
Natural Products as Modulators of Sirtuins




Abstract
:1. Introduction
1.1. Characteristics of Natural Products and Development of Natural Product Databases for Drug Discovery
1.2. Sirtuins as Therapeutic Targets
2. Natural Product Modulators of Sirtuins
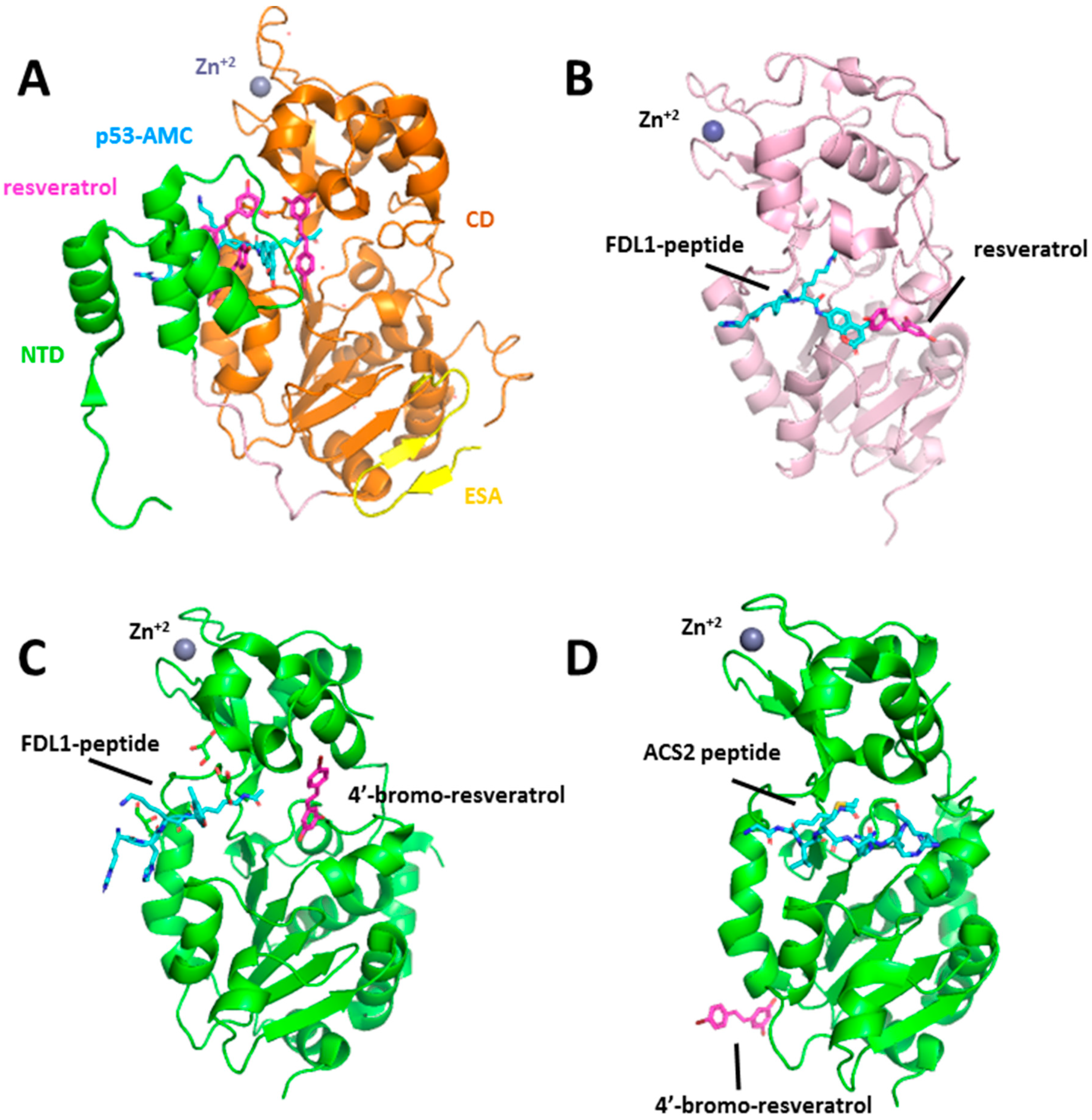
2.1. Stilbenoids: Resveratrol, Piceatannol, and Trans-(−)-ϵ-Viniferin
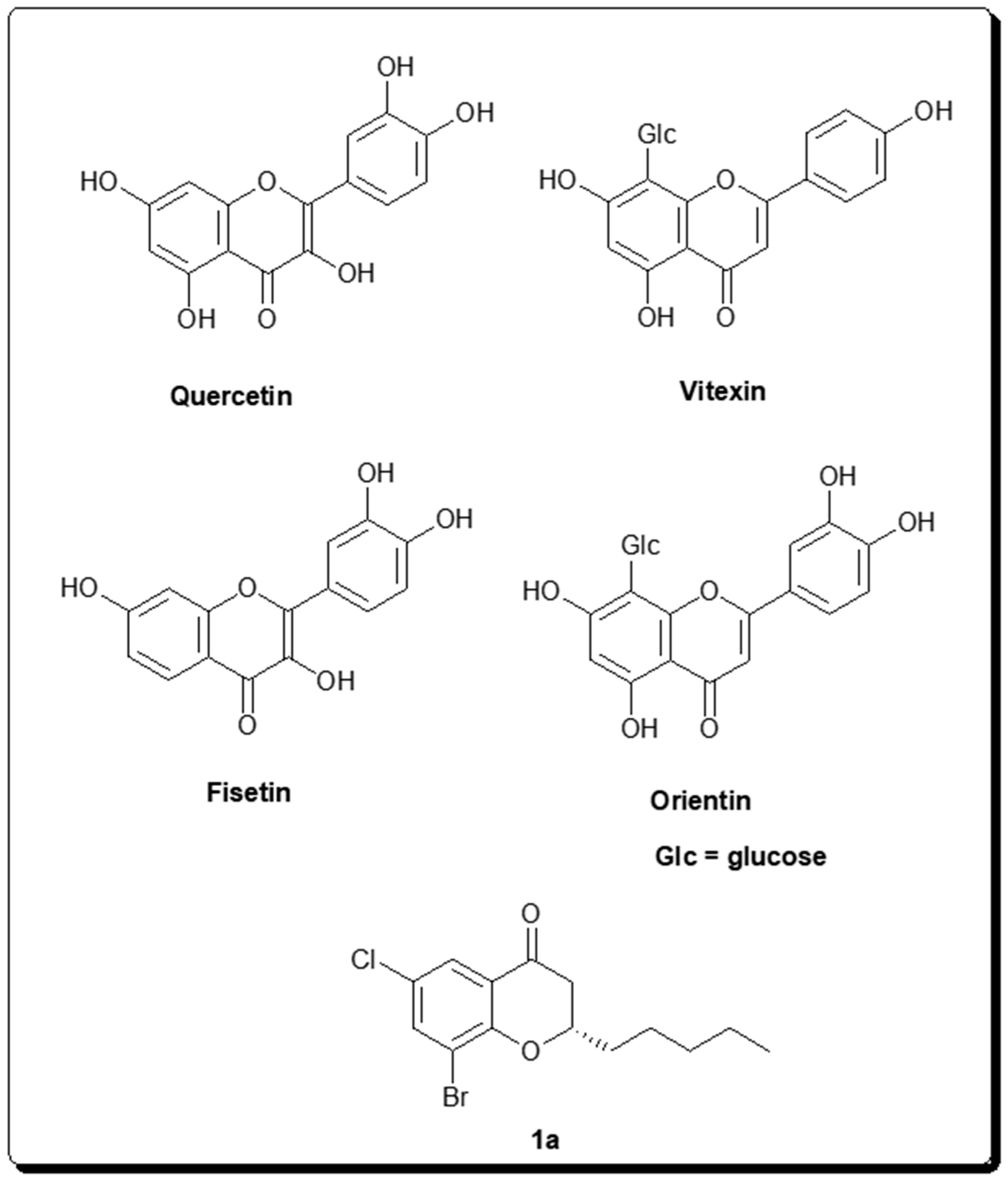
2.2. Chromone-Derived Natural Products
2.3. Structurally Diverse Natural Product Modulators of Sirtuins
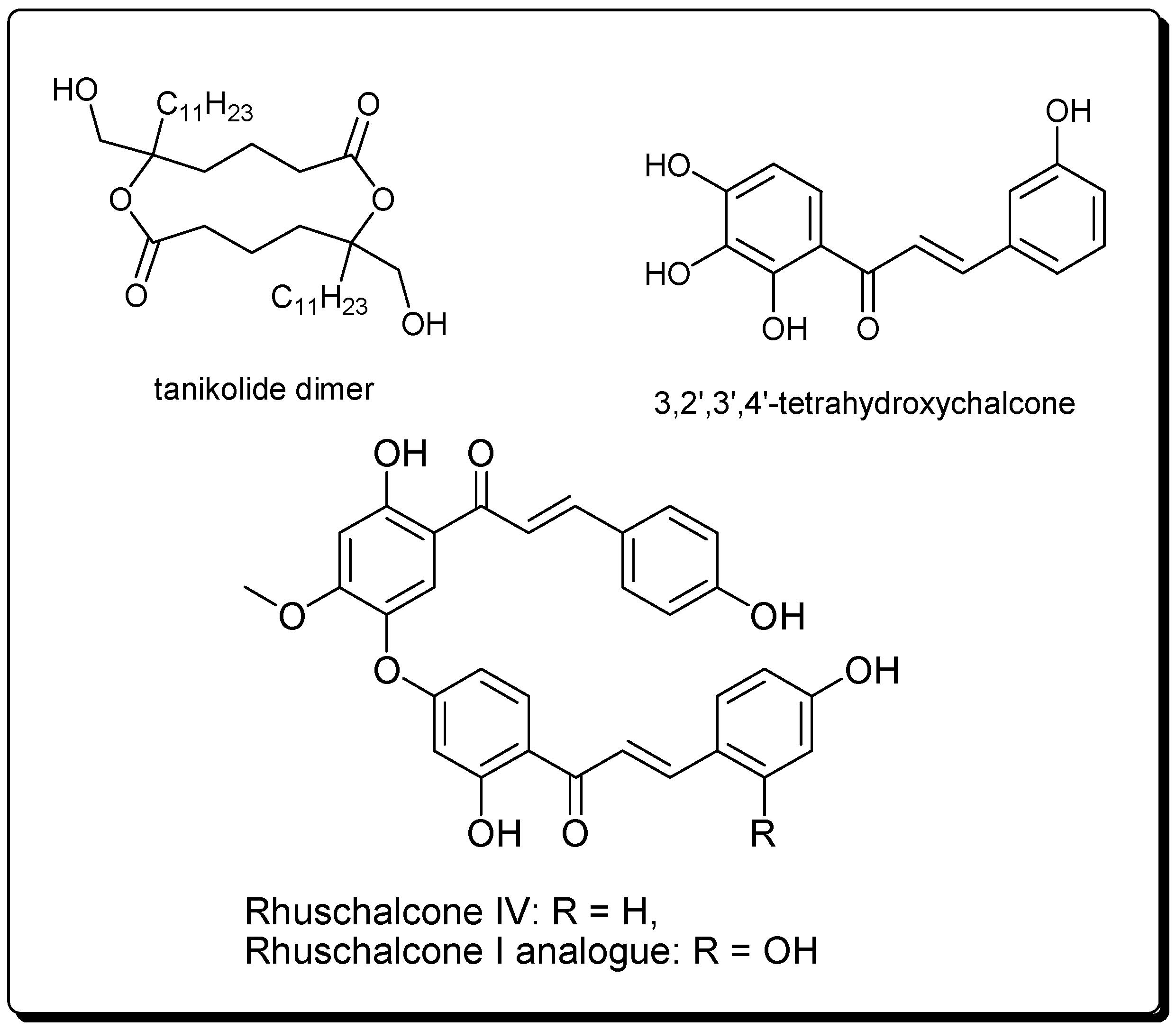
2.3.1. Tanikolide Dimer from the Madagascan Marine Cyanobacterium Lyngbya majuscula
2.3.2. Bichalcones from the African Medicinal Plant Lyngbya majuscula
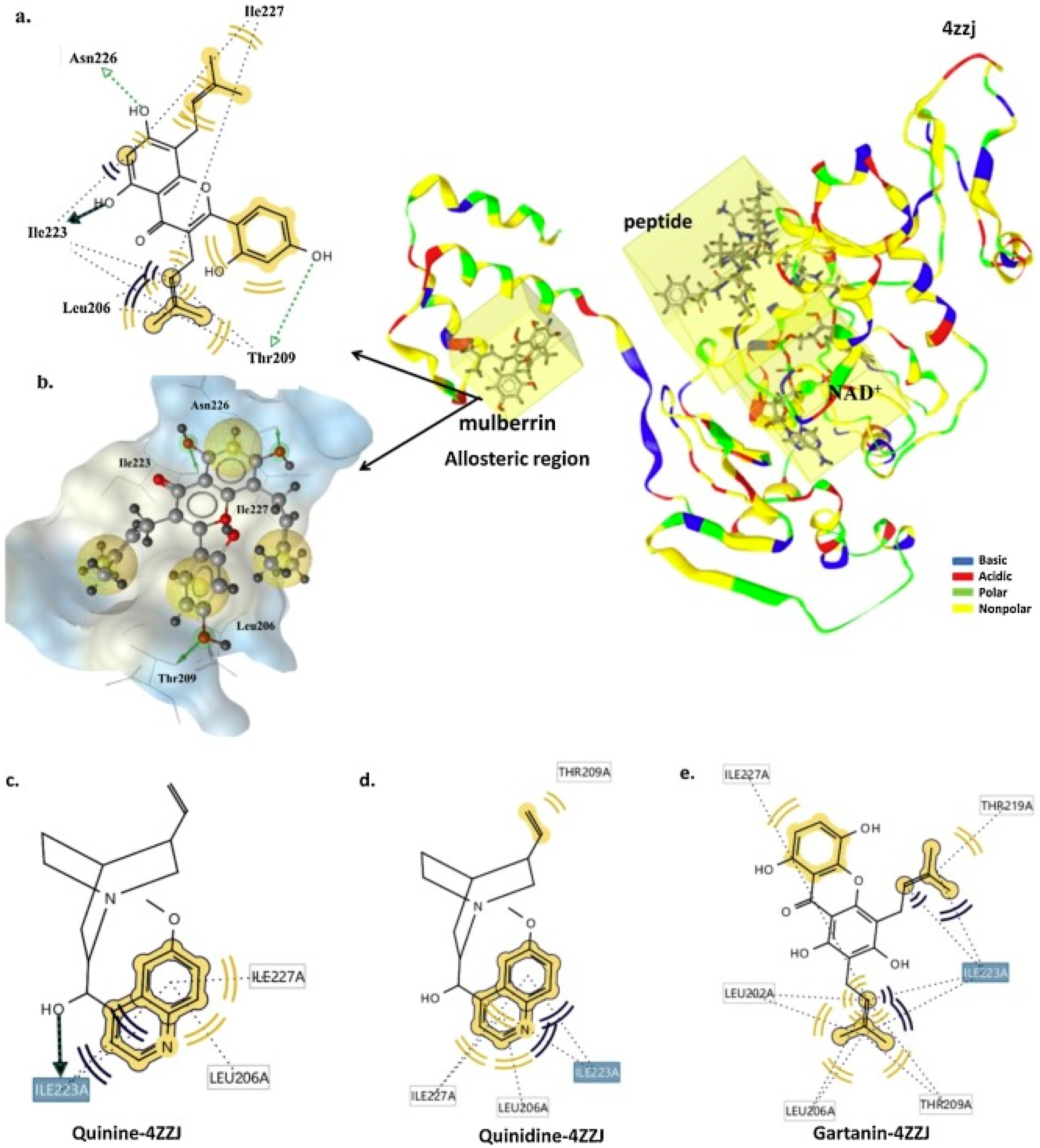
2.3.3. Flavonoids, Alkaloids, and Xanthones from Indonesian Medicinal Plants
3. Therapeutic Importance of Some Naturally Occurring Sirtuin Inhibitors and Modulators
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharm. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef]
- Austin, C.P. The completed human genome: Implications for chemical biology. Curr. Opin. Chem. Biol. 2003, 7, 511–515. [Google Scholar] [CrossRef]
- Collins, F.S.; Morgan, M.; Patrinos, A. The Human Genome Project: Lessons from large-scale biology. Science 2003, 300, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Frearson, J.; Wyatt, P. Drug Discovery in Academia- the third way? Expert Opin. Drug Discov. 2010, 5, 909–919. [Google Scholar] [CrossRef]
- Roy, A.; McDonald, P.R.; Sittampalam, S.; Chaguturu, R. Open access high throughput drug discovery in the public domain: A mount everest in the making. Curr. Pharm. Biotechnol. 2010, 11, 764–778. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, K.H.; Bohm, H.J.; Muller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Entzeroth, M.; Flotow, H.; Condron, P. Overview of high-throughput screening. Curr. Protoc. Pharm. 2009, 44, 9.4.1–9.4.27. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Keseru, G.M.; Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Drewry, D.H.; Macarron, R. Enhancements of screening collections to address areas of unmet medical need: An industry perspective. Curr. Opin. Chem. Biol. 2010, 14, 289–298. [Google Scholar] [CrossRef]
- Jacoby, E.; Schuffenhauer, A.; Popov, M.; Azzaoui, K.; Havill, B.; Schopfer, U.; Engeloch, C.; Stanek, J.; Acklin, P.; Rigollier, P.; et al. Key aspects of the Novartis compound collection enhancement project for the compilation of a comprehensive chemogenomics drug discovery screening collection. Curr. Top. Med. Chem. 2005, 5, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.J.; Eggleston, D.S.; Brinded, K.A.; Hollerton, J.C.; Taylor, N.L.; Readshaw, S.A. Defining and maintaining a high quality screening collection: The GSK experience. Drug Discov. Today 2006, 11, 267–272. [Google Scholar] [CrossRef]
- Sukuru, S.C.; Jenkins, J.L.; Beckwith, R.E.; Scheiber, J.; Bender, A.; Mikhailov, D.; Davies, J.W.; Glick, M. Plate-based diversity selection based on empirical HTS data to enhance the number of hits and their chemical diversity. J. Biomol. Screen. 2009, 14, 690–699. [Google Scholar] [CrossRef]
- Banerjee, P.; Erehman, J.; Gohlke, B.-O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II--a database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [Green Version]
- Mathur, S.; Hoskins, C. Drug development: Lessons from nature. Biomed. Rep. 2017, 6, 612–614. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef]
- Shen, B. A New golden age of natural products drug discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rishton, G.M. Natural products as a robust source of new drugs and drug leads: Past successes and present day issues. Am. J. Cardiol. 2008, 101, 43d–49d. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cai, S.; Zhang, Z.; Chen, Z. High-resolution two-dimensional J-resolved NMR spectroscopy for biological systems. Biophys. J. 2014, 106, 2061–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camp, D.; Davis, R.A.; Campitelli, M.; Ebdon, J.; Quinn, R.J. Drug-like properties: Guiding principles for the design of natural product libraries. J. Nat. Prod. 2012, 75, 72–81. [Google Scholar] [CrossRef]
- Tu, Y.; Jeffries, C.; Ruan, H.; Nelson, C.; Smithson, D.; Shelat, A.A.; Brown, K.M.; Li, X.-C.; Hester, J.P.; Smillie, T.; et al. An automated high-throughput system to fractionate plant natural products for drug discovery. J. Nat. Prod. 2010, 73, 751–754. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, S.; Yu, O. Metabolic engineering of flavonoids in plants and microorganisms. Appl. Microbiol. Biotechnol. 2011, 91, 949–956. [Google Scholar] [CrossRef]
- Wu, J.; Du, G.; Zhou, J.; Chen, J. Systems metabolic engineering of microorganisms to achieve large-scale production of flavonoid scaffolds. J. Biotechnol. 2014, 188, 72–80. [Google Scholar] [CrossRef]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Paddon, C.J.; Keasling, J.D. Semi-synthetic artemisinin: A model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014, 12, 355–367. [Google Scholar] [CrossRef]
- Baker, D.D.; Chu, M.; Oza, U.; Rajgarhia, V. The value of natural products to future pharmaceutical discovery. Nat. Prod. Rep. 2007, 24, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Waldmann, H. Protein structure similarity clustering and natural product structure as guiding principles in drug discovery. Drug Discov. Today 2005, 10, 471–483. [Google Scholar] [CrossRef]
- Kellenberger, E.; Hofmann, A.; Quinn, R.J. Similar interactions of natural products with biosynthetic enzymes and therapeutic targets could explain why nature produces such a large proportion of existing drugs. Nat. Prod. Rep. 2011, 28, 1483–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller-Kuhrt, L. Putting nature back into drug discovery. Nat. Biotechnol. 2003, 21, 602. [Google Scholar] [CrossRef] [PubMed]
- Feher, M.; Schmidt, J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef]
- Dobson, P.D.; Patel, Y.; Kell, D.B. Metabolite-likeness’ as a criterion in the design and selection of pharmaceutical drug libraries. Drug Discov. Today 2009, 14, 31–40. [Google Scholar] [CrossRef]
- Henkel, T.; Brunne, R.M.; Muller, H.; Reichel, F. Statistical Investigation into the Structural Complementarity of Natural Products and Synthetic Compounds. Ang. Chem. Int. Ed. 1999, 38, 643–647. [Google Scholar] [CrossRef]
- Hert, J.; Irwin, J.J.; Laggner, C.; Keiser, M.J.; Shoichet, B.K. Quantifying biogenic bias in screening libraries. Nat. Chem. Biol. 2009, 5, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Suárez, M.; Chen, T.S.; Ting, A.Y. Protein-protein interaction detection in vitro and in cells by proximity biotinylation. J. Am. Chem. Soc. 2008, 130, 9251–9253. [Google Scholar] [CrossRef] [Green Version]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Schiedel, M.; Robaa, D.; Rumpf, T.; Sippl, W.; Jung, M. The current state of NAD(+)-dependent histone deacetylases (Sirtuins) as novel therapeutic targets. Med. Res. Rev. 2018, 38, 147–200. [Google Scholar] [CrossRef] [PubMed]
