C.; Zhong, L.; Li, X.; Wang, S.; Wang, H.; Wei, W.; Sun, Y. Core-shell structured Cu@m-SiO2 and Cu/ZnO@m-SiO2 catalysts for methanol synthesis from CO2 hydrogenation. Catal. Commun. 2016, 84, 56–60. [Google Scholar] [CrossRef]
- Shi, Z.S.; Tan, Q.Q.; Wu, D.F. A novel Core-Shell structured CuIn@SiO2 catalyst for CO2 hydrogenation to methanol. AIChE J. 2019, 65, 1047–1058. [Google Scholar] [CrossRef]
- Kawi, S.; Kathiraser, Y.; Ni, J.; Oemar, U.; Li, Z.; Saw, E.T. Progress in Synthesis of Highly Active and Stable Nickel-Based Catalysts for Carbon Dioxide Reforming of Methane. ChemSusChem 2015, 8, 3556–3575. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Han, B.; Zhang, L.; Xu, L.; Yu, H.; Shi, W. CO2 reforming with methane over small-sized Ni@SiO2 catalysts with unique features of sintering-free and low carbon. Appl. Catal. B Environ. 2018, 235, 26–35. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, F.; Zhu, J.; Han, B.; Fan, W.; Zhao, L.; Cai, W.; Li, Z.; Xu, L.; Yu, H.; et al. CO2 reforming with methane reaction over Ni@SiO2 catalysts coupled by size effect and metal-support interaction. Fuel 2019, 256, 115954. [Google Scholar] [CrossRef]
- Han, B.; Wang, F.; Zhang, L.; Wang, Y.; Fan, W.; Xu, L.; Yu, H.; Li, Z. Syngas production from methane steam reforming and dry reforming reactions over sintering-resistant Ni@SiO2 catalyst. Res. Chem. Intermed. 2020, 46, 1735–1748. [Google Scholar] [CrossRef]
- LeValley, T.L.; Richard, A.R.; Fan, M. The progress in water gas shift and steam reforming hydrogen production technologies—A review. Int. J. Hydrogen Energy 2014, 39, 16983–17000. [Google Scholar] [CrossRef]
- Hwang, K.-R.; Lee, C.-B.; Park, J.-S. Advanced nickel metal catalyst for water-gas shift reaction. J. Power Sources 2011, 196, 1349–1352. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I. Effect of the nature of the support on the catalytic performance of noble metal catalysts for the water-gas shift reaction. Catal. Today 2006, 112, 49–52. [Google Scholar] [CrossRef]
- Ashok, J.; Wai, M.H.; Kawi, S. Nickel-based Catalysts for High-temperature Water Gas Shift Reaction-Methane Suppression. ChemCatChem 2018, 10, 3927–3942. [Google Scholar] [CrossRef]
- Gao, L.; Ta, N.; Dong, J.; Song, T.; Chen, S.; Fu, Q. Facile Transformation of Ni-based Colloids into Highly Stable Nanocatalysts Embedded within h-BN for the Water-Gas Shift Reaction. ChemCatChem 2020, 12, 1556–1561. [Google Scholar] [CrossRef]
- George, S.M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Puurunen, R.L. Surface chemistry of atomic layer deposition: A case study for the trimethylaluminum/water process. J. Appl. Phys. 2005, 97, 121301. [Google Scholar] [CrossRef]
- Hämäläinen, J.; Ritala, M.; Leskelä, M. Atomic Layer Deposition of Noble Metals and Their Oxides. Chem. Mater. 2014, 26, 786–801. [Google Scholar] [CrossRef]
- Hagen, D.J.; Pemble, M.E.; Karppinen, M. Atomic layer deposition of metals: Precursors and film growth. Appl. Phys. Rev. 2019, 6, 041309. [Google Scholar] [CrossRef]
- Elliott, S.D.; Dey, G.; Maimaiti, Y. Classification of processes for the atomic layer deposition of metals based on mechanistic information from density functional theory calculations. J. Chem. Phys. 2017, 146, 052822. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Stair, P.C. Low-Temperature ABC-Type Atomic Layer Deposition: Synthesis of Highly Uniform Ultrafine Supported Metal Nanoparticles. Angew. Chem. Int. Ed. 2010, 49, 2547–2551. [Google Scholar] [CrossRef]
- Stair, P.C. Synthesis of supported catalysts by atomic layer deposition. Top. Catal. 2012, 55, 93–98. [Google Scholar] [CrossRef]
- WANG, H.; LU, J. Atomic Layer Deposition: A Gas Phase Route to Bottom-up Precise Synthesis of Heterogeneous Catalyst. Acta Phys.-Chim. Sin. 2018, 34, 1334–1357. [Google Scholar] [CrossRef]
- Richey, N.E.; De Paula, C.; Bent, S.F. Understanding chemical and physical mechanisms in atomic layer deposition. J. Chem. Phys. 2020, 152, 040902. [Google Scholar] [CrossRef] [PubMed]
- Miikkulainen, V.; Leskelä, M.; Ritala, M.; Puurunen, R.L. Crystallinity of inorganic films grown by atomic layer deposition: Overview and general trends. J. Appl. Phys. 2013, 113, 2. [Google Scholar] [CrossRef]
- Cremers, V.; Puurunen, R.L.; Dendooven, J. Conformality in atomic layer deposition: Current status overview of analysis and modelling. Appl. Phys. Rev. 2019, 6, 021302. [Google Scholar] [CrossRef] [Green Version]
- Onn, T.M.; Küngas, R.; Fornasiero, P.; Huang, K.; Gorte, R.J. Atomic layer deposition on porous materials: Problems with conventional approaches to catalyst and fuel cell electrode preparation. Inorganics 2018, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Dendooven, J.; Devloo-Casier, K.; Levrau, E.; Van Hove, R.; Pulinthanathu Sree, S.; Baklanov, M.R.; Martens, J.A.; Detavernier, C. In Situ Monitoring of Atomic Layer Deposition in Nanoporous Thin Films Using Ellipsometric Porosimetry. Langmuir 2012, 28, 3852–3859. [Google Scholar] [CrossRef]
