
Far-Off Resonance: Multiwavelength Raman Spectroscopy Probing Amide Bands of Amyloid-β-(37–42) Peptide


Abstract
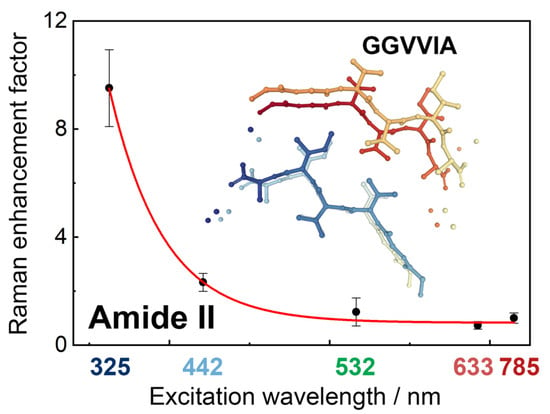
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. UV–Vis Spectroscopy
4.3. FTIR Spectroscopy
4.4. Atomic Force Microscopy
4.5. Raman Spectroscopy
4.6. Spectra Manipulation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bondarev, S.A.; Antonets, K.S.; Kajava, A.V.; Nizhnikov, A.A.; Zhouravleva, G.A. Protein Co-Aggregation Related to Amyloids: Methods of Investigation, Diversity, and Classification. Int. J. Mol. Sci. 2018, 19, 2292. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N.; Hasegawa, K.; Matsuzaki, K.; Naiki, H.; Yanagisawa, K. Environment- and Mutation-Dependent Aggregation Behavior of Alzheimer Amyloid β-Protein. J. Neurochem. 2004, 90, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Groh, N.; Bühler, A.; Huang, C.; Li, K.W.; van Nierop, P.; Smit, A.B.; Fändrich, M.; Baumann, F.; David, D.C. Age-Dependent Protein Aggregation Initiates Amyloid-β Aggregation. Front. Aging Neurosci. 2017, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Y.; Li, Q.; Cheng, H.C.; Du, H.N. β-Sheet Structure Formation of Proteins in Solid State as Revealed by Circular Dichroism Spectroscopy. Biopolym.—Biospectrosc. Sect. 2001, 62, 15–21. [Google Scholar] [CrossRef]
- Sarroukh, R.; Goormaghtigh, E.; Ruysschaert, J.M.; Raussens, V. ATR-FTIR: A “Rejuvenated” Tool to Investigate Amyloid Proteins. Biochim. Biophys. Acta—Biomembr. 2013, 1828, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Wilkosz, N.; Czaja, M.; Seweryn, S.; Skirlińska-Nosek, K.; Szymonski, M.; Lipiec, E.; Sofińska, K. Molecular spectroscopic markers of abnormal protein aggregation. Molecules 2020, 25, 2498. [Google Scholar] [CrossRef]
- Olapedo, S.A.; Xiong, K.; Hong, Z.; Asher, S.A.; Handen, J.; Lednev, I.K. UV resonance Raman investigations of peptide and protein structure and dynamics. Chem. Rev. 2012, 112, 2604–2628. [Google Scholar]
- Jakubek, R.S.; Handen, J.; White, S.E.; Asher, S.A.; Lednev, I.K. Ultraviolet resonance Raman spectroscopic markers for protein structure and dynamics. Trends Anal. Chem. 2018, 103, 223–229. [Google Scholar] [CrossRef]
- Shashilov, V.A.; Sikirzhytski, V.; Popova, L.A.; Lednev, I.K. Quantitative Methods for Structural Characterization of Proteins Based on Deep UV Resonance Raman Spectroscopy. Methods 2010, 52, 23–37. [Google Scholar] [CrossRef]
- Flynn, J.D.; Lee, J.C. Raman Fingerprints of Amyloid Structures. Chem. Commun. 2018, 54, 6983–6986. [Google Scholar] [CrossRef]
- Ignatjev, I.; Proniewicz, E.; Proniewicz, L.M.; Niaura, G. Effect of potential on temperature-dependent SERS spectra of neuromedin B on Cu electrode. Phys. Chem. Chem. Phys. 2013, 15, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Strazdaite, S.; Navakauskas, E.; Kirschner, J.; Sneideris, T.; Niaura, G. Structure Determination of Hen Egg-White Lysozyme Aggregates Adsorbed to Lipid/Water and Air/Water Interfaces. Langmuir 2020, 36, 4766–4775. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Balakrishnan, G.; Spiro, T.G. Protein Secondary Structure from Deep-UV Resonance Raman Spectroscopy. J. Raman Spectrosc. 2006, 37, 277–282. [Google Scholar] [CrossRef]