- Costantini, S.; Sharma, A.; Raucci, R.; Costantini, M.; Autiero, I.; Colonna, G. Genealogy of an ancient protein family: The Sirtuins, a family of disordered members. Bmc Evol. Biol. 2013, 13, 60. [Google Scholar] [CrossRef]
- Lancelot, J.; Caby, S.; Dubois-Abdesselem, F.; Vanderstraete, M.; Trolet, J.; Oliveira, G.; Bracher, F.; Jung, M.; Pierce, R.J. Schistosoma mansoniSirtuins: Characterization and potential as chemotherapeutic targets. PLoS Negl. Trop. Dis. 2013, 7, e2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeberlein, M.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2004, 2, E296. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wang, J.; Fu, J.; Du, L.; Jeong, H.; West, T.; Xiang, L.; Peng, Q.; Hou, Z.; Cai, H.; et al. Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat. Med. 2011, 18, 153–158. [Google Scholar] [CrossRef]
- Jeong, H.; Cohen, D.E.; Cui, L.; Supinski, A.; Savas, J.N.; Mazzulli, J.R.; Yates, J.R., 3rd; Bordone, L.; Guarente, L.; Krainc, D. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 2011, 18, 159–165. [Google Scholar] [CrossRef]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wales, P.; Quinti, L.; Zuo, F.; Moniot, S.; Herisson, F.; Rauf, N.A.; Wang, H.; Silverman, R.B.; Ayata, C.; et al. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS ONE 2015, 10, e0116919. [Google Scholar] [CrossRef] [Green Version]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef] [Green Version]
- Pfister, J.A.; Ma, C.; Morrison, B.E.; D’Mello, S.R. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE 2008, 3, e4090. [Google Scholar] [CrossRef]
- Deng, C.X. SIRT1, is it a tumor promoter or tumor suppressor? Int. J. Biol. Sci. 2009, 5, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Sun, K.; Jiao, S.; Cai, N.; Zhao, X.; Zou, H.; Xie, Y.; Wang, Z.; Zhong, M.; Wei, L. High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Sci. Rep. 2014, 4, 7481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffman, D.M.; Grizzle, W.E.; Bamman, M.M.; Kim, J.S.; Eltoum, I.A.; Elgavish, A.; Nagy, T.R. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007, 67, 6612–6618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, K.R.; Barbieri, A.; Malyarchuk, S.; Saxena, M.; Nedeljkovic-Kurepa, A.; Cameron Mehl, M.; Wang, A.; Gu, X.; Pruitt, K. SIRT1 positively regulates breast cancer associated human aromatase (CYP19A1) expression. Mol. Endocrinol. 2013, 27, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cea, M.; Soncini, D.; Fruscione, F.; Raffaghello, L.; Garuti, A.; Emionite, L.; Moran, E.; Magnone, M.; Zoppoli, G.; Reverberi, D.; et al. Synergistic interactions between HDAC and sirtuin inhibitors in human leukemia cells. PLoS ONE 2011, 6, e22739. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Sengupta, A.; Seerapu, G.P.K.; Nakhi, A.; Shivaji Ramarao, E.V.V.; Bung, N.; Bulusu, G.; Pal, M.; Haldar, D. A novel SIRT1 inhibitor, 4bb induces apoptosis in HCT116 human colon carcinoma cells partially by activating p53. Biochem. Biophys. Res. Comm. 2017, 488, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Jing, H.; Lin, H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 2014, 6, 945–966. [Google Scholar] [CrossRef] [Green Version]
- Alhazzazi, T.Y.; Kamarajan, P.; Verdin, E.; Kapila, Y.L. SIRT3 and cancer: Tumor promoter or suppressor? Biochim. Biophys. Acta 2011, 1816, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Alhazzazi, T.Y.; Kamarajan, P.; Joo, N.; Huang, J.Y.; Verdin, E.; D’Silva, N.J.; Kapila, Y.L. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer 2011, 117, 1670–1678. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyo, M.; Yamamoto, H.; Konno, M.; Colvin, H.; Nishida, N.; Koseki, J.; Kawamoto, K.; Ogawa, H.; Hamabe, A.; Uemura, M.; et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br. J. Cancer 2015, 113, 492–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.M.; Lee, A.; Lee, J.; Haigis, M.C. SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J. Biol. Chem. 2014, 289, 4135–4144. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, Y.; Yamasaki, M.; Sawada, G.; Miyazaki, Y.; Makino, T.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; Takiguchi, S.; Mimori, K.; et al. Downregulation of SIRT4 expression is associated with poor prognosis in esophageal squamous cell carcinoma. Oncology 2016, 90, 347–355. [Google Scholar] [CrossRef]
- George, J.; Ahmad, N. Mitochondrial sirtuins in cancer: Emerging roles and therapeutic potential. Cancer Res. 2016, 76, 2500–2506. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Zuo, Y.; Feng, Y.; Zhang, M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumourbiology 2014, 35, 10699–10705. [Google Scholar] [CrossRef]
- Lai, C.C.; Lin, P.M.; Lin, S.F.; Hsu, C.H.; Lin, H.C.; Hu, M.L.; Hsu, C.M.; Yang, M.Y. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumourbiology 2013, 34, 1847–1854. [Google Scholar] [CrossRef]
- Kugel, S.; Sebastian, C.; Fitamant, J.; Ross, K.N.; Saha, S.K.; Jain, E.; Gladden, A.; Arora, K.S.; Kato, Y.; Rivera, M.N.; et al. SIRT6 Suppresses Pancreatic Cancer through Control of Lin28b. Cell 2016, 165, 1401–1415. [Google Scholar] [CrossRef] [Green Version]
- Ioris, R.M.; Galie, M.; Ramadori, G.; Anderson, J.G.; Charollais, A.; Konstantinidou, G.; Brenachot, X.; Aras, E.; Goga, A.; Ceglia, N.; et al. sirt6 suppresses cancer stem-like capacity in tumors with PI3K activation independently of its deacetylase activity. Cell Rep. 2017, 18, 1858–1868. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Qin, C.Y. Sirt6 suppresses hepatocellular carcinoma cell growth via inhibiting the extracellular signal regulated kinase signaling pathway. Mol. Med. Rep. 2014, 9, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual tumor suppressor and tumor promoter action of sirtuins in determining malignant phenotype. Front. Pharm. 2019, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef] [Green Version]
- van de Ven, R.A.H.; Santos, D.; Haigis, M.C. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol. Med. 2017, 23, 320–331. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, C.; Vassilopoulos, A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell 2017, 16, 1208–1218. [Google Scholar] [CrossRef]
- Watroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Age. Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [Green Version]
- Mendes, K.L.; Lelis, D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins link inflammation and metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef] [Green Version]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Olah, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Comm. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed]
- Sussmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharm. 2015, 79, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilmann, R.; Squitieri, F.; Priller, J.; Saft, C.; Mariotti, C.; Süssmuth, S.; Nemeth, A.; Tabrizi, S.; Quarrell, O.; Craufurd, D.; et al. N02 safety and tolerability of selisistat for the treatment of huntington’s disease: Results from a randomised, double-blind, placebo-controlled phase II trial. J. Neurol. Neurosurg. Psych. 2014, 85, A102. [Google Scholar] [CrossRef]
- Burns, J.; Yokota, T.; Ashihara, H.; Lean, M.E.; Crozier, A. Plant foods and herbal sources of resveratrol. J. Agric. Food Chem. 2002, 50, 3337–3340. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Forstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharm. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Kiskova, T.; Kassayova, M. Resveratrol action on lipid metabolism in cancer. Int. J. Mol. Sci. 2019, 20, 2704. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of resveratrol in cancer therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef] [Green Version]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim. Pol. 2019, 66, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Lopez, M.S.; Dempsey, R.J.; Vemuganti, R. Resveratrol neuroprotection in stroke and traumatic CNS injury. Neurochem. Int. 2015, 89, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertz, M.; Nguyen, G.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Franzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef] [Green Version]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. Biol. Fate Chem. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 2010, 11, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Jin, J.; Cichewicz, R.H.; Hageman, S.A.; Ellis, T.K.; Xiang, L.; Peng, Q.; Jiang, M.; Arbez, N.; Hotaling, K.; et al. trans-(−)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J. Biol. Chem. 2012, 287, 24460–24472. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; DiStefano, P.S.; Fields, S.; et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [Google Scholar] [CrossRef] [Green Version]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Kustigian, L.; Carney, D.; Case, A.; Considine, T.; Hubbard, B.P.; Perni, R.B.; Riera, T.V.; Szczepankiewicz, B.; Vlasuk, G.P.; et al. SIRT1 activation by small molecules: Kinetic and biophysical evidence for direct interaction of enzyme and activator. J. Biol. Chem. 2010, 285, 32695–32703. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Wang, M.; Qiu, X.; Liu, D.; Jiang, H.; Yang, N.; Xu, R.M. Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 2015, 29, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Baur, J.A.; Chen, A.; Miller, C.; Adams, J.K.; Kisielewski, A.; Howitz, K.T.; Zipkin, R.E.; Sinclair, D.A. Design and synthesis of compounds that extend yeast replicative lifespan. Aging Cell 2007, 6, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.T.; Gertz, M.; Steegborn, C. Crystal structures of Sirt3 complexes with 4′-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem. Biol. 2013, 20, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalba, J.M.; de Cabo, R.; Alcain, F.J. A patent review of sirtuin activators: An update. Expert Opin. Ther. Pat. 2012, 22, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Borghi, S.M.; Carvalho, T.T.; Staurengo-Ferrari, L.; Hohmann, M.S.; Pinge-Filho, P.; Casagrande, R.; Verri, W.A., Jr. Vitexin inhibits inflammatory pain in mice by targeting TRPV1, oxidative stress, and cytokines. J. Nat. Prod. 2013, 76, 1141–1149. [Google Scholar] [CrossRef]
- El-Helw, E.A.; Derbala, H.A.; El-Shahawi, M.M.; Salem, M.; Mamdouh, M. Synthesis and In Vitro Antitumor Activity of Novel Chromenones Bearing Benzothiazole Moiety. Russ. J. Bioorg. Chem. 2019, 45, 42–53. [Google Scholar] [CrossRef]
- Rastegari, A.; Nadri, H.; Mahdavi, M.; Moradi, A.; Mirfazli, S.S.; Edraki, N.; Moghadam, F.H.; Larijani, B.; Akbarzadeh, T.; Saeedi, M. Design, synthesis and anti-Alzheimer’s activity of novel 1,2,3-triazole-chromenone carboxamide derivatives. Bioorganic Chem. 2019, 83, 391–401. [Google Scholar] [CrossRef]
- Gaspar, A.; Matos, M.J.; Garrido, J.; Uriarte, E.; Borges, F. Chromone: A valid scaffold in medicinal chemistry. Chem. Rev. 2014, 114, 4960–4992. [Google Scholar] [CrossRef] [Green Version]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Friden-Saxin, M.; Seifert, T.; Landergren, M.R.; Suuronen, T.; Lahtela-Kakkonen, M.; Jarho, E.M.; Luthman, K. Synthesis and evaluation of substituted chroman-4-one and chromone derivatives as sirtuin 2-selective inhibitors. J. Med. Chem 2012, 55, 7104–7113. [Google Scholar] [CrossRef]
- Zhuo, R.; Liu, H.; Liu, N.; Wang, Y. Ligand Fishing: A Remarkable Strategy for Discovering Bioactive Compounds from Complex Mixture of Natural Products. Molecules 2016, 21, 1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, M.; Wilson, D.R.; Fugmann, S.D.; Moaddel, R. Synthesis and characterization of SIRT6 protein coated magnetic beads: Identification of a novel inhibitor of SIRT6 deacetylase from medicinal plant extracts. Anal. Chem. 2011, 83, 7400–7407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Ravichandran, S.; Spelman, K.; Fugmann, S.D.; Moaddel, R. The identification of a novel SIRT6 modulator from Trigonella foenum-graecum using ligand fishing with protein coated magnetic beads. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 968, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Xue, N.; Wu, F.; He, Y.; Zhang, G.; Hu, Z.; Cui, H. Exploration of the Fluorescent Properties and the Modulated Activities against Sirtuin Fluorogenic Assays of Chromenone-Derived Natural Products. Molecules 2018, 23, 1063. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, M.; Andrianasolo, E.H.; Shin, W.K.; Goeger, D.E.; Yokochi, A.; Schemies, J.; Jung, M.; France, D.; Cornell-Kennon, S.; Lee, E.; et al. Structural and synthetic investigations of tanikolide dimer, a SIRT2 selective inhibitor, and tanikolide seco-acid from the Madagascar marine cyanobacterium Lyngbya majuscula. J. Org. Chem. 2009, 74, 5267–5275. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.P.; Milligan, K.E.; Gerwick, W.H. Tanikolide, a toxic and antifungal lactone from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 1999, 62, 1333–1335. [Google Scholar] [CrossRef]
- Kahyo, T.; Ichikawa, S.; Hatanaka, T.; Yamada, M.K.; Setou, M. A novel chalcone polyphenol inhibits the deacetylase activity of SIRT1 and cell growth in HEK293T cells. J. Pharmacol. Sci. 2008, 108, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Karaman, B.; Alhalabi, Z.; Swyter, S.; Mihigo, S.O.; Andrae-Marobela, K.; Jung, M.; Sippl, W.; Ntie-Kang, F. Identification of Bichalcones as Sirtuin Inhibitors by Virtual Screening and In Vitro Testing. Molecules 2018, 23, 416. [Google Scholar] [CrossRef] [Green Version]