- Dendooven, J.; Devloo-Casier, K.; Ide, M.; Grandfield, K.; Kurttepeli, M.; Ludwig, K.F.; Bals, S.; Van Der Voort, P.; Detavernier, C. Atomic layer deposition-based tuning of the pore size in mesoporous thin films studied by in situ grazing incidence small angle X-ray scattering. Nanoscale 2014, 6, 14991–14998. [Google Scholar] [CrossRef] [Green Version]
- Longrie, D.; Deduytsche, D.; Detavernier, C. Reactor concepts for atomic layer deposition on agitated particles: A review. J. Vac. Sci. Technol. A 2013, 32, 010802. [Google Scholar] [CrossRef]
- Wank, J.R.; George, S.M.; Weimer, A.W. Nanocoating individual cohesive boron nitride particles in a fluidized bed by ALD. Powder Technol. 2004, 142, 59–69. [Google Scholar] [CrossRef]
- McCormick, J.; Cloutier, B.; Weimer, A.; George, S. Rotary reactor for atomic layer deposition on large quantities of nanoparticles. J. Vac. Sci. Technol. A 2007, 25, 67–74. [Google Scholar] [CrossRef]
- Longrie, D.; Deduytsche, D.; Haemers, J.; Driesen, K.; Detavernier, C. A rotary reactor for thermal and plasma-enhanced atomic layer deposition on powders and small objects. Surf. Coat. Technol. 2012, 213, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Filez, M.; Poelman, H.; Ramachandran, R.K.; Dendooven, J.; Devloo-Casier, K.; Fonda, E.; Detavernier, C.; Marin, G.B. In situ XAS and XRF study of nanoparticle nucleation during O3-based Pt deposition. Catal. Today 2014, 229, 2–13. [Google Scholar] [CrossRef]
- Masango, S.S.; Peng, L.; Marks, L.D.; Van Duyne, R.P.; Stair, P.C. Nucleation and Growth of Silver Nanoparticles by AB and ABC-Type Atomic Layer Deposition. J. Phys. Chem. C 2014, 118, 17655–17661. [Google Scholar] [CrossRef]
- Feng, H.; Elam, J.W.; Libera, J.A.; Setthapun, W.; Stair, P.C. Palladium Catalysts Synthesized by Atomic Layer Deposition for Methanol Decomposition. Chem. Mater. 2010, 22, 3133–3142. [Google Scholar] [CrossRef]
- Dendooven, J.; Ramachandran, R.K.; Solano, E.; Kurttepeli, M.; Geerts, L.; Heremans, G.; Rongé, J.; Minjauw, M.M.; Dobbelaere, T.; Devloo-Casier, K.; et al. Independent tuning of size and coverage of supported Pt nanoparticles using atomic layer deposition. Nat. Commun. 2017, 8, 1074. [Google Scholar] [CrossRef] [Green Version]
- Sree, S.P.; Dendooven, J.; Masschaele, K.; Hamed, H.M.; Deng, S.; Bals, S.; Detavernier, C.; Martens, J.A. Synthesis of uniformly dispersed anatase nanoparticles inside mesoporous silica thin films via controlled breakup and crystallization of amorphous TiO2 deposited using atomic layer deposition. Nanoscale 2013, 5, 5001–5008. [Google Scholar] [CrossRef]
- Weber, M.J.; Mackus, A.J.; Verheijen, M.A.; Van der Marel, C.; Kessels, W.M. Supported core/shell bimetallic nanoparticles synthesis by atomic layer deposition. Chem. Mater. 2012, 24, 2973–2977. [Google Scholar] [CrossRef]
- Lu, J.; Low, K.-B.; Lei, Y.; Libera, J.A.; Nicholls, A.; Stair, P.C.; Elam, J.W. Toward atomically-precise synthesis of supported bimetallic nanoparticles using atomic layer deposition. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- Singh, J.A.; Thissen, N.F.; Kim, W.-H.; Johnson, H.; Kessels, W.M.; Bol, A.A.; Bent, S.F.; Mackus, A.J. Area-selective atomic layer deposition of metal oxides on noble metals through catalytic oxygen activation. Chem. Mater. 2018, 30, 663–670. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Döhler, D.; Barr, M.; Oks, E.; Wolf, M.; Santinacci, L.; Bachmann, J. Atomic Layer Deposition from Dissolved Precursors. Nano Lett. 2015, 15, 6379–6385. [Google Scholar] [CrossRef] [Green Version]
- Zankowski, S.P.; Van Hoecke, L.; Mattelaer, F.; De Raedt, M.; Richard, O.; Detavernier, C.; Vereecken, P.M. Redox Layer Deposition of Thin Films of MnO2 on Nanostructured Substrates from Aqueous Solutions. Chem. Mater. 2019, 31, 4805–4816. [Google Scholar] [CrossRef]
- Le Monnier, B.P.; Wells, F.; Talebkeikhah, F.; Luterbacher, J.S. Atomic Layer Deposition on Dispersed Materials in Liquid Phase by Stoichiometrically Limited Injections. Adv. Mater. 2019, 31, 1904276. [Google Scholar] [CrossRef] [PubMed]
- Forman, A.J.; Park, J.N.; Tang, W.; Hu, Y.S.; Stucky, G.D.; McFarland, E.W. Silica-Encapsulated Pd Nanoparticles as a Regenerable and Sintering-Resistant Catalyst. Chemcatchem 2010, 2, 1318–1324. [Google Scholar] [CrossRef]
- Vrijburg, W.L.; Van Helden, J.W.A.; Van Hoof, A.J.F.; Friedrich, H.; Groeneveld, E.; Pidko, E.A.; Hensen, E.J.M. Tunable colloidal Ni nanoparticles confined and redistributed in mesoporous silica for CO2 methanation. Catal. Sci. Technol. 2019, 9, 2578–2591. [Google Scholar] [CrossRef] [Green Version]
- Seipenbusch, M.; Binder, A. Structural Stabilization of Metal Nanoparticles by Chemical Vapor Deposition-Applied Silica Coatings. J. Phys. Chem. C 2009, 113, 20606–20610. [Google Scholar] [CrossRef]
- Mao, X.; Foucher, A.; Stach, E.A.; Gorte, R.J. A Study of Support Effects for CH4 and CO Oxidation over Pd Catalysts on ALD-Modified Al2O3. Catal. Lett. 2019, 149, 905–915. [Google Scholar] [CrossRef]
