
Observation of Enhanced Dissociative Photochemistry in the Non-Native Nucleobase 2-Thiouracil

 , and
, and
Abstract
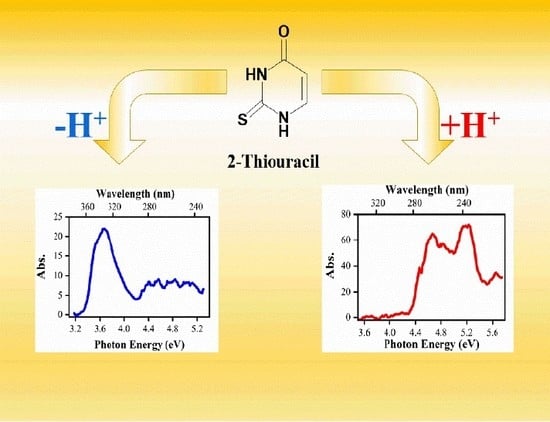
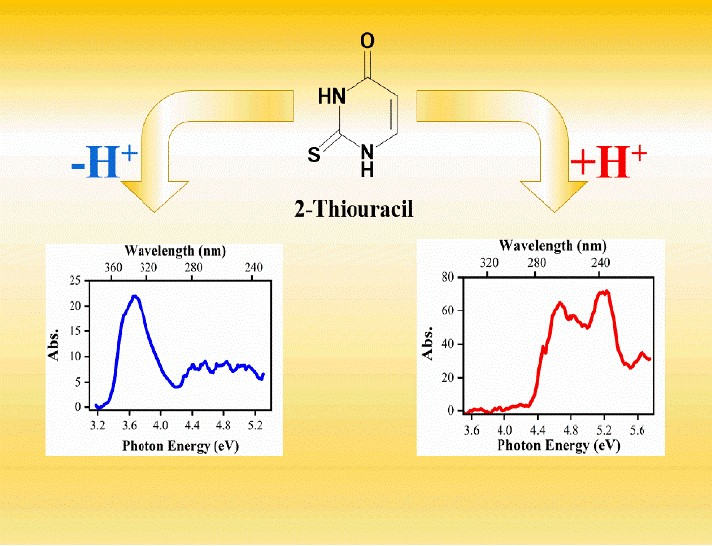
:
1. Introduction
2. Results
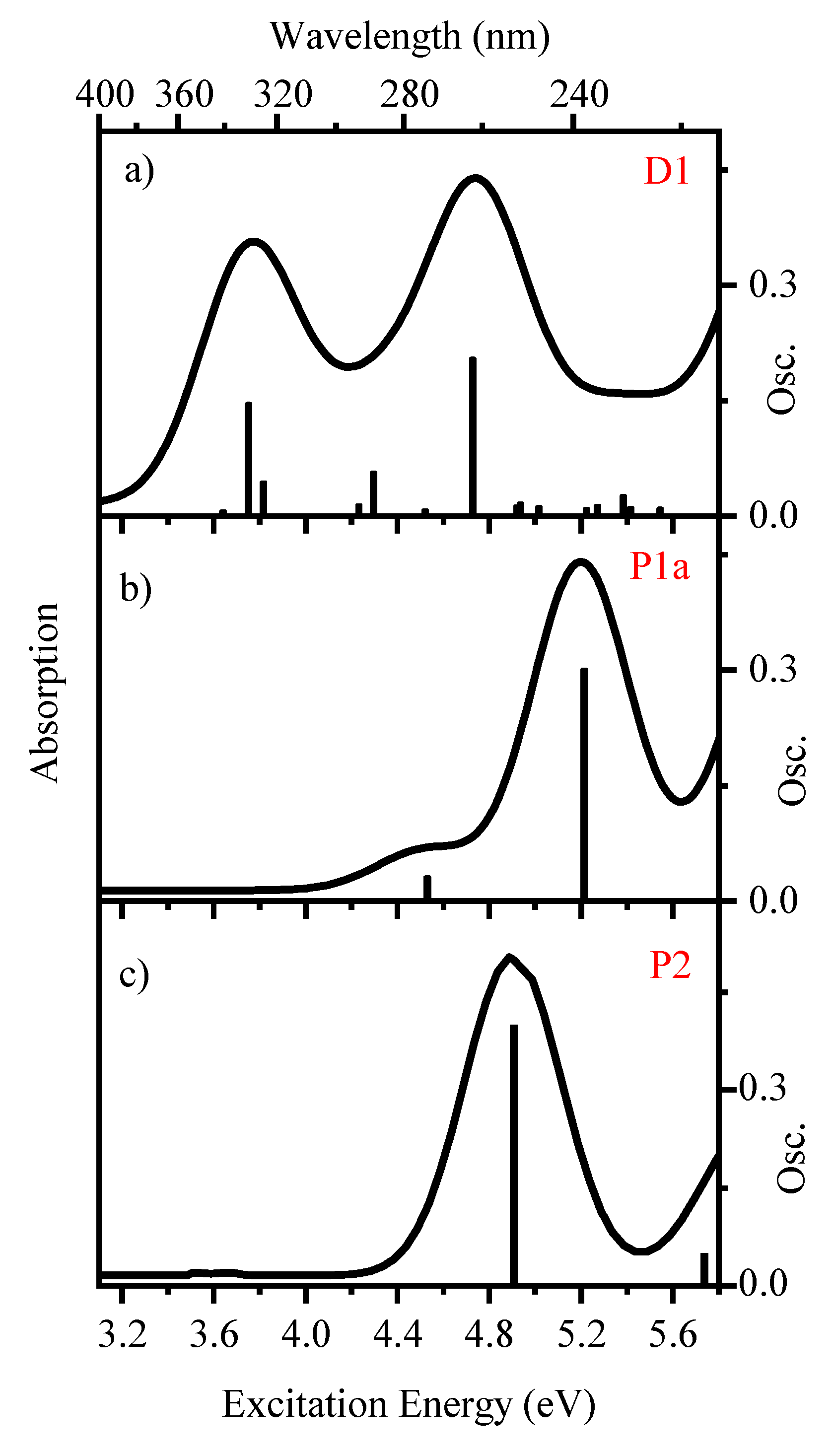
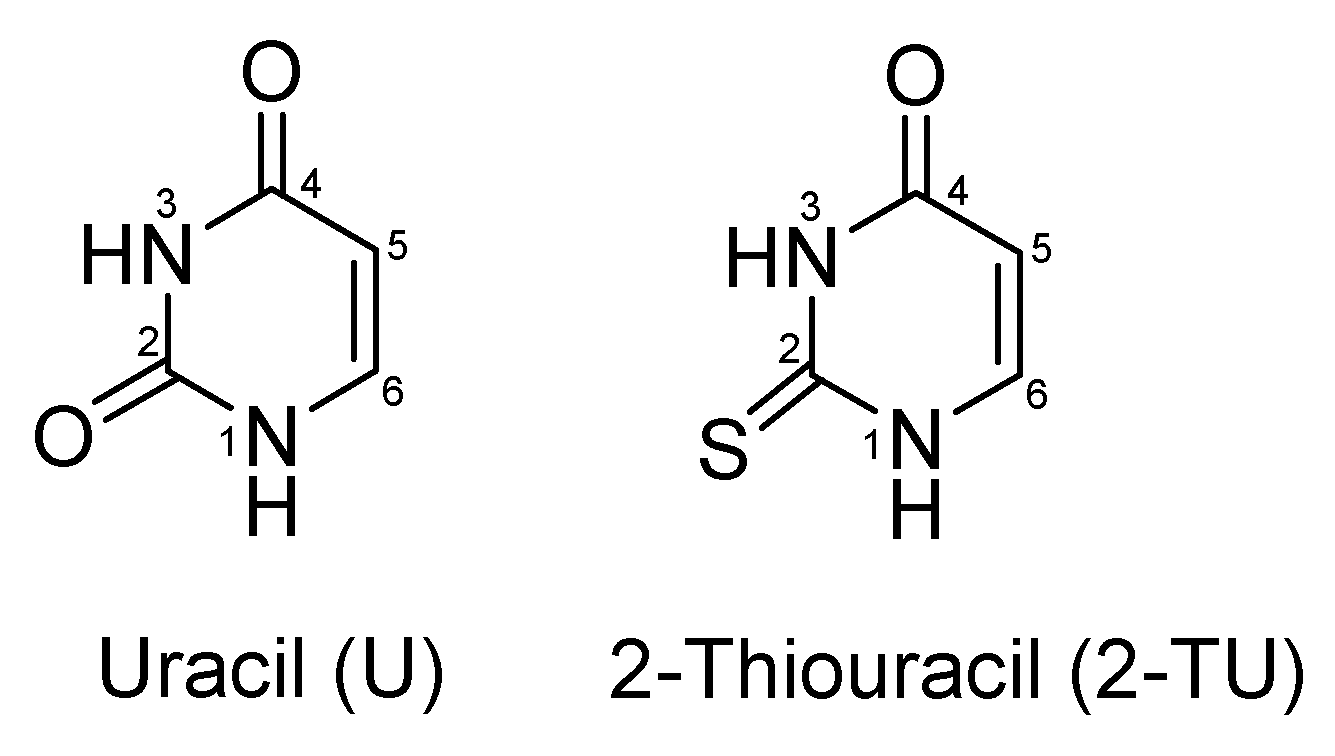