- Masesane, I.B.; Yeboah, S.O.; Liebscher, J.; Mügge, C.; Abegaz, B.M. A bichalcone from the twigs of Rhus pyroides. Phytochemistry 2000, 53, 1005–1008. [Google Scholar] [CrossRef]
- Mdee, L.K.; Yeboah, S.O.; Abegaz, B.M. Rhuschalcones II-VI, five new bichalcones from the root bark of Rhus pyroides. J. Nat. Prod. 2003, 66, 599–604. [Google Scholar] [CrossRef]
- Shetonde, O.; Mdee, L.; Bezabih, M.; Marobela, K.; Mammo, W.; Abegaz, B. The characterization, total synthesis and antiprotozoal activities of novel bichalcones from Rhus pyroides. Planta Med. 2009, 75, SL11. [Google Scholar] [CrossRef]
- Mihigo, S.O.; Mammo, W.; Bezabih, M.; Andrae-Marobela, K.; Abegaz, B.M. Total synthesis, antiprotozoal and cytotoxicity activities of rhuschalcone VI and analogs. Bioorganic Med. Chem. 2010, 18, 2464–2473. [Google Scholar] [CrossRef] [PubMed]
- Arslan, T.; Celik, G.; Celik, H.; Senturk, M.; Yayli, N.; Ekinci, D. Synthesis and Biological Evaluation of Novel Bischalcone Derivatives as Carbonic Anhydrase Inhibitors. Arch. Der Pharm. 2016, 349, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Svenningsen, A.B.; Madsen, K.D.; Liljefors, T.; Stafford, G.I.; van Staden, J.; Jager, A.K. Biflavones from Rhus species with affinity for the GABA(A)/benzodiazepine receptor. J. Ethnopharmacol. 2006, 103, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Azminah, A.; Erlina, L.; Radji, M.; Mun’im, A.; Syahdi, R.R.; Yanuar, A. In silico and in vitro identification of candidate SIRT1 activators from Indonesian medicinal plants compounds database. Comput. Biol. Chem. 2019, 83, 107096. [Google Scholar] [CrossRef]
- Yanuar, A.; Mun’im, A.; Lagho, A.B.A.; Syahdi, R.R.; Rahmat, M.G.; Suhartanto, H. Medicinal Plants Database and Three Dimensional Structure of the Chemical Compounds from Medicinal Plants in Indonesia. Int. J. Comput. Sci. Issue 2011, 8, 180–183. [Google Scholar]
- Naeem, S.; Hylands, P.; Barlow, D. Construction of an Indonesian herbal constituents database and its use in Random Forest modelling in a search for inhibitors of aldose reductase. Bioorganic Med. Chem. 2012, 20, 1251–1258. [Google Scholar] [CrossRef]
- Mulholland, P.J.; Ferry, D.R.; Anderson, D.; Hussain, S.A.; Young, A.M.; Cook, J.E.; Hodgkin, E.; Seymour, L.W.; Kerr, D.J. Pre-clinical and clinical study of QC12, a water-soluble, pro-drug of quercetin. Ann. Oncol. 2001, 12, 245–248. [Google Scholar] [CrossRef]
- Wang, Y.; Zhen, Y.; Wu, X.; Jiang, Q.; Li, X.; Chen, Z.; Zhang, G.; Dong, L. Vitexin protects brain against ischemia/reperfusion injury via modulating mitogen-activated protein kinase and apoptosis signaling in mice. Phytomedicine 2015, 22, 379–384. [Google Scholar] [CrossRef]
- Sun, Z.; Yan, B.; Yu, W.Y.; Yao, X.; Ma, X.; Sheng, G.; Ma, Q. Vitexin attenuates acute doxorubicin cardiotoxicity in rats via the suppression of oxidative stress, inflammation and apoptosis and the activation of FOXO3a. Exp. Med. 2016, 12, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhong, J.; Liu, Y.; Huang, Y.; Luo, F.; Zhou, Y.; Pan, X.; Cao, S.; Zhang, L.; Zhang, Y.; et al. Purified vitexin compound 1, a new neolignan isolated compound, promotes PUMA-dependent apoptosis in colorectal cancer. Cancer Med. 2018, 7, 6158–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, B.P.; Gomes, A.P.; Dai, H.; Li, J.; Case, A.W.; Considine, T.; Riera, T.V.; Lee, J.E.; E, S.Y.; Lamming, D.W.; et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013, 339, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef]
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An Overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar]
- Cappetta, D.; Esposito, G.; Piegari, E.; Russo, R.; Ciuffreda, L.P.; Rivellino, A.; Berrino, L.; Rossi, F.; De Angelis, A.; Urbanek, K. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int. J. Cardiol. 2016, 205, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.C.; Creaser, J.; Rourke, D.A.; Livingston, N.; Harrison, T.K.; Vandenbogaart, E.; Moriguchi, J.; Hamilton, M.A.; Tseng, C.-H.; Fonarow, G.C.; et al. Temporal trends in treatment and outcomes for advanced heart failure with reduced ejection fraction from 1993–2010: Findings from a university referral center. Circ. Heart Fail. 2013, 6, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Metra, M.; Teerlink, J.R. Heart failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- Gu, X.S.; Wang, Z.B.; Ye, Z.; Lei, J.P.; Li, L.; Su, D.F.; Zheng, X. Resveratrol, an activator of SIRT1, upregulates AMPK and improves cardiac function in heart failure. Genet. Mol. Res. 2014, 13, 323–335. [Google Scholar] [CrossRef]
- Rimbaud, S.; Ruiz, M.; Piquereau, J.; Mateo, P.; Fortin, D.; Veksler, V.; Garnier, A.; Ventura-Clapier, R. Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS ONE 2011, 6, e26391. [Google Scholar] [CrossRef] [Green Version]
- Mattison, J.A.; Wang, M.; Bernier, M.; Zhang, J.; Park, S.-S.; Maudsley, S.; An, S.S.; Santhanam, L.; Martin, B.; Faulkner, S.; et al. Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab. 2014, 20, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, S.; Kaga, S.; Zhan, L.; Bagchi, D.; Das, D.K.; Bertelli, A.; Maulik, N. Resveratrol ameliorates myocardial damage by inducing vascular endothelial growth factor-angiogenesis and tyrosine kinase receptor Flk-1. Cell Biochem. Biophys. 2006, 44, 43–49. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Gomez, Y.; Mattison, J.A.; Pearson, K.J.; Martin-Montalvo, A.; Palacios, H.H.; Sossong, A.M.; Ward, T.M.; Younts, C.M.; Lewis, K.; Allard, J.S.; et al. Resveratrol improves adipose insulin signaling and reduces the inflammatory response in adipose tissue of rhesus monkeys on high-fat, high-sugar diet. Cell Metab. 2013, 18, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crandall, J.P.; Oram, V.; Trandafirescu, G.; Reid, M.; Kishore, P.; Hawkins, M.; Cohen, H.W.; Barzilai, N. Pilot study of resveratrol in older adults with impaired glucose tolerance. J. Gerontol A Biol. Sci. Med. Sci. 2012, 67, 1307–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasnyó, P.; Molnár, G.A.; Mohás, M.; Markó, L.; Laczy, B.; Cseh, J.; Mikolás, E.; Szijártó, I.A.; Mérei, A.; Halmai, R.; et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br. J. Nutr. 2011, 106, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Li, J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin. Med. Insights Cardiol. 2016, 10, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Cichoń, N.; Lach, D.; Dziedzic, A.; Bijak, M.; Saluk, J. The inflammatory processes in atherogenesis. Pol. Merkur Lek. 2017, 42, 125–128. [Google Scholar]
- Tang, Y.-L.; Chan, S.-W. A review of the pharmacological effects of piceatannol on cardiovascular diseases. Phytother. Res. 2014, 28, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Fauconneau, B.; Waffo-Teguo, P.; Huguet, F.; Barrier, L.; Decendit, A.; Merillon, J.M. Comparative study of radical scavenger and antioxidant properties of phenolic compounds from Vitis vinifera cell cultures using in vitro tests. Life Sci. 1997, 61, 2103–2110. [Google Scholar] [CrossRef]