- Shen, J.; Hayes, R.E.; Wu, X.; Semagina, N. 100° Temperature Reduction of Wet Methane Combustion: Highly Active Pd-Ni/Al2O3 Catalyst versus Pd/NiAl2O4. ACS Catal. 2015, 5, 2916–2920. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, S.; Ding, Y.; Zhang, L.; Lv, L.; Wang, M.; Wang, S. Pd catalysts supported on Co3O4 with the specified morphologies in CO and CH4 oxidation. Appl. Catal. A Gen. 2017, 532, 95–104. [Google Scholar] [CrossRef]
- Mahara, Y.; Ohyama, J.; Tojo, T.; Murata, K.; Ishikawa, H.; Satsuma, A. Enhanced activity for methane combustion over a Pd/Co/Al2O3 catalyst prepared by a galvanic deposition method. Catal. Sci. Technol. 2016, 6, 4773–4776. [Google Scholar] [CrossRef]
- Gould, T.D.; Lubers, A.M.; Corpuz, A.R.; Weimer, A.W.; Falconer, J.L.; Medlin, J.W. Controlling Nanoscale Properties of Supported Platinum Catalysts through Atomic Layer Deposition. ACS Catal. 2015, 5, 1344–1352. [Google Scholar] [CrossRef]
- Grillo, F.; Van Bui, H.; Moulijn, J.A.; Kreutzer, M.T.; Van Ommen, J.R. Understanding and Controlling the Aggregative Growth of Platinum Nanoparticles in Atomic Layer Deposition: An Avenue to Size Selection. J. Phys. Chem. Lett. 2017, 8, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Lubers, A.M.; Muhich, C.L.; Anderson, K.M.; Weimer, A.W. Mechanistic studies for depositing highly dispersed Pt nanoparticles on carbon by use of trimethyl(methylcyclopentadienyl)platinum(IV) reactions with O2 and H2. J. Nanopart. Res. 2015, 17, 179. [Google Scholar] [CrossRef]
- Gong, T.; Huang, Y.; Qin, L.; Zhang, W.; Li, J.; Hui, L.; Feng, H. Atomic layer deposited Palladium nanoparticle catalysts supported on Titanium dioxide modified MCM-41 for selective hydrogenation of acetylene. Appl. Surf. Sci. 2019, 495, 143495. [Google Scholar] [CrossRef]
- Kim, I.S.; Li, Z.; Zheng, J.; Platero-Prats, A.E.; Mavrandonakis, A.; Pellizzeri, S.; Ferrandon, M.; Vjunov, A.; Gallington, L.C.; Webber, T.E. Sinter-Resistant Platinum Catalyst Supported by Metal-Organic Framework. Angew. Chem. Int. Ed. 2018, 57, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.D.; Lubers, A.M.; Neltner, B.T.; Carrier, J.V.; Weimer, A.W.; Falconer, J.L.; Will Medlin, J. Synthesis of supported Ni catalysts by atomic layer deposition. J. Catal. 2013, 303, 9–15. [Google Scholar] [CrossRef]
- Vang, R.T.; Honkala, K.; Dahl, S.; Vestergaard, E.K.; Schnadt, J.; Lægsgaard, E.; Clausen, B.S.; Nørskov, J.K.; Besenbacher, F. Ethylene dissociation on flat and stepped Ni(111): A combined STM and DFT study. Surf. Sci. 2006, 600, 66–77. [Google Scholar] [CrossRef]
- Martin, G.A. The kinetics of the catalytic hydrogenolysis of ethane over Ni/SiO2. J. Catal. 1979, 60, 345–355. [Google Scholar] [CrossRef]
- Bengaard, H.S.; Nørskov, J.K.; Sehested, J.; Clausen, B.; Nielsen, L.; Molenbroek, A.; Rostrup-Nielsen, J. Steam reforming and graphite formation on Ni catalysts. J. Catal. 2002, 209, 365–384. [Google Scholar] [CrossRef]
- Kim, D.H.; Sim, J.K.; Lee, J.; Seo, H.O.; Jeong, M.-G.; Kim, Y.D.; Kim, S.H. Carbon dioxide reforming of methane over mesoporous Ni/SiO2. Fuel 2013, 112, 111–116. [Google Scholar] [CrossRef]
- Shang, Z.; Li, S.; Li, L.; Liu, G.; Liang, X. Highly active and stable alumina supported nickel nanoparticle catalysts for dry reforming of methane. Appl. Catal. B Environ. 2017, 201, 302–309. [Google Scholar] [CrossRef]
- Shang, Z.; Li, S.; Wang, Q.; Gu, X.; Liang, X. Nano-engineered nickel catalysts supported on 4-channel α-Al2O3 hollow fibers for dry reforming of methane. AIChE J. 2018, 64, 2625–2631. [Google Scholar] [CrossRef]
- Wang, G.; Luo, F.; Cao, K.; Zhang, Y.; Li, J.; Zhao, F.; Chen, R.; Hong, J. Effect of Ni Content of Ni/γ-Al2O3 Catalysts Prepared by the Atomic Layer Deposition Method on CO2 Reforming of Methane. Energy Technol. 2019, 7, 1800359. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, J.; Zhu, C.; Li, L.; Li, Q.; Ding, Y.; Yang, W. The Enhanced Catalytic Performance and Stability of Rh/γ-Al2O3 Catalyst Synthesized by Atomic Layer Deposition (ALD) for Methane Dry Reforming. Materials 2018, 11, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Shang, Z.; Xiao, Z.; Wang, L.; Liang, X.; Liu, G. Steam reforming of n-dodecane over mesoporous alumina supported nickel catalysts: Effects of metal-support interaction on nickel catalysts. Int. J. Hydrogen Energy 2019, 44, 6965–6977. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Liu, C.-J.; Zeng, Z.; Zhang, H.; Zhou, C.; Jia, X.; Yang, Y. Formation of monometallic Au and Pd and bimetallic Au-Pd nanoparticles confined in mesopores via Ar glow-discharge plasma reduction and their catalytic applications in aerobic oxidation of benzyl alcohol. J. Catal. 2012, 289, 105–117. [Google Scholar] [CrossRef]
- Christensen, S.T.; Feng, H.; Libera, J.L.; Guo, N.; Miller, J.T.; Stair, P.C.; Elam, J.W. Supported Ru-Pt bimetallic nanoparticle catalysts prepared by atomic layer deposition. Nano Lett. 2010, 10, 3047–3051. [Google Scholar] [CrossRef]