- Kurouski, D.; Van Duyne, R.P.; Lednev, I.K. Exploring the structure and formation mechanism of amyloid fibrils by Raman spectroscopy: A review. Analyst 2015, 140, 4967–4980. [Google Scholar] [CrossRef]
- Popova, L.A.; Kodali, R.; Wetzel, R.; Lednev, I.K. Structural variations in the cross-β core of amyloid β fibrils revealed by deep UV resonance Raman spectroscopy. J. Am. Chem. Soc. 2010, 132, 6324–6328. [Google Scholar] [CrossRef]
- Flynn, J.D.; Jiang, Z.; Lee, J.C. Segmental 13C-labeling and Raman microspectroscopy of α-synuclein amyloid formation. Angew. Chem. Int. Ed. 2018, 57, 17069–17072. [Google Scholar] [CrossRef]
- Ishigaki, M.; Morimoto, K.; Chatani, E.; Ozaki, Y. Exploration of insulin amyloid polymorphism using Raman spectroscopy and imaging. Biophys. J. 2020, 118, 2997–3007. [Google Scholar] [CrossRef]
- Rivas-Arancibia, S.; Rodriguez-Martinez, E.; Badillo-Ramirez, I.; López-González, U.; Saniger, J.M. Structural changes of amyloid beta in hippocampus of rats exposed to ozone: A Raman spectroscopy study. Front. Mol. Neurosci. 2017, 10, 137. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Lu, H.P. Revealing the secondary structural changes of amyloid β peptide by probing the spectral fingerprint characters. J. Raman Spectrosc. 2013, 44, 670–674. [Google Scholar] [CrossRef]
- Chi, Z.; Chen, X.G.; Holtz, J.S.W.W.; Asher, S.A. UV Resonance Raman-Selective Amide Vibrational Enhancement: Quantitative Methodology for Determining Protein Secondary Structure. Biochemistry 1998, 37, 2854–2864. [Google Scholar] [CrossRef]
- Roach, C.A.; Simpson, J.V.; JiJi, R.D. Evolution of quantitative methods in protein secondary structure determination via deep-ultraviolet resonance Raman spectroscopy. Analyst 2012, 137, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Frushour, B.G.; Koenig, J.L. Raman Spectroscopic Study of Mechanically Deformed Poly-L-Alanine. Biopolymers 1974, 13, 455–474. [Google Scholar] [CrossRef]
- Harada, I.; Sugawara, Y.; Matsuura, H.; Shimanouchi, T. Preresonance Raman Spectra of Simple Amides Using Ultraviolet Lasers. J. Raman Spectrosc. 1975, 4, 91–98. [Google Scholar] [CrossRef]
- Catalini, S.; Rossi, B.; Foggi, P.; Masciovecchio, C.; Bruni, F. Aqueous Solvation of Glutathione Probed by UV Resonance Raman Spectroscopy. J. Mol. Liq. 2019, 283, 537–547. [Google Scholar] [CrossRef]
- Myshakina, N.S.; Ahmed, Z.; Asher, S.A. Dependence of amide vibrations on hydrogen bonding. J. Phys. Chem. B 2008, 112, 11873–11877. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic Structures of Amyloid Cross-β Spines Reveal Varied Steric Zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Chang, L.K.; Zhao, J.H.; Liu, H.L.; Liu, K.T.; Chen, J.T.; Tsai, W.B.; Ho, Y. Molecular Dynamics Simulations to Investigate the Structural Stability and Aggregation Behavior of the GGVVIA Oligomers Derived from Amyloid β Peptide. J. Biomol. Struct. Dyn. 2009, 26, 731–740. [Google Scholar] [CrossRef]
- Tzotzos, S.; Doig, A.J. Amyloidogenic Sequences in Native Protein Structures. Protein Sci. 2010, 19, 327–348. [Google Scholar] [CrossRef] [Green Version]
- Barth, A. Infrared Spectroscopy of Proteins. Biochim. Biophys. Acta—Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [Green Version]