- Hung, L.M.; Chen, J.K.; Lee, R.S.; Liang, H.C.; Su, M.J. Beneficial effects of astringinin, a resveratrol analogue, on the ischemia and reperfusion damage in rat heart. Free Radic. Biol. Med. 2001, 30, 877–883. [Google Scholar] [CrossRef]
- Kershaw, J.; Kim, K.-H. The Therapeutic Potential of Piceatannol, a Natural Stilbene, in Metabolic Diseases: A Review. J. Med. Food 2017, 20, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Hijona, E.; Aguirre, L.; Pérez-Matute, P.; Villanueva-Millán, M.J.; Mosqueda-Solis, A.; Hasnaoui, M.; Nepveu, F.; Senard, J.M.; Bujanda, L.; Aldámiz-Echevarría, L.; et al. Limited beneficial effects of piceatannol supplementation on obesity complications in the obese Zucker rat: Gut microbiota, metabolic, endocrine, and cardiac aspects. J. Physiol. Biochem. 2016, 72, 567–582. [Google Scholar] [CrossRef]
- Tung, Y.-C.; Lin, Y.-H.; Chen, H.-J.; Chou, S.-C.; Cheng, A.-C.; Kalyanam, N.; Ho, C.-T.; Pan, M.-H. Piceatannol Exerts Anti-Obesity Effects in C57BL/6 Mice through Modulating Adipogenic Proteins and Gut Microbiota. Molecules (BaselSwitz.) 2016, 21, 1419. [Google Scholar] [CrossRef] [Green Version]
- Minakawa, M.; Miura, Y.; Yagasaki, K. Piceatannol, a resveratrol derivative, promotes glucose uptake through glucose transporter 4 translocation to plasma membrane in L6 myocytes and suppresses blood glucose levels in type 2 diabetic model db/db mice. Biochem. Biophys. Res. Commun. 2012, 422, 469–475. [Google Scholar] [CrossRef]
- Uchida-Maruki, H.; Inagaki, H.; Ito, R.; Kurita, I.; Sai, M.; Ito, T. Piceatannol lowers the blood glucose level in diabetic mice. Biol. Pharm. Bull. 2015, 38, 629–633. [Google Scholar] [CrossRef] [Green Version]
- Oritani, Y.; Okitsu, T.; Nishimura, E.; Sai, M.; Ito, T.; Takeuchi, S. Enhanced glucose tolerance by intravascularly administered piceatannol in freely moving healthy rats. Biochem. Biophys. Res. Commun. 2016, 470, 753–758. [Google Scholar] [CrossRef]
- Seyed, M.A.; Jantan, I.; Bukhari, S.N.A.; Vijayaraghavan, K. A Comprehensive Review on the Chemotherapeutic Potential of Piceatannol for Cancer Treatment, with Mechanistic Insights. J. Agric. Food Chem. 2016, 64, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, M.; Fraiz, N.; Cano, E.; Orallo, F. (−)-Trans-epsilon-viniferin, a polyphenol present in wines, is an inhibitor of noradrenaline and 5-hydroxytryptamine uptake and of monoamine oxidase activity. Eur. J. Pharmacol. 2006, 542, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Vion, E.; Page, G.; Bourdeaud, E.; Paccalin, M.; Guillard, J.; Rioux Bilan, A. Trans ε-viniferin is an amyloid-β disaggregating and anti-inflammatory drug in a mouse primary cellular model of Alzheimer’s disease. Mol. Cell Neurosci. 2018, 88, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Jiang, Y.; Zhang, B.; Yang, H.; Ma, T. Resveratrol dimer trans-ε-viniferin prevents rotaviral diarrhea in mice by inhibition of the intestinal calcium-activated chloride channel. Pharm. Res. 2018, 129, 453–461. [Google Scholar] [CrossRef]
- Maher, P.; Dargusch, R.; Ehren, J.L.; Okada, S.; Sharma, K.; Schubert, D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS ONE 2011, 6, e21226. [Google Scholar] [CrossRef]
- Syed, D.N.; Adhami, V.M.; Khan, M.I.; Mukhtar, H. Inhibition of Akt/mTOR signaling by the dietary flavonoid fisetin. Anticancer Agents Med. Chem. 2013, 13, 995–1001. [Google Scholar] [CrossRef]
- Khan, N.; Syed, D.N.; Ahmad, N.; Mukhtar, H. Fisetin: A dietary antioxidant for health promotion. Antioxid. Redox Signal. 2013, 19, 151–162. [Google Scholar] [CrossRef]
- Lall, R.K.; Adhami, V.M.; Mukhtar, H. Dietary flavonoid fisetin for cancer prevention and treatment. Mol. Nutr. Food Res. 2016, 60, 1396–1405. [Google Scholar] [CrossRef]
- Tripathi, R.; Samadder, T.; Gupta, S.; Surolia, A.; Shaha, C. Anticancer activity of a combination of cisplatin and fisetin in embryonal carcinoma cells and xenograft tumors. Mol. Cancer 2011, 10, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Miles, S.L.; McFarland, M.; Niles, R.M. Molecular and physiological actions of quercetin: Need for clinical trials to assess its benefits in human disease. Nutr. Rev. 2014, 72, 720–734. [Google Scholar] [CrossRef]
- Yuan, Z.-P.; Chen, L.-J.; Fan, L.-Y.; Tang, M.-H.; Yang, G.-L.; Yang, H.-S.; Du, X.-B.; Wang, G.-Q.; Yao, W.-X.; Zhao, Q.-M.; et al. Liposomal quercetin efficiently suppresses growth of solid tumors in murine models. Clin. Cancer Res. 2006, 12, 3193–3199. [Google Scholar] [CrossRef] [Green Version]
- Ferry, D.R.; Smith, A.; Malkhandi, J.; Fyfe, D.W.; deTakats, P.G.; Anderson, D.; Baker, J.; Kerr, D.J. Phase I clinical trial of the flavonoid quercetin: Pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin. Cancer Res. 1996, 2, 659–668. [Google Scholar] [PubMed]
- Mazloom, Z.; Abdollahzadeh, S.M.; Dabbaghmanesh, M.H.; Rezaianzadeh, A. The Effect of Quercetin Supplementation on Oxidative Stress, Glycemic Control, Lipid Profile and Insulin Resistance in Type 2 Diabetes: A Randomized Clinical Trial. J. Health Sci. Surveill. Syst. 2014, 2, 8–14. [Google Scholar]
- Zahedi, M.; Ghiasvand, R.; Feizi, A.; Asgari, G.; Darvish, L. Does Quercetin Improve Cardiovascular Risk factors and Inflammatory Biomarkers in Women with Type 2 Diabetes: A Double-blind Randomized Controlled Clinical Trial. Int. J. Prev. Med. 2013, 4, 777–785. [Google Scholar] [PubMed]
- Rezvan, N.; Moini, A.; Janani, L.; Mohammad, K.; Saedisomeolia, A.; Nourbakhsh, M.; Gorgani-Firuzjaee, S.; Mazaherioun, M.; Hosseinzadeh-Attar, M.J. Effects of Quercetin on Adiponectin-Mediated Insulin Sensitivity in Polycystic Ovary Syndrome: A Randomized Placebo-Controlled Double-Blind Clinical Trial. Horm. Metab. Res. 2017, 49, 115–121. [Google Scholar] [CrossRef]
- Serban, M.-C.; Sahebkar, A.; Zanchetti, A.; Mikhailidis, D.P.; Howard, G.; Antal, D.; Andrica, F.; Ahmed, A.; Aronow, W.S.; Muntner, P.; et al. Effects of Quercetin on Blood Pressure: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2016, 5, e002713. [Google Scholar] [CrossRef] [Green Version]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Quehenberger, O.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging (Albany Ny) 2017, 9, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.; Ren, G.; Deng, C.; Zhang, J.; Luo, X.; Wu, X.; Mani, S.; Dou, W.; Wang, Z. C-glycosyl flavonoid orientin improves chemically induced inflammatory bowel disease in mice. J. Funct. Foods 2016, 21, 418–430. [Google Scholar] [CrossRef]