- Enache, D.I.; Edwards, J.K.; Landon, P.; Solsona-Espriu, B.; Carley, A.F.; Herzing, A.A.; Watanabe, M.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Solvent-free oxidation of primary alcohols to aldehydes using Au-Pd/TiO2 catalysts. Science 2006, 311, 362–365. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Yan, H.; Yi, H.; Lu, J. Precisely-controlled synthesis of Au@ Pd core-shell bimetallic catalyst via atomic layer deposition for selective oxidation of benzyl alcohol. J. Catal. 2015, 324, 59–68. [Google Scholar] [CrossRef]
- Ramachandran, R.K.; Dendooven, J.; Filez, M.; Galvita, V.V.; Poelman, H.; Solano, E.; Minjauw, M.M.; Devloo-Casier, K.; Fonda, E.; Hermida-Merino, D. Atomic layer deposition route to tailor nanoalloys of noble and non-noble metals. ACS Nano 2016, 10, 8770–8777. [Google Scholar] [CrossRef]
- Filez, M.; Redekop, E.A.; Poelman, H.; Galvita, V.V.; Meledina, M.; Turner, S.; Van Tendeloo, G.; Detavernier, C.; Marin, G.B. One-pot synthesis of Pt catalysts based on layered double hydroxides: An application in propane dehydrogenation. Catal. Sci. Technol. 2016, 6, 1863–1869. [Google Scholar] [CrossRef]
- Sun, P.; Siddiqi, G.; Vining, W.C.; Chi, M.; Bell, A.T. Novel Pt/Mg(In)(Al)O catalysts for ethane and propane dehydrogenation. J. Catal. 2011, 282, 165–174. [Google Scholar] [CrossRef]
- Camacho-Bunquin, J.; Ferrandon, M.S.; Sohn, H.; Kropf, A.J.; Yang, C.; Wen, J.; Hackler, R.A.; Liu, C.; Celik, G.; Marshall, C.L.; et al. Atomically Precise Strategy to a PtZn Alloy Nanocluster Catalyst for the Deep Dehydrogenation of n-Butane to 1,3-Butadiene. ACS Catal. 2018, 8, 10058–10063. [Google Scholar] [CrossRef]
- Wang, S.; Xu, D.; Zhu, D.; Zhao, B.; Guan, H.; Qin, Y.; Wu, B.; Yang, Y.; Li, Y. Elucidating the restructuring-induced highly active bimetallic Pt-Co/KL catalyst for the aromatization of n-heptane. Chem. Commun. 2020, 56, 892–895. [Google Scholar] [CrossRef] [PubMed]
- García-Diéguez, M.; Finocchio, E.; Larrubia, M.Á.; Alemany, L.J.; Busca, G. Characterization of alumina-supported Pt, Ni and PtNi alloy catalysts for the dry reforming of methane. J. Catal. 2010, 274, 11–20. [Google Scholar] [CrossRef]
- García-Diéguez, M.; Pieta, I.S.; Herrera, M.C.; Larrubia, M.A.; Alemany, L.J. Improved Pt-Ni nanocatalysts for dry reforming of methane. Appl. Catal. A Gen. 2010, 377, 191–199. [Google Scholar] [CrossRef]
- Pawelec, B.; Damyanova, S.; Arishtirova, K.; Fierro, J.L.G.; Petrov, L. Structural and surface features of PtNi catalysts for reforming of methane with CO2. Appl. Catal. A Gen. 2007, 323, 188–201. [Google Scholar] [CrossRef]
- De Miguel, S.R.; Vilella, I.M.J.; Maina, S.P.; San José-Alonso, D.; Román-Martínez, M.C.; Illán-Gómez, M.J. Influence of Pt addition to Ni catalysts on the catalytic performance for long term dry reforming of methane. Appl. Catal. A Gen. 2012, 435–436, 10–18. [Google Scholar] [CrossRef]
- Gould, T.D.; Montemore, M.M.; Lubers, A.M.; Ellis, L.D.; Weimer, A.W.; Falconer, J.L.; Medlin, J.W. Enhanced dry reforming of methane on Ni and Ni-Pt catalysts synthesized by atomic layer deposition. Appl. Catal. A Gen. 2015, 492, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.; Du, H.; Hu, Y.; Yan, H.; Jiang, H.-L.; Lu, J. Precisely Controlled Porous Alumina Overcoating on Pd Catalyst by Atomic Layer Deposition: Enhanced Selectivity and Durability in Hydrogenation of 1,3-Butadiene. ACS Catal. 2015, 5, 2735–2739. [Google Scholar] [CrossRef]
- O’Neill, B.J.; Sener, C.; Jackson, D.H.; Kuech, T.F.; Dumesic, J.A. Control of Thickness and Chemical Properties of Atomic Layer Deposition Overcoats for Stabilizing Cu/γ-Al2O3 Catalysts. ChemSusChem 2014, 7, 3247–3251. [Google Scholar] [CrossRef]
- Lu, J.; Liu, B.; Greeley, J.P.; Feng, Z.; Libera, J.A.; Lei, Y.; Bedzyk, M.J.; Stair, P.C.; Elam, J.W. Porous Alumina Protective Coatings on Palladium Nanoparticles by Self-Poisoned Atomic Layer Deposition. Chem. Mater. 2012, 24, 2047–2055. [Google Scholar] [CrossRef]
- Lu, J.; Fu, B.; Kung, M.C.; Xiao, G.; Elam, J.W.; Kung, H.H.; Stair, P.C. Coking-and sintering-resistant palladium catalysts achieved through atomic layer deposition. Science 2012, 335, 1205–1208. [Google Scholar] [PubMed]
- Feng, H.; Lu, J.; Stair, P.C.; Elam, J.W. Alumina Over-coating on Pd Nanoparticle Catalysts by Atomic Layer Deposition: Enhanced Stability and Reactivity. Catal. Lett. 2011, 141, 512–517. [Google Scholar] [CrossRef]
- Solano, E.; Dendooven, J.; Ramachandran, R.K.; Van de Kerckhove, K.; Dobbelaere, T.; Hermida-Merino, D.; Detavernier, C. Key role of surface oxidation and reduction processes in the coarsening of Pt nanoparticles. Nanoscale 2017, 9, 13159–13170. [Google Scholar] [CrossRef] [PubMed]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gu, X.-K.; Canlas, C.; Kropf, A.J.; Aich, P.; Greeley, J.P.; Elam, J.W.; Meyers, R.J.; Dumesic, J.A.; Stair, P.C.; et al. Atomic Layer Deposition Overcoating: Tuning Catalyst Selectivity for Biomass Conversion. Angew. Chem. Int. Ed. 2014, 53, 12132–12136. [Google Scholar] [CrossRef]