- Owen, T. Fundamentals of Modern UV-Visible Spectroscopy; Hewlett-Packard Co.: Palo Alto, CA, USA, 1996. [Google Scholar] [CrossRef]
- Sharma, B.; Bykov, S.V.; Asher, S.A. UV Resonance Raman Investigation of Electronic Transitions in α-Helical and Polyproline II-Like Conformations. J. Phys. Chem. B 2008, 112, 11762–11769. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, G.; Hu, Y.; Case, M.A.; Spiro, T.G. Microsecond Melting of a Folding Intermediate in a Coiled-Coil Peptide, Monitored by T-Jump/UV Raman Spectroscopy. J. Phys. Chem. B 2006, 110, 19877–19883. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer-Stenner, R.; Eker, F.; Huang, Q.; Griebenow, K.; Mroz, P.A.; Kozlowski, P.M. Structure Analysis of Dipeptides in Water by Exploring and Utilizing the Structural Sensitivity of Amide III by Polarized Visible Raman, FTIR-spectroscopy and DFT Based Normal Coordinate Analysis. J. Phys. Chem. B 2002, 106, 4294–4304. [Google Scholar] [CrossRef]
- Ridgley, D.M.; Claunch, E.C.; Barone, J.R. Characterization of Large Amyloid Fibers and Tapes with Fourier Transform Infrared (FT-IR) and Raman Spectroscopy. Appl. Spectrosc. 2013, 67, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Purrello, R.; Jordan, T.; Spiro, T.G. UVRR Spectroscopy of the Peptide Bond. 1. Amide S, a Nonhelical Structure Marker, is a CαH Bending Mode. J. Am. Chem. Soc. 1991, 113, 6359–6368. [Google Scholar] [CrossRef]
- Misiūnas, A.; Talaikyte, Z.; Niaura, G.; Razumas, V.; Nylander, T. Thermomyces Lanuginosus Lipase in the Liquid-Crystalline Phases of Aqueous Phytantriol: X-ray Diffraction and Vibrational Spectroscopic Studies. Biophys. Chem. 2008, 134, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.; Pflüger, F.; Nsangou, M.; Ghomi, M. Vibrational analysis of amino acids and short peptides in hydrated media. IV. Amino Acids with Hydrophobic Side Chains: L-Alanine, L-Valine, and L-Isoleucine. J. Phys. Chem. B 2009, 113, 3169–3178. [Google Scholar] [CrossRef]
- Bartlett, G.J.; Choudhary, A.; Raines, R.T.; Woolfson, D.N. N→π* interactions in proteins. Nat. Chem. Biol. 2010, 6, 615–620. [Google Scholar] [CrossRef]
- Newberry, R.W.; Bartlett, G.J.; VanVeller, B.; Woolfson, D.N.; Raines, R.T. Signatures of n→π* interactions in proteins. Protein Sci. 2014, 23, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.G.; Asher, S.; Schweitzer-Stenner, R.; Mirkin, N.G.; Krimm, S. UV Raman Determination of the ππ* Excited State Geometry of N-Methylacetamide: Vibrational Enhancement Pattern. J. Am. Chem. Soc. 1995, 117, 2884–2895. [Google Scholar] [CrossRef]
- Mayne, L.C.; Hudson, B. Resonance Raman spectroscopy of N-methylacetamide: Overtones and combinations of the C−N stretch (amide II’) and effect of solvation on the C=O stretch (amide I) intensity. J. Phys. Chem. 1991, 95, 2962–2967. [Google Scholar] [CrossRef]