- Nayak, V.; Uma, P. Antioxidant and Radioprotective Effects of Ocimum Flavonoids Orientin and Vicenin in Escherichia coli. Def. Sci. J. 2006, 56. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Feng, S.; Cen, Y.; Yang, Y.; Wang, L. Study on the antibacterial and antiviral activity compositions of Trollium chinensis Bunge. J. Zhejiang Univ. (Sci. Ed.) 2004, 31, 412–415. [Google Scholar]
- Li, Y.L.; Ma, S.C.; Yang, Y.T.; Ye, S.M.; But, P.P. Antiviral activities of flavonoids and organic acid from Trollius chinensis Bunge. J. Ethnopharmacol. 2002, 79, 365–368. [Google Scholar] [CrossRef]
- Boominathan, S.P.; Sarangan, G.; Srikakulapu, S.; Rajesh, S.; Duraipandian, C.; Srikanth, P.; Jo, L. Antiviral activity of bioassay guided fractionation of plumbago zeylanica roots against herpes simplex virus type 2. World J. Pharm. Sci. 2014, 3, 1003–1017. [Google Scholar]
- Yoo, H.; Ku, S.K.; Lee, T.; Bae, J.S. Orientin inhibits HMGB1-induced inflammatory responses in HUVECs and in murine polymicrobial sepsis. Inflammation 2014, 37, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ku, S.K.; Bae, J.S. Vascular barrier protective effects of orientin and isoorientin in LPS-induced inflammation in vitro and in vivo. Vasc. Pharm. 2014, 62, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S. Inhibitory Effect of Orientin on Secretory Group IIA Phospholipase A2. Inflammation 2015, 38, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- An, F.; Wang, S.; Tian, Q.; Zhu, D. Effects of orientin and vitexin from Trollius chinensis on the growth and apoptosis of esophageal cancer EC-109 cells. Oncol. Lett. 2015, 10, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Nagai, S.; Matsumoto, C.; Shibano, M.; Fujimori, K. Suppression of Fatty Acid and Triglyceride Synthesis by the Flavonoid Orientin through Decrease of C/EBPδ Expression and Inhibition of PI3K/Akt-FOXO1 Signaling in Adipocytes. Nutrients 2018, 10, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, V.; Devi, P.U. Protection of mouse bone marrow against radiation-induced chromosome damage and stem cell death by the ocimum flavonoids orientin and vicenin. Radiat. Res. 2005, 163, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Uma Devi, P.; Ganasoundari, A.; Rao, B.S.; Srinivasan, K.K. In vivo radioprotection by ocimum flavonoids: Survival of mice. Radiat. Res. 1999, 151, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.Y.; Ling, A.P.; Koh, R.Y.; Wong, Y.P.; Say, Y.H. A Review on Medicinal Properties of Orientin. Adv. Pharm. Sci. 2016, 2016, 4104595. [Google Scholar] [CrossRef] [Green Version]
- Kwon, G.T.; Jung, J.I.; Song, H.R.; Woo, E.Y.; Jun, J.G.; Kim, J.K.; Her, S.; Park, J.H. Piceatannol inhibits migration and invasion of prostate cancer cells: Possible mediation by decreased interleukin-6 signaling. J. Nutr. Biochem. 2012, 23, 228–238. [Google Scholar] [CrossRef]
- Rimando, A.M.; Nagmani, R.; Feller, D.R.; Yokoyama, W. Pterostilbene, a new agonist for the peroxisome proliferator-activated receptor alpha-isoform, lowers plasma lipoproteins and cholesterol in hypercholesterolemic hamsters. J. Agric. Food Chem. 2005, 53, 3403–3407. [Google Scholar] [CrossRef]
- Hamdy, A.A.; Ibrahem, M.A. Management of aphthous ulceration with topical quercetin: A randomized clinical trial. J. Contemp Dent. Pr. 2010, 11, E009–E016. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.A.; Henson, D.A.; Austin, M.D.; Jin, F.; Nieman, D.C. Quercetin supplementation and upper respiratory tract infection: A randomized community clinical trial. Pharm. Res. 2010, 62, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Amirchaghmaghi, M.; Delavarian, Z.; Iranshahi, M.; Shakeri, M.T.; Mosannen Mozafari, P.; Mohammadpour, A.H.; Farazi, F.; Iranshahy, M. A Randomized Placebo-controlled Double Blind Clinical Trial of Quercetin for Treatment of Oral Lichen Planus. J. Dent. Res. Dent. Clin Dent. Prospect. 2015, 9, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.M.; Vestergaard, P.F.; Clasen, B.F.; Radko, Y.; Christensen, L.P.; Stødkilde-Jørgensen, H.; Møller, N.; Jessen, N.; Pedersen, S.B.; Jørgensen, J.O. High-dose resveratrol supplementation in obese men: An investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013, 62, 1186–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.V.; Martinez, M.; Stamos, M.J.; Moyer, M.P.; Planutis, K.; Hope, C.; Holcombe, R.F. Results of a phase I pilot clinical trial examining the effect of plant-derived resveratrol and grape powder on Wnt pathway target gene expression in colonic mucosa and colon cancer. Cancer Manag. Res. 2009, 1, 25–37. [Google Scholar] [PubMed]
- Magyar, K.; Halmosi, R.; Palfi, A.; Feher, G.; Czopf, L.; Fulop, A.; Battyany, I.; Sumegi, B.; Toth, K.; Szabados, E. Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin. Hemorheol. Microcirc. 2012, 50, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ma, T.; Fan, B.; Yang, L.; Han, C.; Luo, J.; Kong, L. Protective effect of trans-δ-viniferin against high glucose-induced oxidative stress in human umbilical vein endothelial cells through the SIRT1 pathway. Free Radic. Res. 2016, 50, 68–83. [Google Scholar] [CrossRef]
- Lee, C.Y.; Chien, Y.S.; Chiu, T.H.; Huang, W.W.; Lu, C.C.; Chiang, J.H.; Yang, J.S. Apoptosis triggered by vitexin in U937 human leukemia cells via a mitochondrial signaling pathway. Oncol. Rep. 2012, 28, 1883–1888. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Min, J.W.; Kong, W.L.; He, X.H.; Li, J.X.; Peng, B.W. A review on the pharmacological effects of vitexin and isovitexin. Fitoterapia 2016, 115, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, B.C.; Kim, J.H.; Sim, G.S.; Lee, D.H.; Lee, K.E.; Yun, Y.P.; Pyo, H.B. The isolation and antioxidative effects of vitexin from Acer palmatum. Arch. Pharm. Res. 2005, 28, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B.; Kim, J.H.; Cha, Y.S.; Kim, M.; Song, S.B.; Cha, D.S.; Jeon, H.; Eun, J.S.; Han, S.; Kim, D.K. Lifespan Extending and Stress Resistant Properties of Vitexin from Vigna angularis in Caenorhabditis elegans. Biomolecules (Seoul) 2015, 23, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Melo, G.O.; Muzitano, M.F.; Legora-Machado, A.; Almeida, T.A.; De Oliveira, D.B.; Kaiser, C.R.; Koatz, V.L.; Costa, S.S. C-glycosylflavones from the aerial parts of Eleusine indica inhibit LPS-induced mouse lung inflammation. Planta Med. 2005, 71, 362–363. [Google Scholar] [CrossRef]
- Kang, I.; Choi, S.; Ha, T.J.; Choi, M.; Wi, H.R.; Lee, B.W.; Lee, M. Effects of Mung Bean (Vigna radiata L.) Ethanol Extracts Decrease Proinflammatory Cytokine-Induced Lipogenesis in the KK-Ay Diabese Mouse Model. J. Med. Food 2015, 18, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Flores, G.; Dastmalchi, K.; Dabo, A.J.; Whalen, K.; Pedraza-Peñalosa, P.; Foronjy, R.F.; D’Armiento, J.M.; Kennelly, E.J. Antioxidants of therapeutic relevance in COPD from the neotropical blueberry Anthopterus wardii. Food Chem. 2012, 131, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Dos Reis, G.O.; Vicente, G.; de Carvalho, F.K.; Heller, M.; Micke, G.A.; Pizzolatti, M.G.; Fröde, T.S. Croton antisyphiliticus Mart. attenuates the inflammatory response to carrageenan-induced pleurisy in mice. Inflammopharmacology 2014, 22, 115–126. [Google Scholar] [CrossRef]