- Fu, B.; Lu, J.; Stair, P.C.; Xiao, G.; Kung, M.C.; Kung, H.H. Oxidative dehydrogenation of ethane over alumina-supported Pd catalysts. Effect of alumina overlayer. J. Catal. 2013, 297, 289–295. [Google Scholar] [CrossRef]
- Zaera, F. The New Materials Science of Catalysis: Toward Controlling Selectivity by Designing the Structure of the Active Site. J. Phys. Chem. Lett. 2010, 1, 621–627. [Google Scholar] [CrossRef]
- Nilsen, O.; Klepper, K.; Nielsen, H.; Fjellvaag, H. Deposition of Organic- Inorganic Hybrid Materials by Atomic Layer Deposition. ECS Trans. 2019, 16, 3–14. [Google Scholar] [CrossRef]
- Sundberg, P.; Karppinen, M. Organic and inorganic-organic thin film structures by molecular layer deposition: A. review. Beilstein J. Nanotechnol. 2014, 5, 1104–1136. [Google Scholar] [CrossRef]
- George, S.M.; Lee, B.H.; Yoon, B.; Abdulagatov, A.I.; Hall, R.A. Metalcones: Hybrid organic-inorganic films fabricated using atomic and molecular layer deposition techniques. J. Nanosci. Nanotechnol. 2011, 11, 7948–7955. [Google Scholar] [CrossRef]
- Gould, T.D.; Izar, A.; Weimer, A.W.; Falconer, J.L.; Medlin, J.W. Stabilizing Ni Catalysts by Molecular Layer Deposition for Harsh, Dry Reforming Conditions. ACS Catal. 2014, 4, 2714–2717. [Google Scholar] [CrossRef]
- Kint, J.; Mattelaer, F.; Vandenbroucke, S.S.T.; Muriqi, A.; Minjauw, M.M.; Nisula, M.; Vereecken, P.M.; Nolan, M.; Dendooven, J.; Detavernier, C. Molecular Layer Deposition of “Magnesicone”, a Magnesium-based Hybrid Material. Chem. Mater. 2020, 32, 4451–4466. [Google Scholar] [CrossRef]
- Van de Kerckhove, K.; Dendooven, J.; Detavernier, C. Annealing of thin “Tincone” films, a tin-based hybrid material deposited by molecular layer deposition, in reducing, inert, and oxidizing atmospheres. J. Vac. Sci. Technol. A 2018, 36, 051506. [Google Scholar] [CrossRef]
- Van de Kerckhove, K.; Mattelaer, F.; Dendooven, J.; Detavernier, C. Molecular layer deposition of “vanadicone”, a vanadium-based hybrid material, as an electrode for lithium-ion batteries. Dalton Trans. 2017, 46, 4542–4553. [Google Scholar] [CrossRef]
- Bergsman, D.S.; Baker, J.G.; Closser, R.G.; MacIsaac, C.; Lillethorup, M.; Strickler, A.L.; Azarnouche, L.; Godet, L.; Bent, S.F. Structurally Stable Manganese Alkoxide Films Grown by Hybrid Molecular Layer Deposition for Electrochemical Applications. Adv. Funct. Mater. 2019, 29, 1904129. [Google Scholar] [CrossRef]
- Ahvenniemi, E.; Karppinen, M. ALD/MLD processes for Mn and Co based hybrid thin films. Dalton Trans. 2016, 45, 10730–10735. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Yu, M.; Li, J.; Jiang, Y.-B.; Weimer, A.W. Ultra-thin microporous-mesoporous metal oxide films prepared by molecular layer deposition (MLD). Chem. Commun. 2009, 45, 7140–7142. [Google Scholar] [CrossRef]
- Liang, X.; Evanko, B.W.; Izar, A.; King, D.M.; Jiang, Y.-B.; Weimer, A.W. Ultrathin highly porous alumina films prepared by alucone ABC molecular layer deposition (MLD). Microporous Mesoporous Mater. 2013, 168, 178–182. [Google Scholar] [CrossRef]
- Van de Kerckhove, K.; Barr, M.K.S.; Santinacci, L.; Vereecken, P.M.; Dendooven, J.; Detavernier, C. The transformation behaviour of “alucones”, deposited by molecular layer deposition, in nanoporous Al2O3 layers. Dalton Trans. 2018, 47, 5860–5870. [Google Scholar] [CrossRef]
- Cui, W.; Li, S.; Wang, D.; Deng, Y.; Chen, Y. High reactivity and sintering resistance of CH4 oxidation over modified Pd/Al2O3. Catal. Commun. 2019, 119, 86–90. [Google Scholar] [CrossRef]
- Onn, T.M.; Arroyo-Ramirez, L.; Monai, M.; Oh, T.-S.; Talati, M.; Fornasiero, P.; Gorte, R.J.; Khader, M.M. Modification of Pd/CeO2 catalyst by Atomic Layer Deposition of ZrO2. Appl. Catal. B Environ. 2016, 197, 280–285. [Google Scholar] [CrossRef]
- Onn, T.M.; Zhang, S.; Arroyo-Ramirez, L.; Chung, Y.-C.; Graham, G.W.; Pan, X.; Gorte, R.J. Improved Thermal Stability and Methane-Oxidation Activity of Pd/Al2O3 Catalysts by Atomic Layer Deposition of ZrO2. ACS Catal. 2015, 5, 5696–5701. [Google Scholar] [CrossRef]
- Chen, C.; Yeh, Y.-H.; Cargnello, M.; Murray, C.B.; Fornasiero, P.; Gorte, R.J. Methane Oxidation on Pd@ZrO2/Si-Al2O3 Is Enhanced by Surface Reduction of ZrO2. ACS Catal. 2014, 4, 3902–3909. [Google Scholar] [CrossRef]
- Fornasiero, P.; Balducci, G.; Di Monte, R.; Kašpar, J.; Sergo, V.; Gubitosa, G.; Ferrero, A.; Graziani, M. Modification of the Redox Behaviour of CeO2 Induced by Structural Doping with ZrO2. J. Catal. 1996, 164, 173–183. [Google Scholar] [CrossRef]
- Gong, Y.; Palacio, D.; Song, X.; Patel, R.L.; Liang, X.; Zhao, X.; Goodenough, J.B.; Huang, K. Stabilizing Nanostructured Solid Oxide Fuel Cell Cathode with Atomic Layer Deposition. Nano Lett. 2013, 13, 4340–4345. [Google Scholar] [CrossRef]
- Liu, K.; Wang, A.; Zhang, T. Recent advances in preferential oxidation of CO reaction over platinum group metal catalysts. ACS Catal. 2012, 2, 1165–1178. [Google Scholar] [CrossRef]