- Punihaole, D.; Jakubek, R.S.; Dahlburg, E.M.; Hong, Z.; Myshakina, N.S.; Geib, S.; Asher, S.A. UV Resonance Raman Investigation of the Aqueous Solvation Dependence of Primary Amide Vibrations. J. Phys. Chem. B 2015, 119, 3931–3939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.D.; McGlinchey, R.P.; Walker, R.L.; Lee, J.C. Structural Features of α-Synuclein Amyloid Fibrils Revealed by Raman Spectroscopy. J. Biol. Chem. 2018, 293, 767–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, K.; Fan, Z.; Sun, L.; Makam, P.; Tian, Z.; Ruegsegger, M.; Shaham-Niv, S.; Hansford, D.; Aizen, R.; Galster, S.; et al. Quantum confined peptide assemblies with tunable visible to near infrared spectral range. Nat. Commun. 2018, 9, 3217. [Google Scholar] [CrossRef] [PubMed]
- Pinotsi, D.; Grisanti, L.; Mahou, P.; Gebauer, R.; Kaminski, C.F.; Hassanali, A.; Kaminski Schierle, G.S. Proton transfer and structure-specific fluorescence in hydrogen bond-rich protein structures. J. Am. Chem. Soc. 2016, 138, 3046–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, F.T.S.; Schierle, G.S.K.; Kumita, J.R.; Bertoncini, C.W.; Dobson, C.M.; Kaminski, C.F. Protein amyloids develop an intrinsic fluorescence signature during aggregation. Analyst 2013, 138, 2156–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapshina, N.; Shishkin, I.I.; Nandi, R.; Noskov, R.E.; Barhom, H.; Joseph, S.; Apter, B.; Ellenbogen, T.; Natan, A.; Ginzburg, P.; et al. Bioinspired amyloid nanodots with visible fluorescence. Adv. Opt. Mater. 2019, 7, 1801400. [Google Scholar] [CrossRef]
- Sirangelo, I.; Borriello, M.; Irace, G.; Iannuzzi, C. Intrinsic blue-green fluorescence in amyloid fibrils. AIMS Biophys. 2018, 5, 155–165. [Google Scholar] [CrossRef]
- Pansieri, J.; Josserand, V.; Lee, S.-J.; Rongier, A.; Imbert, D.; Sallanon, M.M.; Kövari, E.; Dane, T.G.; Vendrely, C.; Chaix-Pluchery, O.; et al. Ultraviolet-visible-near-infrared optical properties of amyloid fibrils shed light on amyloidogenesis. Nat. Photon. 2019, 13, 473–479. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Wavenumber, cm−1 | Assignment | References |
|---|---|---|
| 1124 | r(CH3), δ(CCH) | [37] |
| 1231 | AIII3; β-sheet | [9,20,33,34] |
| 1264 | AIII2 | [33] |
| 1292 | AIII1 | [33] |
| 1320 | tCH2, wCH2 | [33] |
| 1346 | δCH2 | [36] |
| 1396 | AS | [9,20,35] |
| 1435 | δCH2 | [36] |
| 1446 | δCH2 | [36] |
| 1463 | δCH2, δCH3 | [36] |
| 1558 | AII | [9,13,20,32] |
| 1664 | AI; β-sheet | [13,20,34] |
| Laser Line, nm | Power at Sample, mW | Grating, Lines/mm | Objective, Mag. (NA); Spectral Range |
|---|---|---|---|
| 325 | 4.3 | 3600 | Thor Labs, 15× (0.32); UV |
| 442 | 53 | 2400 | Leica, 5×, (0.12); Vis |
| 633 | 9.4 | 1200 | Leica, 5×, (0.12); Vis |
| 785 | 92 | 1200 | Olympus, 50× (0.65); IR |
| 830 | 166 | 830 | Olympus, 50× (0.65); IR |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talaikis, M.; Strazdaitė, S.; Žiaunys, M.; Niaura, G. Far-Off Resonance: Multiwavelength Raman Spectroscopy Probing Amide Bands of Amyloid-β-(37–42) Peptide. Molecules 2020, 25, 3556. https://doi.org/10.3390/molecules25153556
Talaikis M, Strazdaitė S, Žiaunys M, Niaura G. Far-Off Resonance: Multiwavelength Raman Spectroscopy Probing Amide Bands of Amyloid-β-(37–42) Peptide. Molecules. 2020; 25(15):3556. https://doi.org/10.3390/molecules25153556
Chicago/Turabian StyleTalaikis, Martynas, Simona Strazdaitė, Mantas Žiaunys, and Gediminas Niaura. 2020. "Far-Off Resonance: Multiwavelength Raman Spectroscopy Probing Amide Bands of Amyloid-β-(37–42) Peptide" Molecules 25, no. 15: 3556. https://doi.org/10.3390/molecules25153556