- Huang, S.T.; Chen, C.T.; Chieng, K.T.; Huang, S.H.; Chiang, B.H.; Wang, L.F.; Kuo, H.S.; Lin, C.M. Inhibitory effects of a rice hull constituent on tumor necrosis factor alpha, prostaglandin E2, and cyclooxygenase-2 production in lipopolysaccharide-activated mouse macrophages. Ann. N. Y. Acad. Sci. 2005, 1042, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.M.; Huang, S.T.; Liang, Y.C.; Lin, M.S.; Shih, C.M.; Chang, Y.C.; Chen, T.Y.; Chen, C.T. Isovitexin suppresses lipopolysaccharide-mediated inducible nitric oxide synthase through inhibition of NF-kappa B in mouse macrophages. Planta Med. 2005, 71, 748–753. [Google Scholar] [CrossRef]
- Rosa, S.I.; Rios-Santos, F.; Balogun, S.O.; Martins, D.T. Vitexin reduces neutrophil migration to inflammatory focus by down-regulating pro-inflammatory mediators via inhibition of p38, ERK1/2 and JNK pathway. Phytomedicine 2016, 23, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, M.; Paul, S.; Jakhar, R.; Kang, S.C. Potential role of vitexin in alleviating heat stress-induced cytotoxicity: Regulatory effect of Hsp90 on ER stress-mediated autophagy. Life Sci. 2015, 142, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Min, J.W.; Hu, J.J.; He, M.; Sanchez, R.M.; Huang, W.X.; Liu, Y.Q.; Bsoul, N.B.; Han, S.; Yin, J.; Liu, W.H.; et al. Vitexin reduces hypoxia-ischemia neonatal brain injury by the inhibition of HIF-1alpha in a rat pup model. Neuropharmacology 2015, 99, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Kim, S.Y.; Hong, Y.M.; Jo, D.G.; Lee, J.Y.; Shim, S.M.; Chung, C.W.; Seo, S.J.; Yoo, Y.J.; Koh, J.Y.; et al. Essential role of E2-25K/Hip-2 in mediating amyloid-beta neurotoxicity. Mol. Cell 2003, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Xu, Y.Q.; Wu, J.C.; Hang, P.Z.; Wang, Y.; Wang, C.; Wu, J.W.; Qi, J.C.; Zhang, Y.; Du, Z.M. Vitexin protects against cardiac hypertrophy via inhibiting calcineurin and CaMKII signaling pathways. Naunyn Schmiedebergs Arch. Pharm. 2013, 386, 747–755. [Google Scholar] [CrossRef]
- Piccinelli, A.L.; García Mesa, M.; Armenteros, D.M.; Alfonso, M.A.; Arevalo, A.C.; Campone, L.; Rastrelli, L. HPLC-PDA-MS and NMR characterization of C-glycosyl flavones in a hydroalcoholic extract of Citrus aurantifolia leaves with antiplatelet activity. J. Agric. Food Chem. 2008, 56, 1574–1581. [Google Scholar] [CrossRef]
- Choo, C.Y.; Sulong, N.Y.; Man, F.; Wong, T.W. Vitexin and isovitexin from the Leaves of Ficus deltoidea with in-vivo α-glucosidase inhibition. J. Ethnopharmacol. 2012, 142, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Galleano, M.; Calabro, V.; Prince, P.D.; Litterio, M.C.; Piotrkowski, B.; Vazquez-Prieto, M.A.; Miatello, R.M.; Oteiza, P.I.; Fraga, C.G. Flavonoids and metabolic syndrome. Ann. N. Y. Acad. Sci. 2012, 1259, 87–94. [Google Scholar] [CrossRef]
- Gaitan, E.; Lindsay, R.H.; Reichert, R.D.; Ingbar, S.H.; Cooksey, R.C.; Legan, J.; Meydrech, E.F.; Hill, J.; Kubota, K. Antithyroid and goitrogenic effects of millet: Role of C-glycosylflavones. J. Clin. Endocrinol. Metab. 1989, 68, 707–714. [Google Scholar] [CrossRef]
- Schloms, L.; Swart, A.C. Rooibos flavonoids inhibit the activity of key adrenal steroidogenic enzymes, modulating steroid hormone levels in H295R cells. Molecules 2014, 19, 3681–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quílez, A.; Berenguer, B.; Gilardoni, G.; Souccar, C.; de Mendonça, S.; Oliveira, L.F.; Martín-Calero, M.J.; Vidari, G. Anti-secretory, anti-inflammatory and anti-Helicobacter pylori activities of several fractions isolated from Piper carpunya Ruiz & Pav. J. Ethnopharmacol. 2010, 128, 583–589. [Google Scholar] [CrossRef]
- Krcatović, E.; Rusak, G.; Bezić, N.; Krajacić, M. Inhibition of tobacco mosaic virus infection by quercetin and vitexin. Acta Virol. 2008, 52, 119–124. [Google Scholar] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound (PCID *) | Health Benefits | Mode of Action | References |
|---|---|---|---|
| Fisetin (5281614) | Antiaging and against cardiovascular disease | -Demonstrates senotherapeutic activity in mice and in human tissues. -Reduces the levels of the cyclin-dependent kinase 5 (Cdk5) activator p35 cleavage product, p25, in both control and Alzheimer’s disease brains. -Elevates the levels of p25 relative to p35, causing dysregulation of Cdk5 activity leading to neuroinflammation and neurodegeneration. | [109,177,178] |
| Anticarcinogenic agent, chemopreventive/ chemotherapeutic agent against cancer | -Activates caspases-7 and -9. -Increases expression of proapoptotic protein Bak and induces its mitochondrial oligomerization. -Inhibits of Akt/mTOR signaling | [166,168,169] | |
| Antioxidant agent | Not well known | [167] | |
| Antidiabetic | Lowers methylglyoxal dependent protein glycation | [165] | |
| Orientin (5281675) | Anti-inflammatory | -Decreases the activity of myeloperoxidase (MPO) and the production of cytokines in rats. -Inhibits the nuclear translocation of nuclear factor kappa B (NF-κB) p65, the activity of NF-κB-luciferase, and the expression of NF-κB target genes. -Attenuates experimental inflammatory bowel disease (IBD) via suppression of TLR4 and inactivation of NF-κB and MAPK pathways in rats. | [179] |
| Antioxidant and Antiaging | Reduces the H2O2-induced β-galactosidase activity | [180] | |
| Antiviral and antibacterial agent | -Shows moderate or potent antiviral activity against Para 3 virus. -The flavonoid mixture (containing orientin, rutin, quercetin, and kaempferol) at the maximum nontoxic dose of 0.048 μg/mL was shown to fully inhibit Herpes Simplex Virus Type 2 (HSV-2) of different viral titre (1, 10, 100 TCID50) on Hep-2 cells. | [181,182,183] | |
| Anti-inflammatory agent | -Inhibits the high mobility group box-1 (HMGB1) protein level in lipopolysaccharide- (LPS-) induced umbilical vein endothelial cells (HUVECs) as well as the HMGB1-mediated cytoskeletal rearrangements. -Suppresses LPS-induced membrane disruption, migration of monocytes, expression of cell adhesion molecules (CAMs), and LPS-induced EPCR detaching. | [184,185,186] | |
| Anticancer effects | -Regulates the apoptosis-related gene expression of p53 and bcl-2. -May serve as therapeutic agents for the treatment of esophageal cancer. | [187] | |
| Weight loss | -Represses the accumulation of intracellular triglyceride (TG) in mouse adipocyte 3T3-L1 cells. -Decreases the mRNA levels of the genes involved in adipogenesis, lipogenesis, lipolysis, and TG synthesis, and reduces the release of glycerol. -Lowers the expression of CCAAT/enhancer binding protein (C/EBP) δ in the early stage of adipogenesis, leading to a decrease in the expression of the adipogenic master transcription factors such as peroxisome proliferator-activated receptor (PPAR) γ and C/EBP. | [188] | |