- Korotkikh, O.; Farrauto, R. Selective catalytic oxidation of CO in H2: Fuel cell applications. Catal. Today 2000, 62, 249–254. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Y.; Wang, J.; Liu, K.; Wang, X.; Wang, A.; Zhang, T. IrFeOx/SiO2—A highly active catalyst for preferential CO oxidation in H2. Int. J. Hydrogen Energy 2010, 35, 3065–3071. [Google Scholar] [CrossRef]
- Lin, J.; Qiao, B.; Liu, J.; Huang, Y.; Wang, A.; Li, L.; Zhang, W.; Allard, L.F.; Wang, X.; Zhang, T. Design of a highly active Ir/Fe (OH)x catalyst: Versatile application of Pt-group metals for the preferential oxidation of carbon monoxide. Angew. Chem. Int. Ed. 2012, 51, 2920–2924. [Google Scholar] [CrossRef]
- Wang, C.; Yao, Q.; Cao, L.; Li, J.; Chen, S.; Lu, J. Precise Tailoring of Ir-FeOx Interfaces for Improved Catalytic Performance in Preferential Oxidation of Carbon Monoxide in Hydrogen. J. Phys. Chem. C 2019, 123, 29262–29270. [Google Scholar] [CrossRef]
- Asundi, A.S.; Hoffman, A.S.; Bothra, P.; Boubnov, A.; Vila, F.D.; Yang, N.; Singh, J.A.; Zeng, L.; Raiford, J.A.; Abild-Pedersen, F.; et al. Understanding Structure-Property Relationships of MoO3-Promoted Rh Catalysts for Syngas Conversion to Alcohols. J. Am. Chem. Soc. 2019, 141, 19655–19668. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Boahene, P.E.; Hu, Y.; Dalai, A.; Wang, H. Atomic Layer Deposition ZnO Over-Coated Cu/SiO2 Catalysts for Methanol Synthesis from CO2 Hydrogenation. Catalysts 2019, 9, 922. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Kang, Y.; Li, H.; Li, H. CO2 conversion to synthesis gas via DRM on the durable Al2O3/Ni/Al2O3 sandwich catalyst with high activity and stability. Green Chem. 2018, 20, 2781–2787. [Google Scholar] [CrossRef]
- Cao, K.; Gong, M.; Yang, J.; Cai, J.; Chu, S.; Chen, Z.; Shan, B.; Chen, R. Nickel catalyst with atomically-thin meshed cobalt coating for improved durability in dry reforming of methane. J. Catal. 2019, 373, 351–360. [Google Scholar] [CrossRef]
- Xu, L.; Wang, F.; Chen, M.; Fan, X.; Yang, H.; Nie, D.; Qi, L. Alkaline-promoted Co-Ni bimetal ordered mesoporous catalysts with enhanced coke-resistant performance toward CO2 reforming of CH4. J. CO2 Util. 2017, 18, 1–14. [Google Scholar] [CrossRef]
- AlSabban, B.; Falivene, L.; Kozlov, S.M.; Aguilar-Tapia, A.; Ould-Chikh, S.; Hazemann, J.-L.; Cavallo, L.; Basset, J.-M.; Takanabe, K. In-operando elucidation of bimetallic CoNi nanoparticles during high-temperature CH4/CO2 reaction. Appl. Catal. B Environ. 2017, 213, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Kawi, S. Highly carbon-resistant Ni-Co/SiO2 catalysts derived from phyllosilicates for dry reforming of methane. J. CO2 Util. 2017, 18, 345–352. [Google Scholar] [CrossRef]
- Al-Fatesh, A.S.; Arafat, Y.; Atia, H.; Ibrahim, A.A.; Ha, Q.L.M.; Schneider, M.; M-Pohl, M.; Fakeeha, A.H. CO2-reforming of methane to produce syngas over Co-Ni/SBA-15 catalyst: Effect of support modifiers (Mg, La and Sc) on catalytic stability. J. CO2 Util. 2017, 21, 395–404. [Google Scholar] [CrossRef]
- Yang, N.; Medford, A.J.; Liu, X.; Studt, F.; Bligaard, T.; Bent, S.F.; Nørskov, J.K. Intrinsic Selectivity and Structure Sensitivity of Rhodium Catalysts for C2+ Oxygenate Production. J. Am. Chem. Soc. 2016, 138, 3705–3714. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.; Nørskov, J.K. Step sites in syngas catalysis. Top. Catal. 2006, 40, 45–48. [Google Scholar] [CrossRef]
- Wang, B.; Liang, D.; Zhang, R.; Ling, L. Crystal Facet Dependence for the Selectivity of C2 Species over Co2C Catalysts in the Fischer-Tropsch Synthesis. J. Phys. Chem. C 2018, 122, 29249–29258. [Google Scholar] [CrossRef]
- Cao, K.; Cai, J.; Chen, R. Inherently Selective Atomic Layer Deposition and Applications. Chem. Mater. 2020, 32, 2195–2207. [Google Scholar] [CrossRef]
- Cao, K.; Zhu, Q.; Shan, B.; Chen, R. Controlled Synthesis of Pd/Pt Core Shell Nanoparticles Using Area-selective Atomic Layer Deposition. Sci. Rep. 2015, 5, 8470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zyulkov, I.; Krishtab, M.; De Gendt, S.; Armini, S. Selective Ru ALD as a Catalyst for Sub-Seven-Nanometer Bottom-Up Metal Interconnects. ACS Appl. Mater. Interfaces 2017, 9, 31031–31041. [Google Scholar] [CrossRef]
- Hashemi, F.S.M.; Bent, S.F. Sequential Regeneration of Self-Assembled Monolayers for Highly Selective Atomic Layer Deposition. Adv. Mater. Interfaces 2016, 3, 1600464. [Google Scholar] [CrossRef]
- Mameli, A.; Merkx, M.J.M.; Karasulu, B.; Roozeboom, F.; Kessels, W.M.M.; Mackus, A.J.M. Area-Selective Atomic Layer Deposition of SiO2 Using Acetylacetone as a Chemoselective Inhibitor in an ABC-Type Cycle. ACS Nano 2017, 11, 9303–9311. [Google Scholar] [CrossRef] [PubMed]
- Minaye Hashemi, F.S.; Prasittichai, C.; Bent, S.F. Self-Correcting Process for High Quality Patterning by Atomic Layer Deposition. ACS Nano 2015, 9, 8710–8717. [Google Scholar] [CrossRef]
- Vos, M.F.J.; Chopra, S.N.; Verheijen, M.A.; Ekerdt, J.G.; Agarwal, S.; Kessels, W.M.M.; Mackus, A.J.M. Area-Selective Deposition of Ruthenium by Combining Atomic Layer Deposition and Selective Etching. Chem. Mater. 2019, 31, 3878–3882. [Google Scholar] [CrossRef] [Green Version]
- Song, S.K.; Saare, H.; Parsons, G.N. Integrated Isothermal Atomic Layer Deposition/Atomic Layer Etching Supercycles for Area-Selective Deposition of TiO2. Chem. Mater. 2019, 31, 4793–4804. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, Q.; Lang, Y.; Cao, K.; Chu, S.; Shan, B.; Chen, R. Oxide-nanotrap-anchored platinum nanoparticles with high activity and sintering resistance by area-selective atomic layer deposition. Angew. Chem. 2017, 129, 1670–1674. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, S.; Gao, Z.; Li, Y.; Zhang, Q.; Xiang, Q.; Qin, Y. The precise decoration of Pt nanoparticles with Fe oxide by atomic layer deposition for the selective hydrogenation of cinnamaldehyde. Appl. Catal. B Environ. 2017, 218, 591–599. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.; Yao, Q.; Yan, H.; Li, J.; Lu, J. Precisely Applying TiO2 Overcoat on Supported Au Catalysts Using Atomic Layer Deposition for Understanding the Reaction Mechanism and Improved Activity in CO Oxidation. J. Phys. Chem. C 2016, 120, 478–486. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, J.; Cao, K.; Gong, M.; Lang, Y.; Liu, X.; Chu, S.; Shan, B.; Chen, R. Selective Passivation of Pt Nanoparticles with Enhanced Sintering Resistance and Activity toward CO Oxidation via Atomic Layer Deposition. ACS Appl. Nano Mater. 2018, 1, 522–530. [Google Scholar] [CrossRef]
- Ding, L.; Yi, H.; Zhang, W.; You, R.; Cao, T.; Yang, J.; Lu, J.; Huang, W. Activating Edge Sites on Pd Catalysts for Selective Hydrogenation of Acetylene via Selective Ga2O3 Decoration. ACS Catal. 2016, 6, 3700–3707. [Google Scholar] [CrossRef]
- Cao, K.; Shi, L.; Gong, M.; Cai, J.; Liu, X.; Chu, S.; Lang, Y.; Shan, B.; Chen, R. Nanofence Stabilized Platinum Nanoparticles Catalyst via Facet-Selective Atomic Layer Deposition. Small 2017, 13, 1700648. [Google Scholar] [CrossRef]
- Wen, Y.; Cai, J.; Zhang, J.; Yang, J.; Shi, L.; Cao, K.; Chen, R.; Shan, B. Edge-Selective Growth of MCp2 (M = Fe, Co, and Ni) Precursors on Pt Nanoparticles in Atomic Layer Deposition: A Combined Theoretical and Experimental Study. Chem. Mater. 2019, 31, 101–111. [Google Scholar] [CrossRef]
- Wang, H.; Gu, X.-K.; Zheng, X.; Pan, H.; Zhu, J.; Chen, S.; Cao, L.; Li, W.-X.; Lu, J. Disentangling the size-dependent geometric and electronic effects of palladium nanocatalysts beyond selectivity. Sci. Adv. 2019, 5, eaat6413. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Cao, K.; Gong, M.; Shan, B.; Chen, R. Atomically decorating of MnOx on palladium nanoparticles towards selective oxidation of benzyl alcohol with high yield. J. Catal. 2020, 386, 60–69. [Google Scholar] [CrossRef]
- Mackus, A.J.M.; Schneider, J.R.; MacIsaac, C.; Baker, J.G.; Bent, S.F. Synthesis of Doped, Ternary, and Quaternary Materials by Atomic Layer Deposition: A Review. Chem. Mater. 2019, 31, 1142–1183. [Google Scholar] [CrossRef]
- Fu, L.; Cheng, G.; Luo, W. Colloidal synthesis of monodisperse trimetallic IrNiFe nanoparticles as highly active bifunctional electrocatalysts for acidic overall water splitting. J. Mater. Chem. A 2017, 5, 24836–24841. [Google Scholar] [CrossRef]
- Li, J.; Luo, Z.; He, F.; Zuo, Y.; Zhang, C.; Liu, J.; Yu, X.; Du, R.; Zhang, T.; Infante-Carrió, M.F.; et al. Colloidal Ni-Co-Sn nanoparticles as efficient electrocatalysts for the methanol oxidation reaction. J. Mater. Chem. A 2018, 6, 22915–22924. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Zheng, P.; Zheng, A.-G.; Li, H.-F.; Xia, G.-F.; Li, M.-F.; Zheng, R.-Y.; Xu, B.-Q. Noble-metal efficient Pt-Ir-Co/SiO2 catalyst for selective hydrogenolytic ring opening of methylcyclopentane. Catal. Today 2018, 316, 162–170. [Google Scholar] [CrossRef]
- Müller, P.; Hermans, I. Applications of Modulation Excitation Spectroscopy in Heterogeneous Catalysis. Ind. Eng. Chem. Res. 2017, 56, 1123–1136. [Google Scholar] [CrossRef]
- Ferri, D.; Newton, M.A.; Nachtegaal, M. Modulation Excitation X-Ray Absorption Spectroscopy to Probe Surface Species on Heterogeneous Catalysts. Top. Catal. 2011, 54, 1070. [Google Scholar] [CrossRef]
- Gaur, A.; Hartmann Dabros, T.M.; Høj, M.; Boubnov, A.; Prüssmann, T.; Jelic, J.; Studt, F.; Jensen, A.D.; Grunwaldt, J.-D. Probing the Active Sites of MoS2 Based Hydrotreating Catalysts Using Modulation Excitation Spectroscopy. ACS Catal. 2019, 9, 2568–2579. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.; Collins, S.E. Selective detection of reaction intermediates using concentration-modulation excitation DRIFT spectroscopy. Catal. Today 2013, 205, 34–40. [Google Scholar] [CrossRef]
- Nuguid, R.J.G.; Ferri, D.; Marberger, A.; Nachtegaal, M.; Kröcher, O. Modulated Excitation Raman Spectroscopy of V2O5/TiO2: Mechanistic Insights into the Selective Catalytic Reduction of NO with NH3. ACS Catal. 2019, 9, 6814–6820. [Google Scholar] [CrossRef]
- Ferri, D.; Newton, M.A.; Di Michiel, M.; Chiarello, G.L.; Yoon, S.; Lu, Y.; Andrieux, J. Revealing the Dynamic Structure of Complex Solid Catalysts Using Modulated Excitation X-ray Diffraction. Angew. Chem. Int. Ed. 2014, 53, 8890–8894. [Google Scholar] [CrossRef]