| Protection against bone marrow damage | Reduces chromosomal aberration cells in bone marrow | [189,190] | |
| Other (e.g., vasodilatation, cardioprotective, radioprotective, neuroprotective, antidepressant-like, antiadipogenesis, antinociceptive, etc., effects) | -Inhibits the protein expressions of C/EBPα and PPARγ -Reduces acetic acidinduced writhing and capsaicin- and glutamate-induced pain in mice -Orientin was shown to be 20-fold more potent than the typical painkiller, acetylsalicylic acid (aspirin), and 3.5-fold more dynamic than the common anti-inflammatory drug, indomethacin | [191] | |
| Piceatannol (667639) | Anticancer effects | -Inhibits migration and invasion of prostate cancer cells, possibly mediated by decreased interleukin-6 signaling. -Inhibits COX-1/2 and CSN-associated kinase. | [161,192] |
| Metabolic diseases | -Inhibits adipogenesis. -Blocks mitotic clonal expansion. -Inhibits insulin signaling due to non-competitive binding to insulin receptor. -Lowers lipid accumulation during late stages of differentiation. -Etc. | [155] | |
| Cardiovascular diseases | -Activates a nuclear receptor, peroxisome proliferator-activated receptor alpha (PPAR-α) isoform, on rat hepatoma (H4IIEC3 cells) in vitro. -Lowers lipid and lipoprotein in vivo. -Etc. | [152,193] | |
| Quercetin (5280343) | Cancer treatment | Tyrosine kinase inhibition in vivo | [171,172] |
| Treatment of colitis and gastric ulcer | Not well known | [194] | |
| Treatment of respiratory tract infection | Not well known | [195] | |
| Treatment of type 2 diabetes | Was shown to be beneficial in improving the antioxidant status of patients with type 2 diabetes while having no other significant effect on glycemic control and lipid profile | [173,174,175] | |
| Treatment of high blood pressure | -Mode of action still under investigation | [176] | |
| Treatment of oral lichen planus | -Restriction of cyto-kines including IL12, INFγ, INF α, IL8, cyclooxi-genase 2 and prostaglandin E | [196] | |
| Other health benefits | Not well known | [170] | |
| Resveratrol (445154) | Obesity treatment | Sirtuin modulation | [197] |
| Colon cancer prevention | Inhibits a key signaling pathway involved in colon cancer initiation, the Wnt pathway, in vitro. | [198] | |
| Cardioprotection | Decreases low-density lipoprotein (LDL) oxidation, and functions as a direct free radical scavenger. | [199] | |
| trans-(−)-ϵ-Viniferin (5315232) | Anti-inflammatory, antioxidant, platelet antiaggregatory and anticarcinogenic properties | Monoamine oxidase activity | [162] |
| Prevention of rotaviral diarrhea | Inhibition of the intestinal calcium-activated chloride channel | [164] | |
| Potential for the treatment of Alzheimer’s disease. | Induces the disaggregation of amyloid β (Aβ) peptide | [163] | |
| Potential for the treatment of diabetes. | Inhibition of high glucose-induced apoptosis by maintaining Ca2+ and preserving mitochondrial membrane potential (MMP) levels. | [200] | |
| Vitexin (5280441) | Anticancer effects | -Regulates the apoptosis-related gene expression of p53 and bcl-2 -May serve as therapeutic agents for the treatment of esophageal cancer -Targeting cell apoptosis in U937 cells | [187,201] |
| Anti-oxidant effects | Subdues oxygen free radical and protecting the antioxidant enzyme activity in cells and the sulfhydryl in the red cell membrane protein. | [202,203] | |
| Lifespan extending and stress resistant properties | Reduces intracellular reactive oxygen species (ROS) accumulation in a dose-dependent manner | [204] | |
| Anti-inflammatory effects | -Inhibit IL-1β, IL-6, IL-8, IL-17, and IL-33 -Inhibit tumor necrosis factor-α (TNF-α) secretion -Inhibit COX-2 -Inhibit NF-κB activation -Inhibit iNOS (inducible nitric oxide synthase) -Inhibit NO, PGE2, monocyte chemoattractant protein-1 (MCP-1), and neutrophil influx -Increase in IL-10 and reduce the expression of p-p38, p-ERK and p-JNK. | [105,205,206,207,208,209,210,211] | |
| Anti-neoplastic effects | -Promotes autophagy through the up-regulation of Hsp90 expression and subsequent activation of endoplasmic reticulum (ER)-stress -Hsp90 may be involved in a new signaling pathway with anti-neoplastic effects of vitexin | [212] | |
| Protective effects against neurological and psychiatric diseases (e.g., hypoxia and ischemia injury, Alzheimer’s disease, learning, cognition and depression, Anti-nociceptive activity, Other neurological and psychiatric disorders, etc.) | -Helps to maintain blood-brain barrier (BBB) integrity and attenuate brain oedema with down-regulated HIF1-α and VEGF -Contributes to the protection against cerebral I/R injury, and it up-regulated p-ERK1/2, down-regulated p-JNK and p-P38, as well as increased Bcl-2 and decreased Bax expression in the cortex and hippocampus. -As a mediator of Aβ toxicity, ubiquitin- ligating enzyme E2-25 K/Hip-2 plays a role in the pathogenesis of AD | [129,201,213,214,215] | |
| Protective activity in cardiovascular system | -Inhibits the isoproterenol-induced increase in resting intracellular free calcium as well as expression of the calcium downstream effectors calcineurin- NFATc3 and phosphorylated calmodulin kinase II (CaMKII) both in vitro and in vivo -Vitexin-containing lime leaf significantly inhibited platelet aggregation in a concentration-dependent manner | [216,217] | |
| Protective effects against endocrine and metabolic disease (e.g., diabetes, obesity, other endocrine and metabolic diseases, anti-thyroid effect, etc.) | -Reduces postprandial blood glucose both in sucrose loaded normoglycemic mice and sucrose induced diabetic rats -Improves adipocyte functionality related to regulating PPARγ, carnitine palmitoyltransferase-1 and enzymes involved in lipogenesis -Inhibits thyroid peroxidase (TPO) activity -Inhibits the activities of P450 17α-hydroxylase/17 (CYP17A1), deoxycortisol conversion by P450 11β-hydroxylase (CYP11B1), but not P450 21-hydroxylase (CYP21A2) | [218,219,220,221] | |
| Anti-microbial and anti-viral effects | -Activity against anti-Helicobacter pylori effect is probably due to its activities on anti-MPO enzyme and inhibition of H+, K+-ATPase activity -Vitexin also exerts anti-phytoviral activity against Tobacco mosaic virus (TMV) | [222,223] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karaman Mayack, B.; Sippl, W.; Ntie-Kang, F. Natural Products as Modulators of Sirtuins. Molecules 2020, 25, 3287. https://doi.org/10.3390/molecules25143287
Karaman Mayack B, Sippl W, Ntie-Kang F. Natural Products as Modulators of Sirtuins. Molecules. 2020; 25(14):3287. https://doi.org/10.3390/molecules25143287
Chicago/Turabian StyleKaraman Mayack, Berin, Wolfgang Sippl, and Fidele Ntie-Kang. 2020. "Natural Products as Modulators of Sirtuins" Molecules 25, no. 14: 3287. https://doi.org/10.3390/molecules25143287