- Morgan, K.; Maguire, N.; Fushimi, R.; Gleaves, J.T.; Goguet, A.; Harold, M.P.; Kondratenko, E.V.; Menon, U.; Schuurman, Y.; Yablonsky, G.S. Forty years of temporal analysis of products. Catal. Sci. Technol. 2017, 7, 2416–2439. [Google Scholar] [CrossRef] [Green Version]
- Ledesma, C.; Yang, J.; Chen, D.; Holmen, A. Recent Approaches in Mechanistic and Kinetic Studies of Catalytic Reactions Using SSITKA Technique. ACS Catal. 2014, 4, 4527–4547. [Google Scholar] [CrossRef]
- Zugic, B.; Wang, L.; Heine, C.; Zakharov, D.N.; Lechner, B.A.J.; Stach, E.A.; Biener, J.; Salmeron, M.; Madix, R.J.; Friend, C.M. Dynamic restructuring drives catalytic activity on nanoporous gold-silver alloy catalysts. Nat. Mater. 2017, 16, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Van Ommen, J.R.; Kooijman, D.; Niet, M.d.; Talebi, M.; Goulas, A. Continuous production of nanostructured particles using spatial atomic layer deposition. J. Vac. Sci. Technol. A 2015, 33, 021513. [Google Scholar] [CrossRef] [Green Version]
- Poodt, P.; Cameron, D.C.; Dickey, E.; George, S.M.; Kuznetsov, V.; Parsons, G.N.; Roozeboom, F.; Sundaram, G.; Vermeer, A. Spatial atomic layer deposition: A route towards further industrialization of atomic layer deposition. J. Vac. Sci. Technol. A 2012, 30, 010802. [Google Scholar] [CrossRef] [Green Version]






















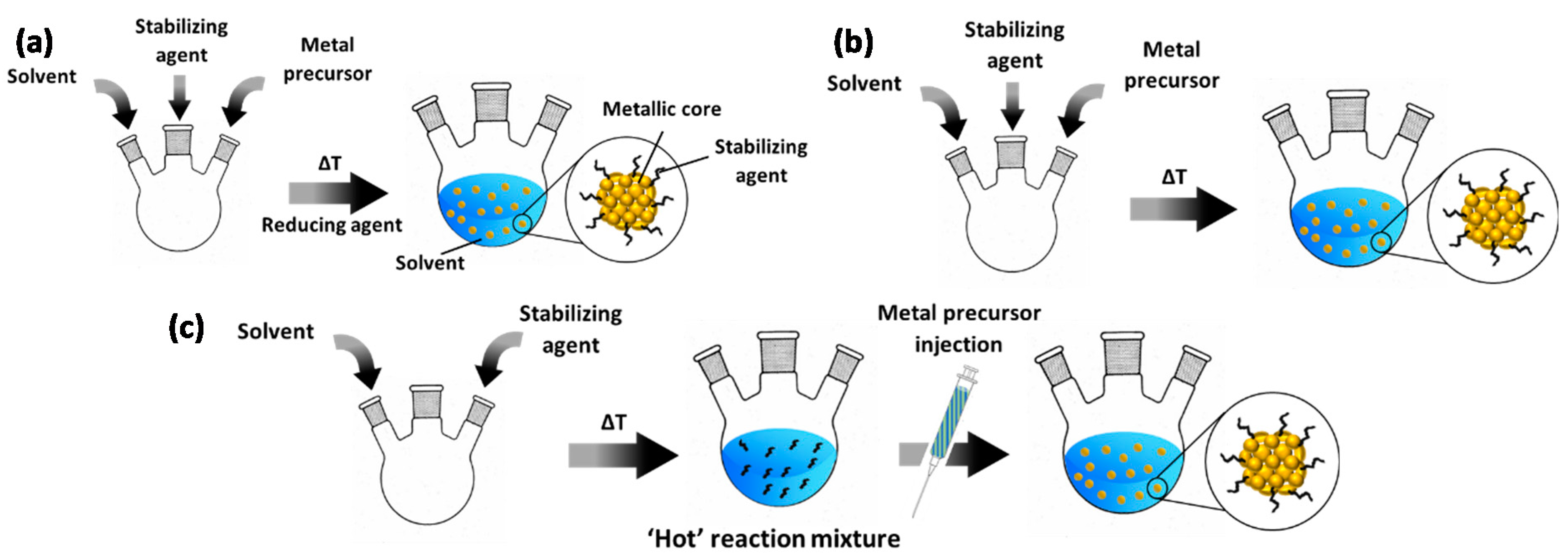{kind=link}
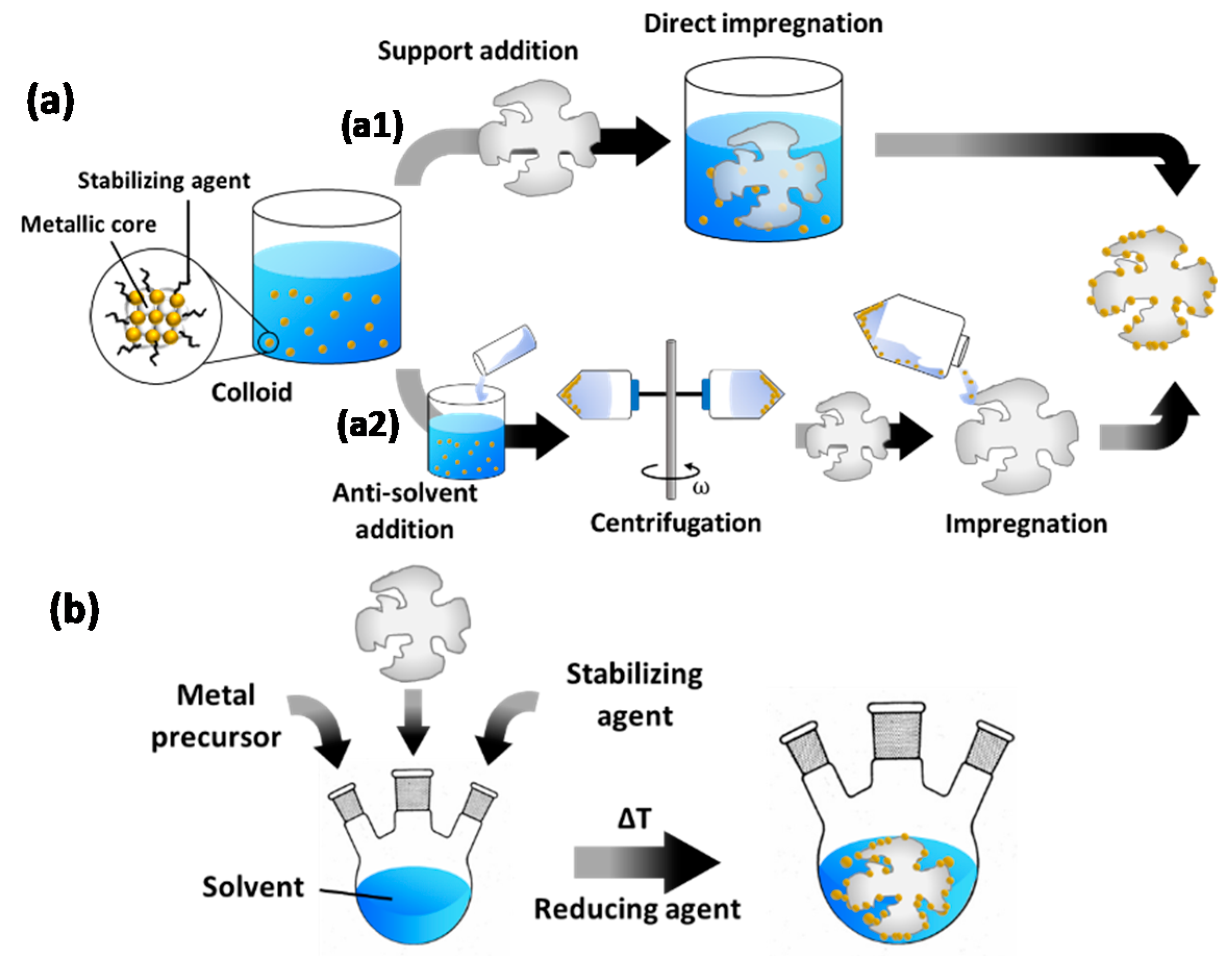{kind=link}
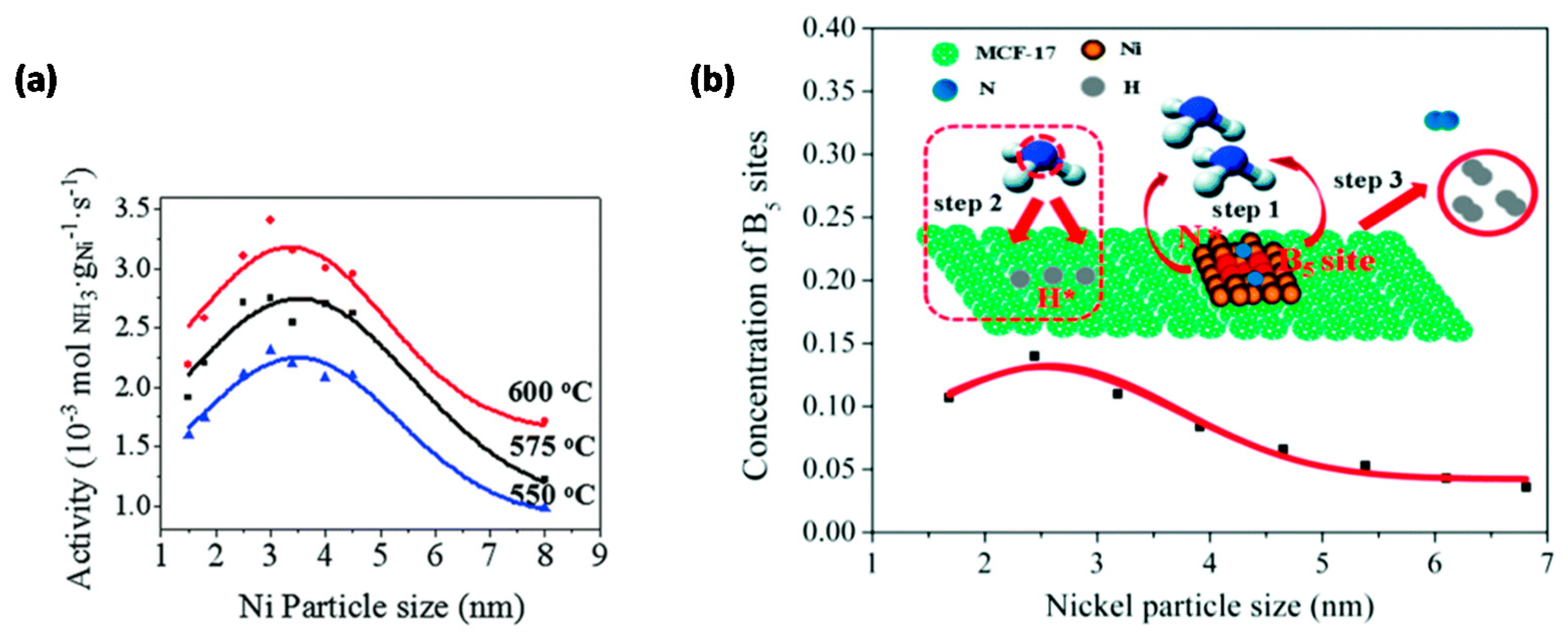{kind=link}
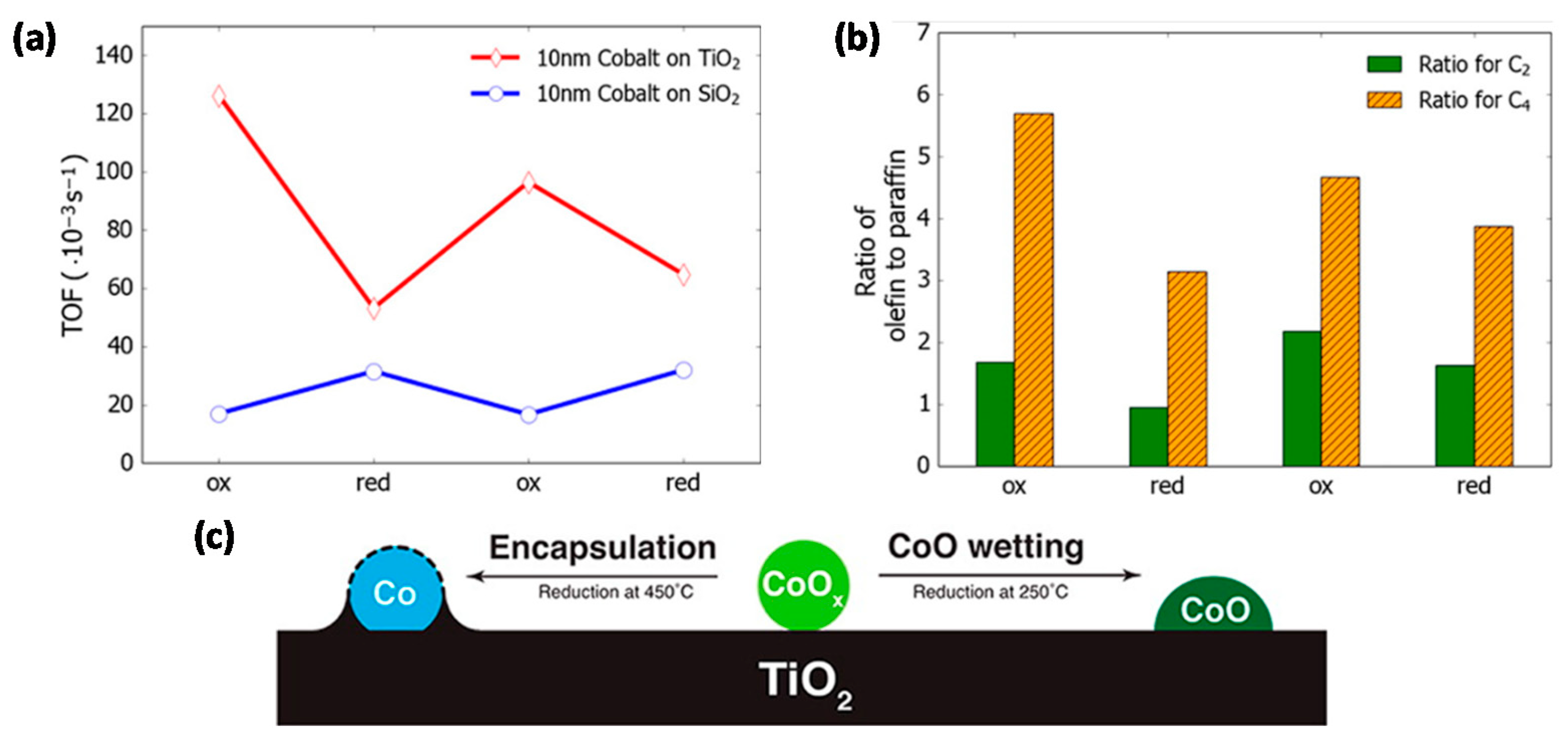{kind=link}
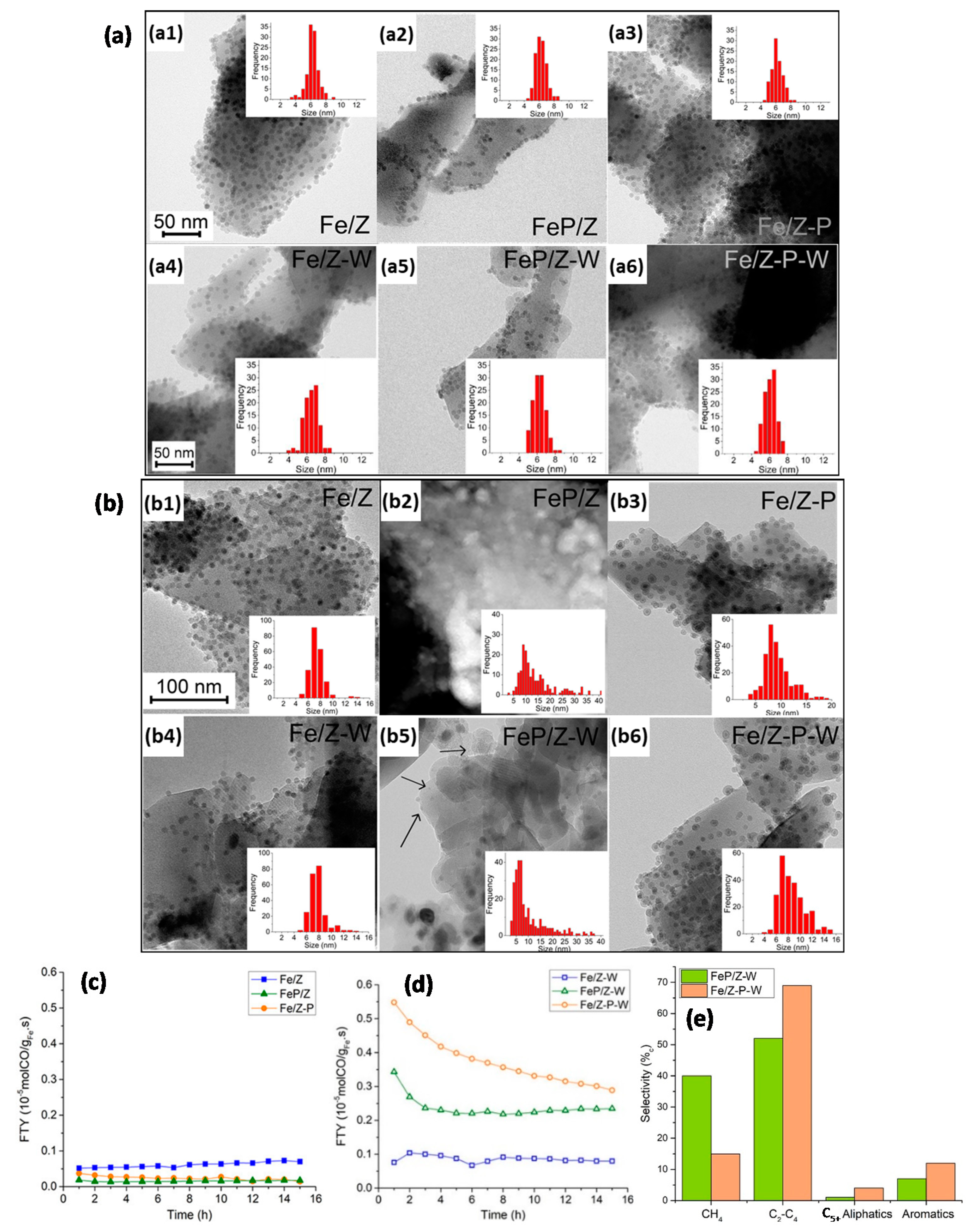{kind=link}
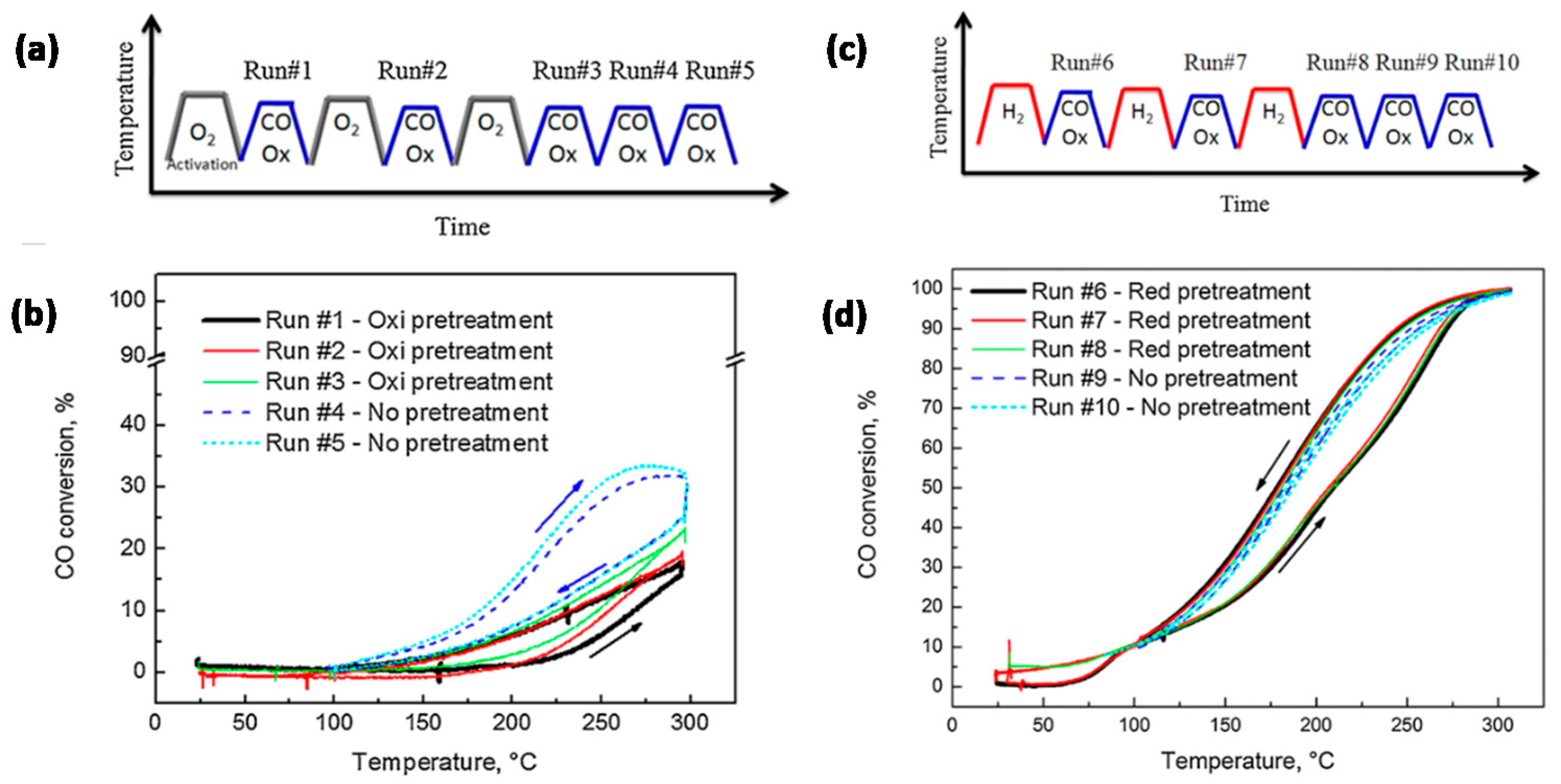{kind=link}
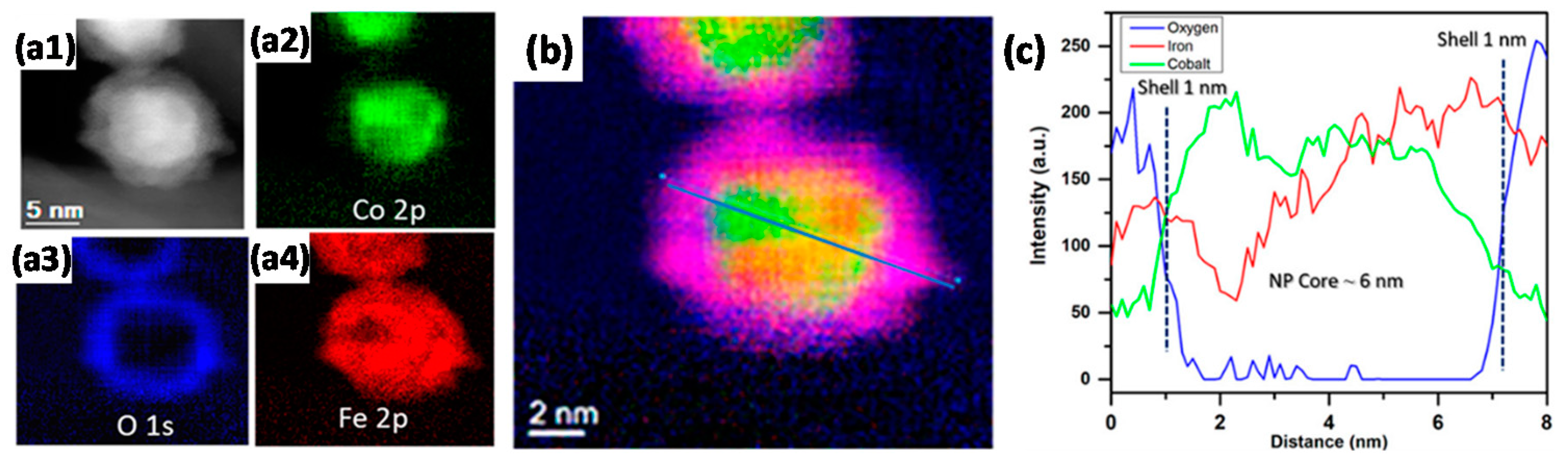{kind=link}
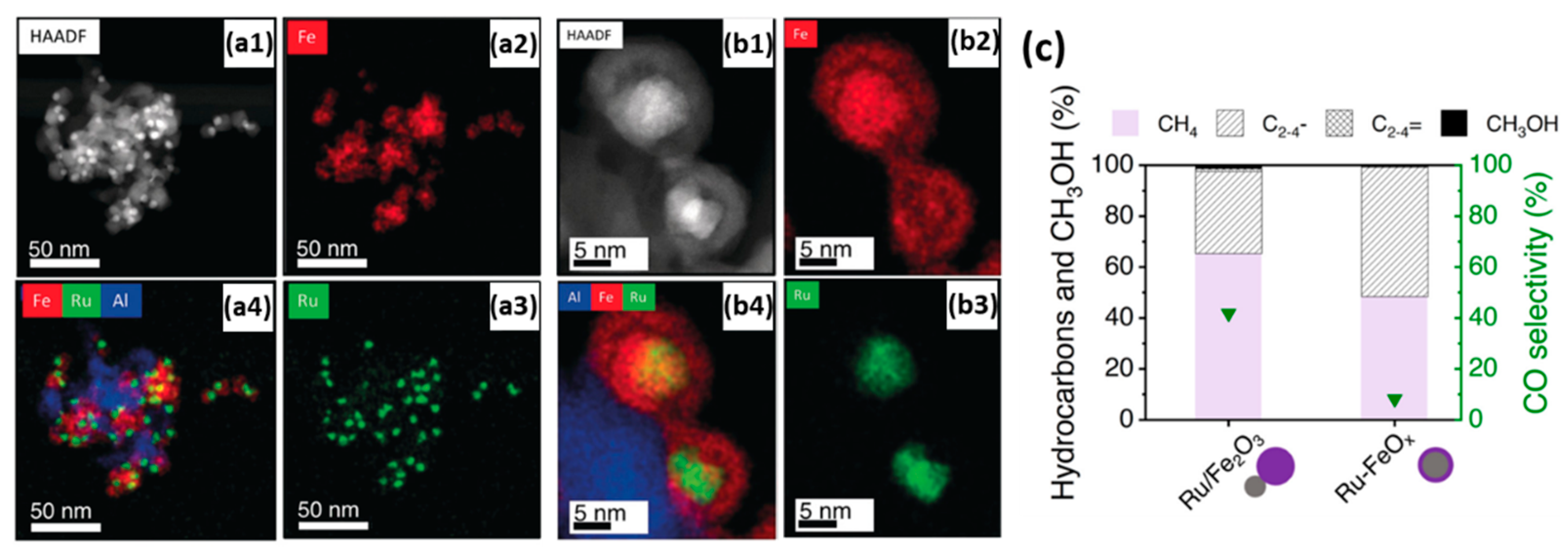{kind=link}
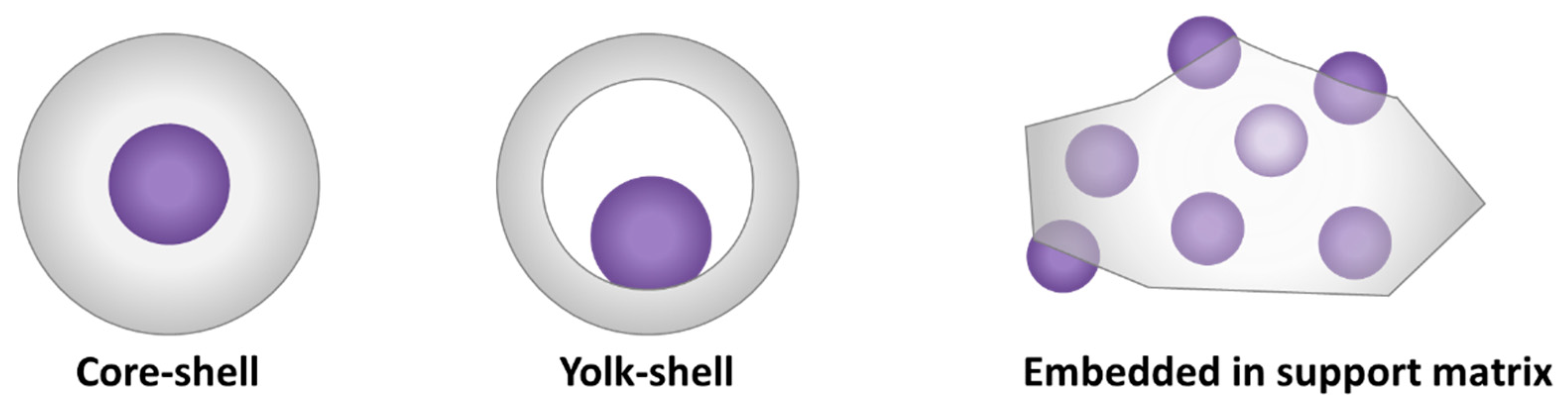{kind=link}
{kind=link}
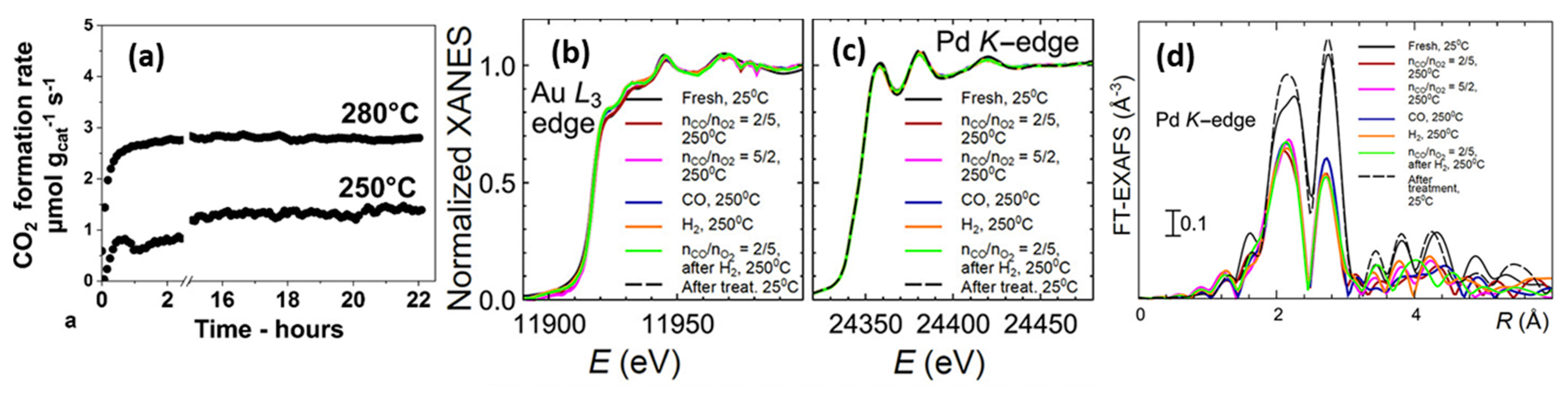{kind=link}
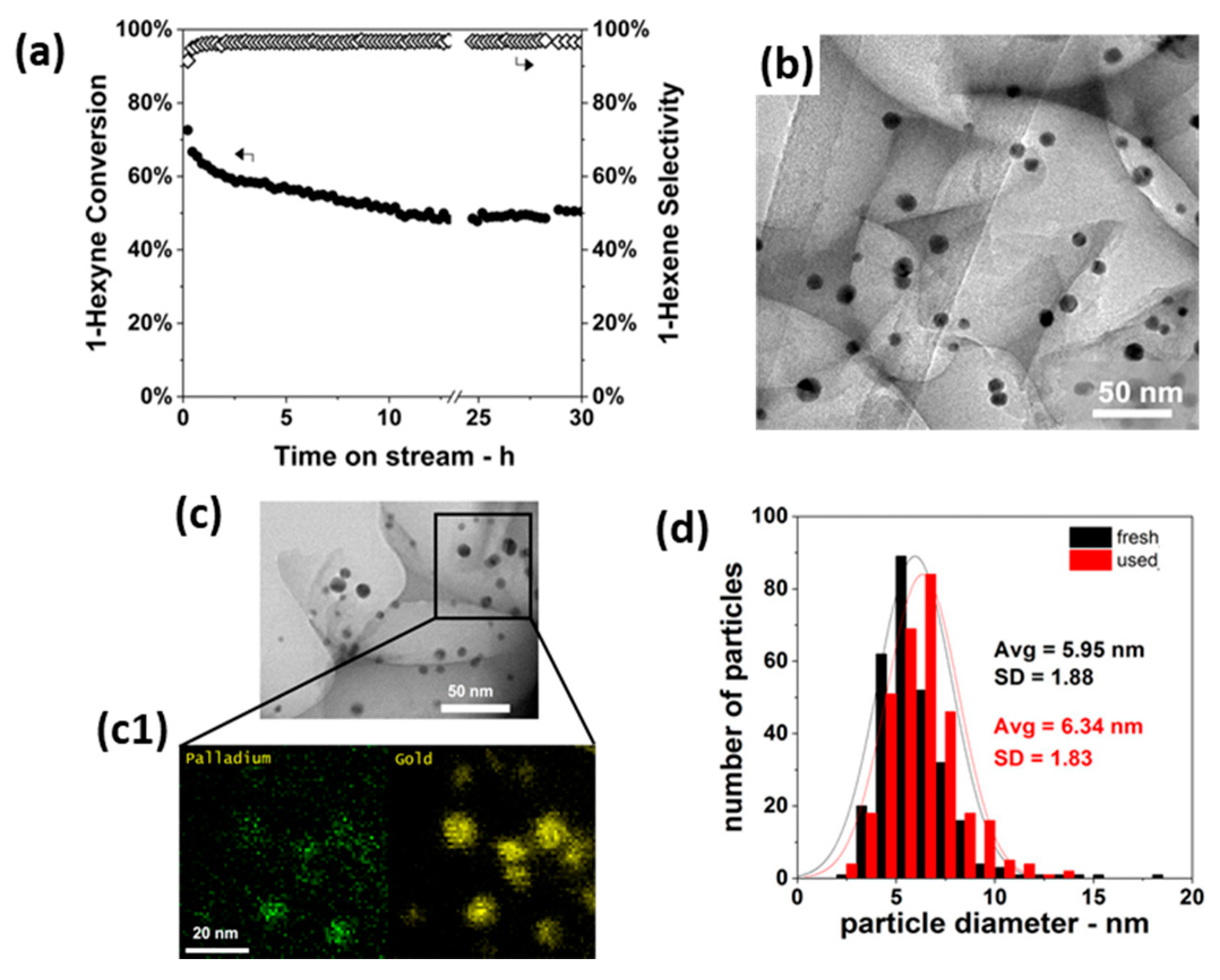{kind=link}
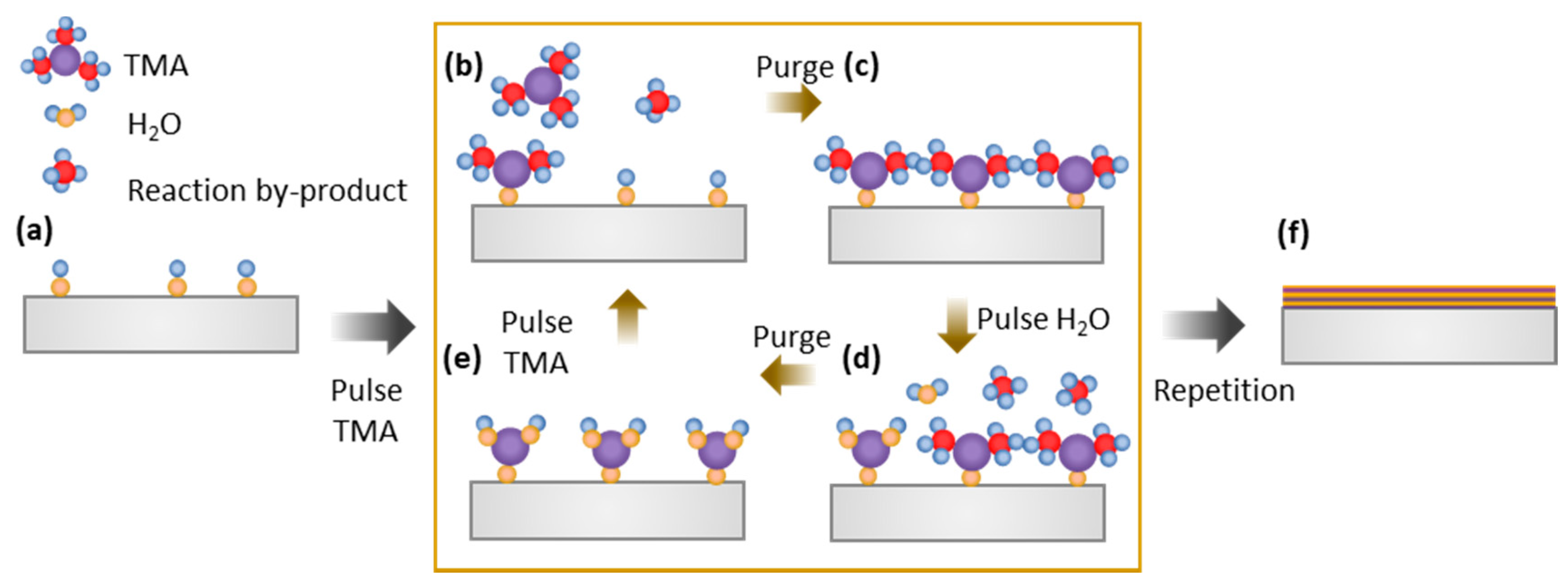{kind=link}
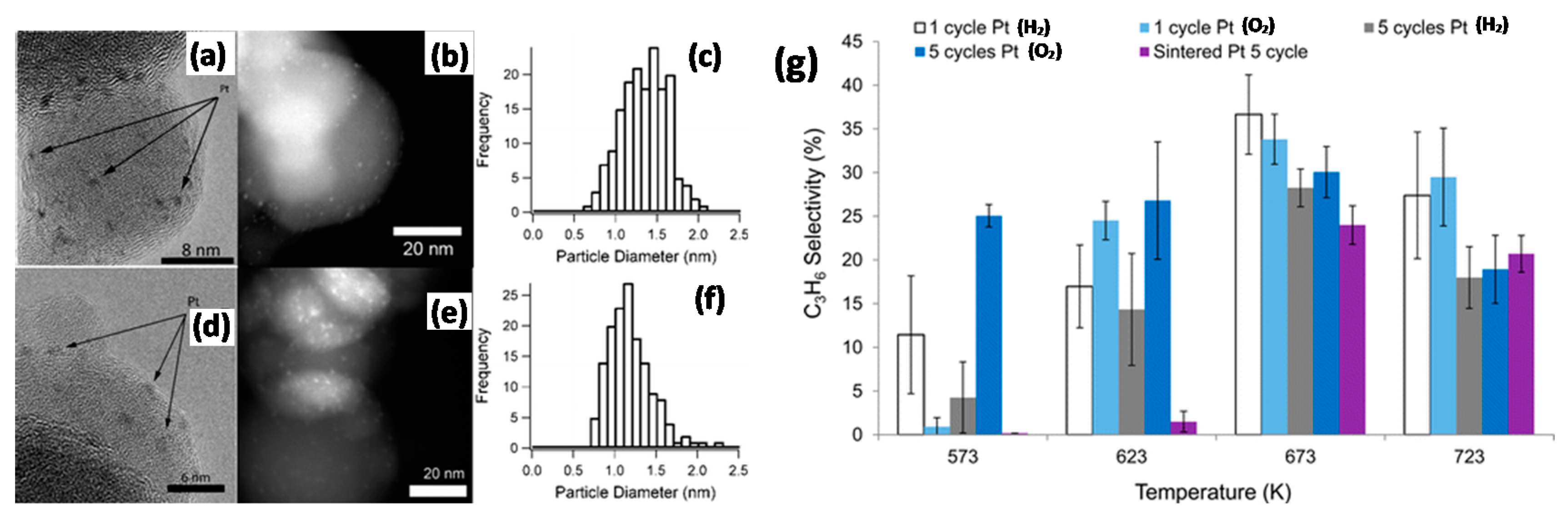{kind=link}
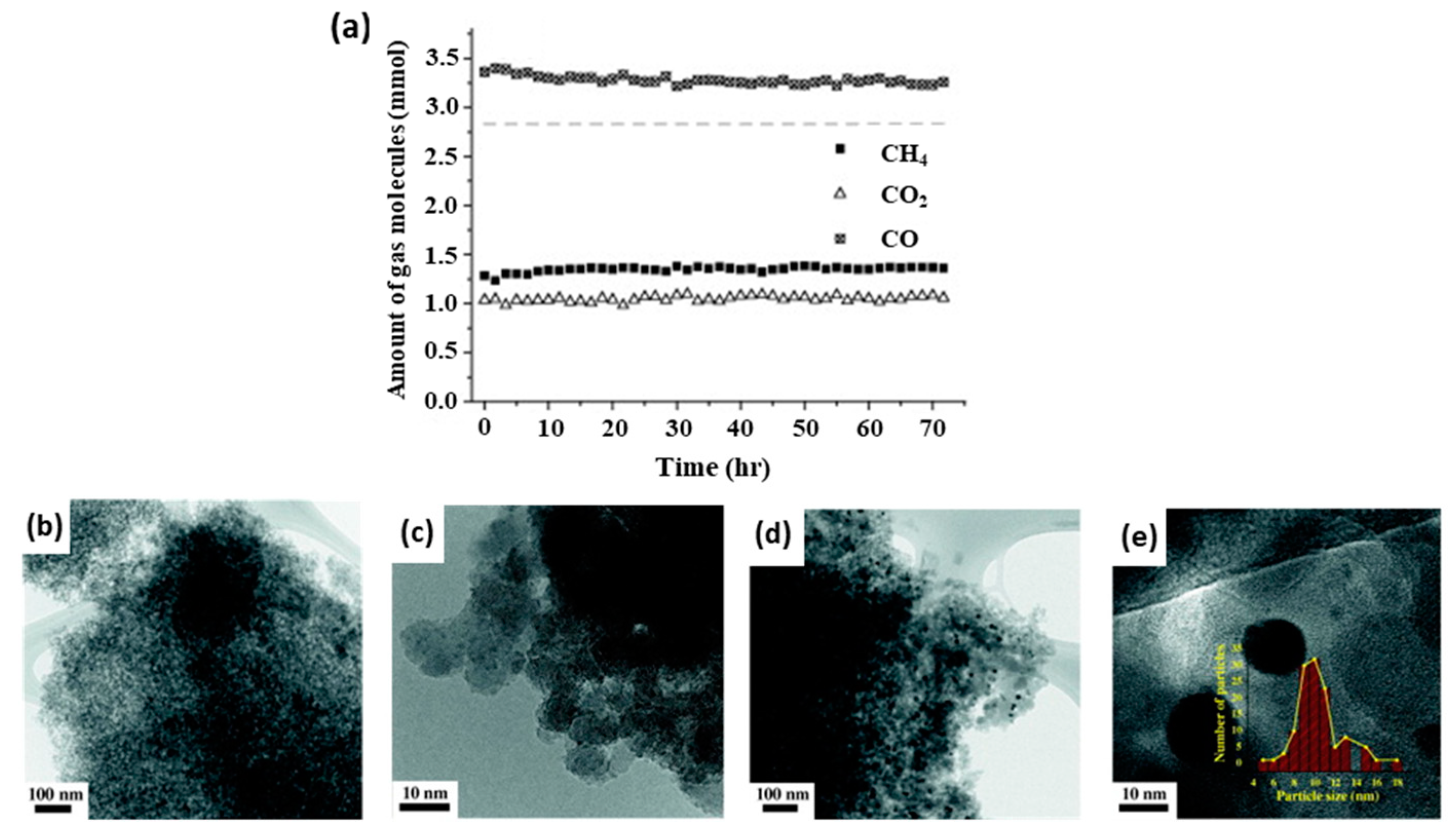{kind=link}
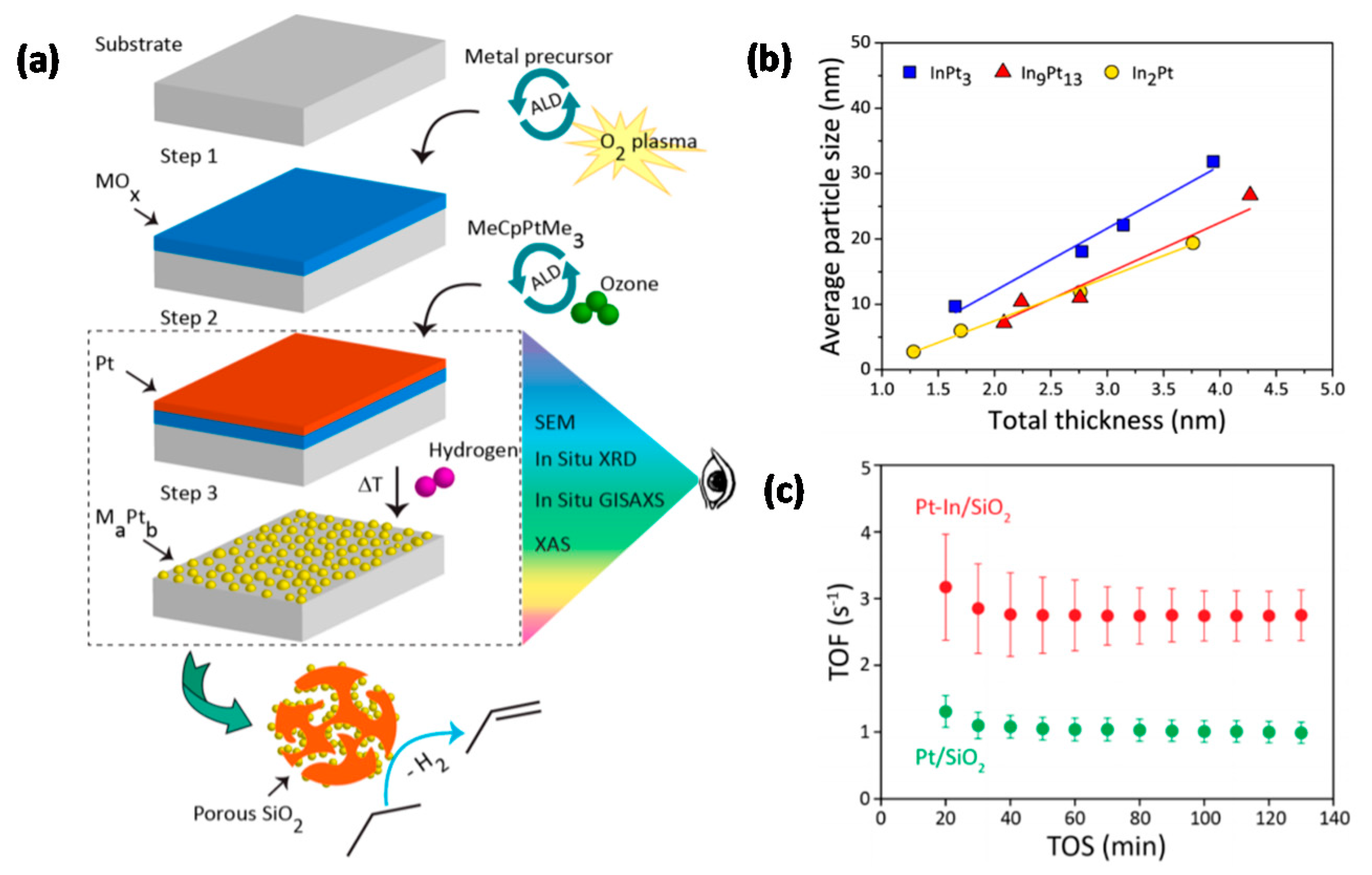{kind=link}
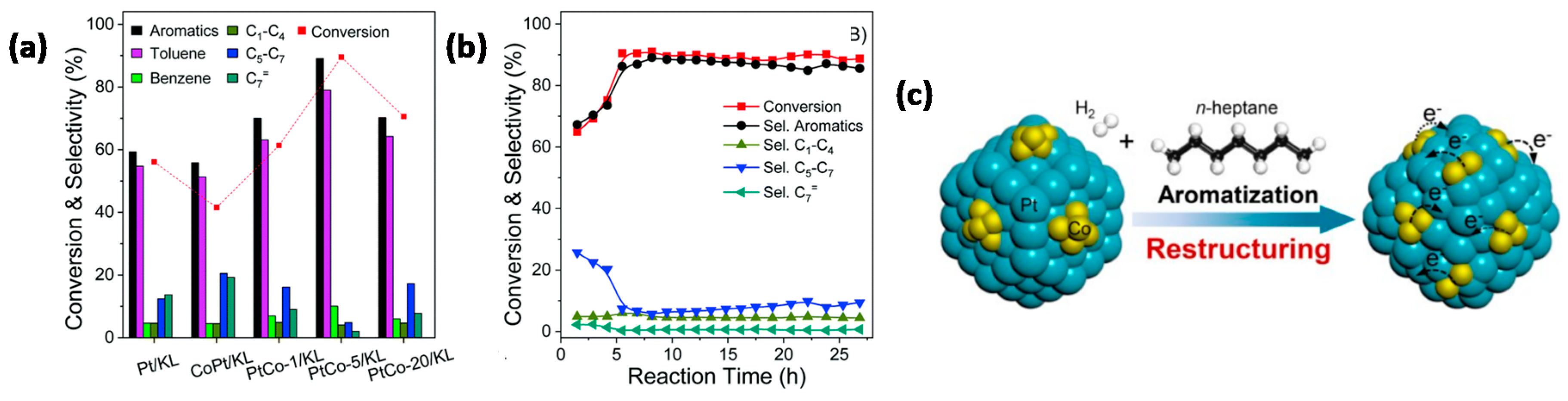{kind=link}
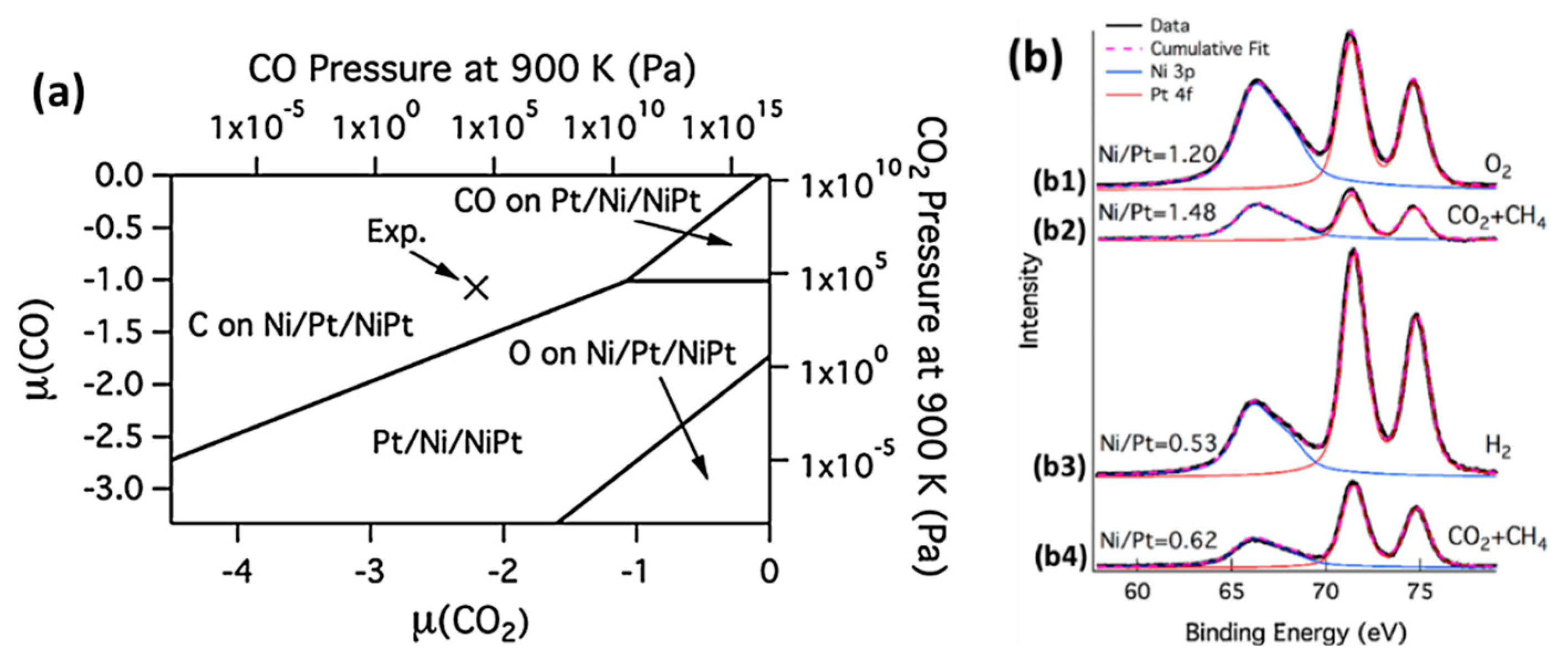{kind=link}
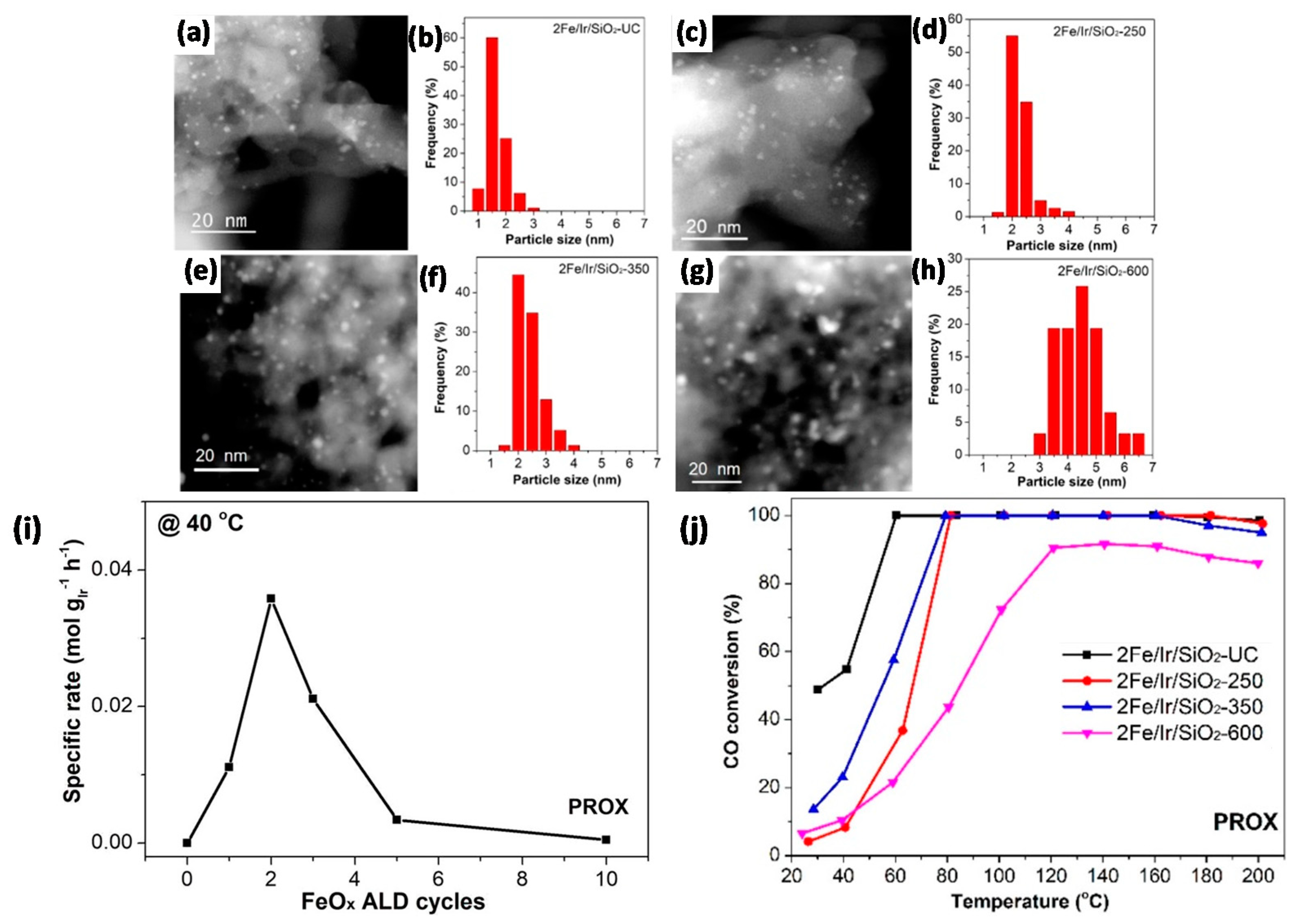{kind=link}
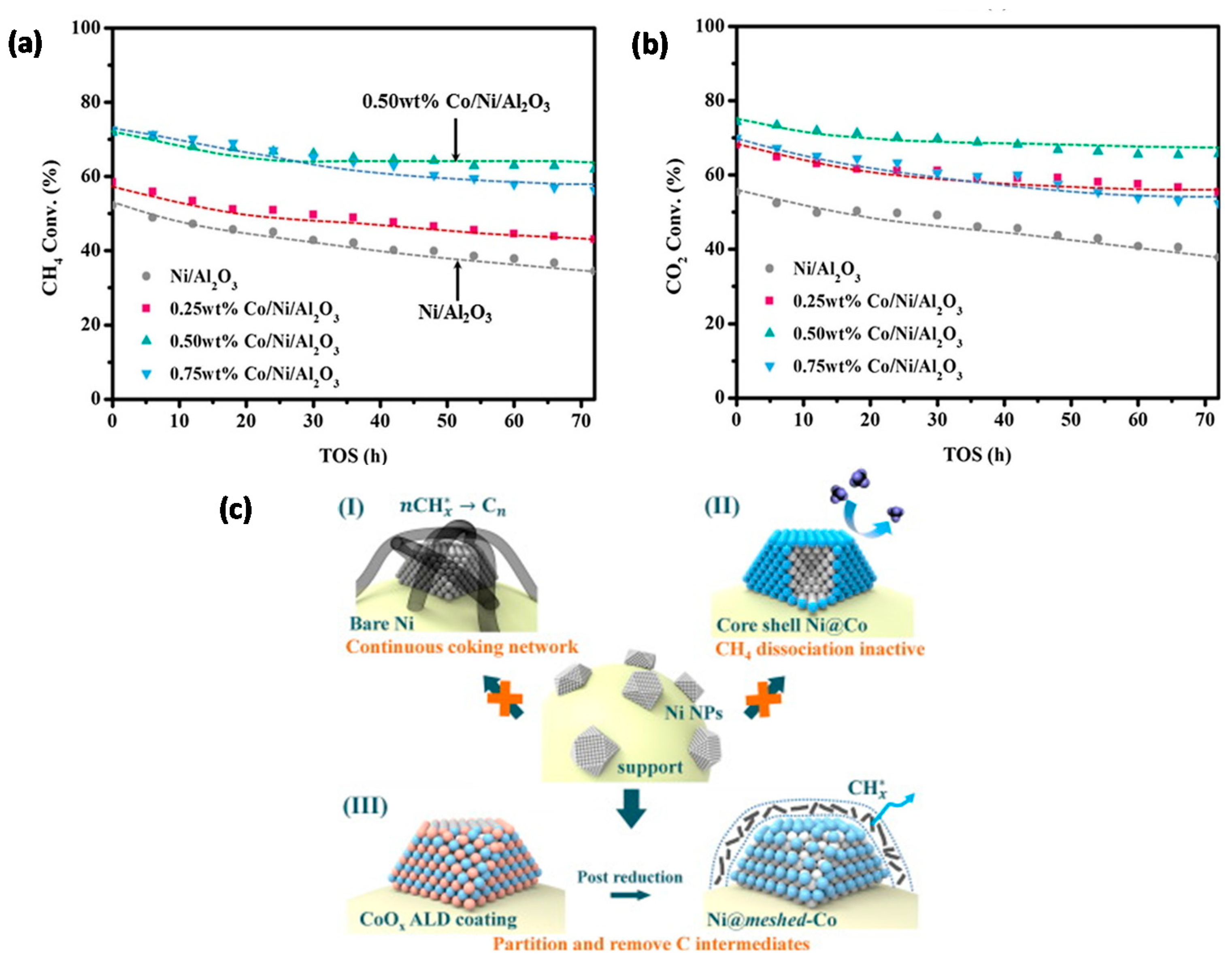{kind=link}
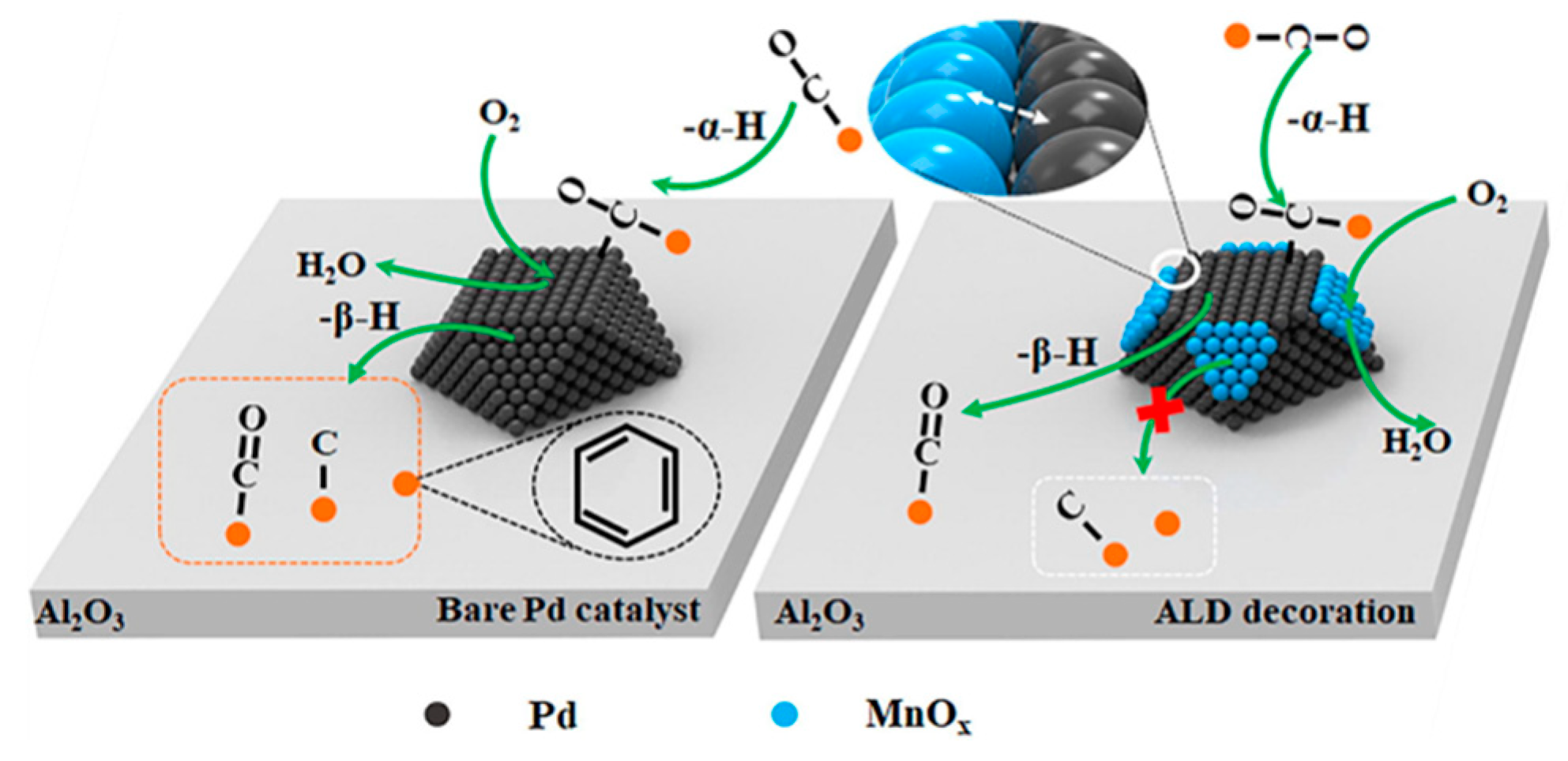{kind=link}
| Colloidal Synthesis | ALD | |||
|---|---|---|---|---|
| + | − | + | − | |
| Equipment | Simple equipment (hot plate, stirrer, inert gas/vacuum line) | Requires dedicated setups/reactors 1; (vacuum equipment) 2; deposition onto porous supports requires dedicated equipment to assure adequate exposure of all surfaces | ||
| Chemicals: -precursor | Broad choice of precursor type | Requires volatile precursors1 | ||
| -solvent | Requires solvent (removal) | No solvent (removal) required 2 | ||
| -stabilizing agent | Removal of stabilizing agent required by thermal process | No stabilizing agent removal required; thermal activation processes not per se necessary 3 | ||
| Control of: -size | NP size easily tunable down to ~nm size | Å-level control of coatings/NP size | ||
| -composition | Can prepare bi/tri/multimetallic NPs of virtually any elements, provided precursors exist | Creation of bi/tri/multimetallic NPs requires precursors with compatible reaction chemistry | ||
| -coating | Embedding restricted to colloids | Oxide overlayer deposition possible | ||
| Deposition: -on support | Requires a NP deposition step 4 | Direct deposition on substrate | ||
| -selective | Not selective | Chemical selectivity (area-selective ALD) | ||
| -conformal | Deposition into high surface area materials depends on several factors: e.g., synthesis method, pore size, NP size | Deposition possible into mesoporous materials as long as the pore diameter is larger than the size of the precursor molecule | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Coster, V.; Poelman, H.; Dendooven, J.; Detavernier, C.; Galvita, V.V. Designing Nanoparticles and Nanoalloys for Gas-Phase Catalysis with Controlled Surface Reactivity Using Colloidal Synthesis and Atomic Layer Deposition. Molecules 2020, 25, 3735. https://doi.org/10.3390/molecules25163735
De Coster V, Poelman H, Dendooven J, Detavernier C, Galvita VV. Designing Nanoparticles and Nanoalloys for Gas-Phase Catalysis with Controlled Surface Reactivity Using Colloidal Synthesis and Atomic Layer Deposition. Molecules. 2020; 25(16):3735. https://doi.org/10.3390/molecules25163735
Chicago/Turabian StyleDe Coster, Valentijn, Hilde Poelman, Jolien Dendooven, Christophe Detavernier, and Vladimir V. Galvita. 2020. "Designing Nanoparticles and Nanoalloys for Gas-Phase Catalysis with Controlled Surface Reactivity Using Colloidal Synthesis and Atomic Layer Deposition" Molecules 25, no. 16: 3735. https://doi.org/10.3390/molecules25163735