2.1. Geometric Structures and Time-Dependent Density Functional Theory (TDDFT) Calculations [2-TU-H]− and [2-TU·H]+
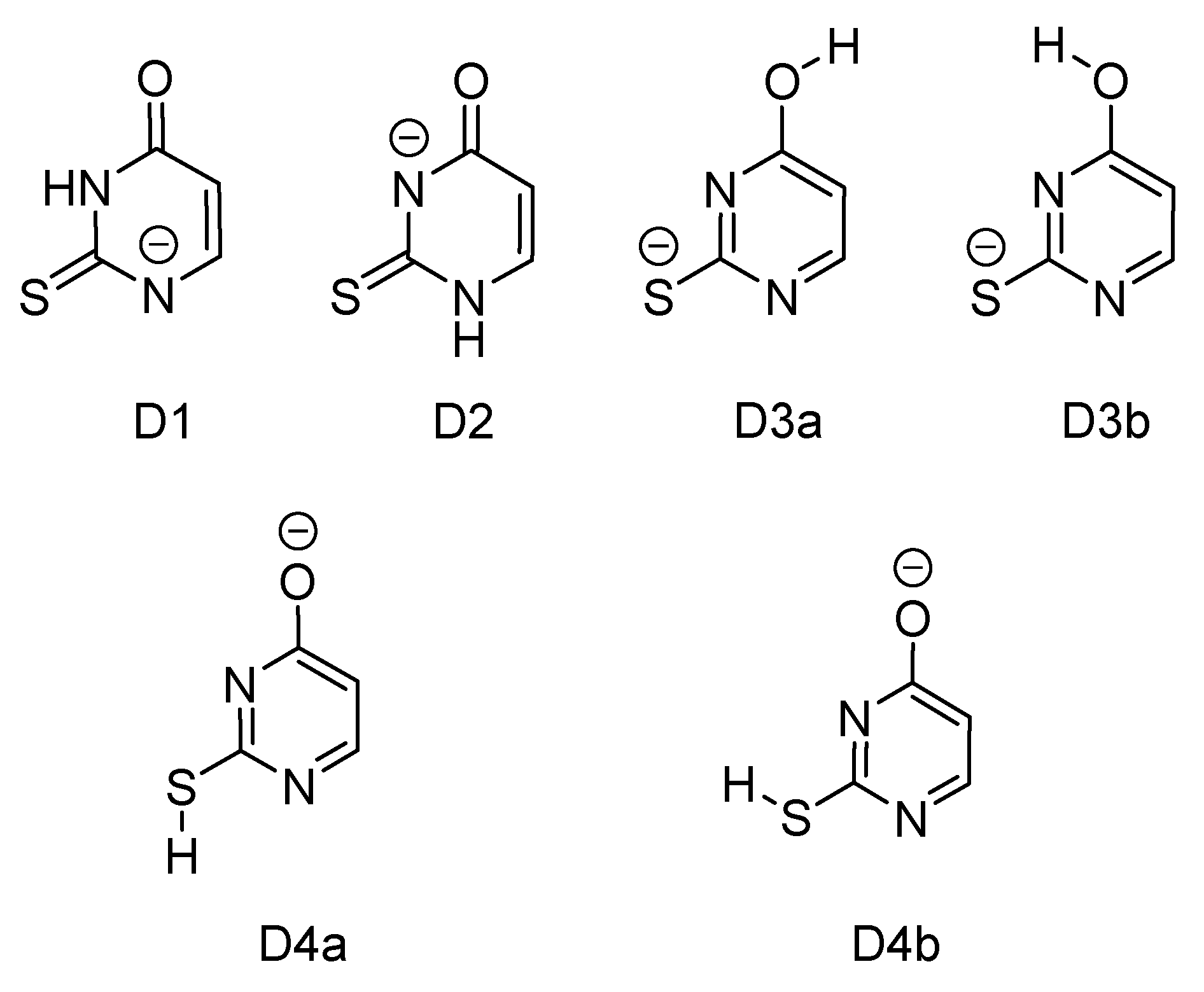
2.2. Deprotonated 2-Thiouracil
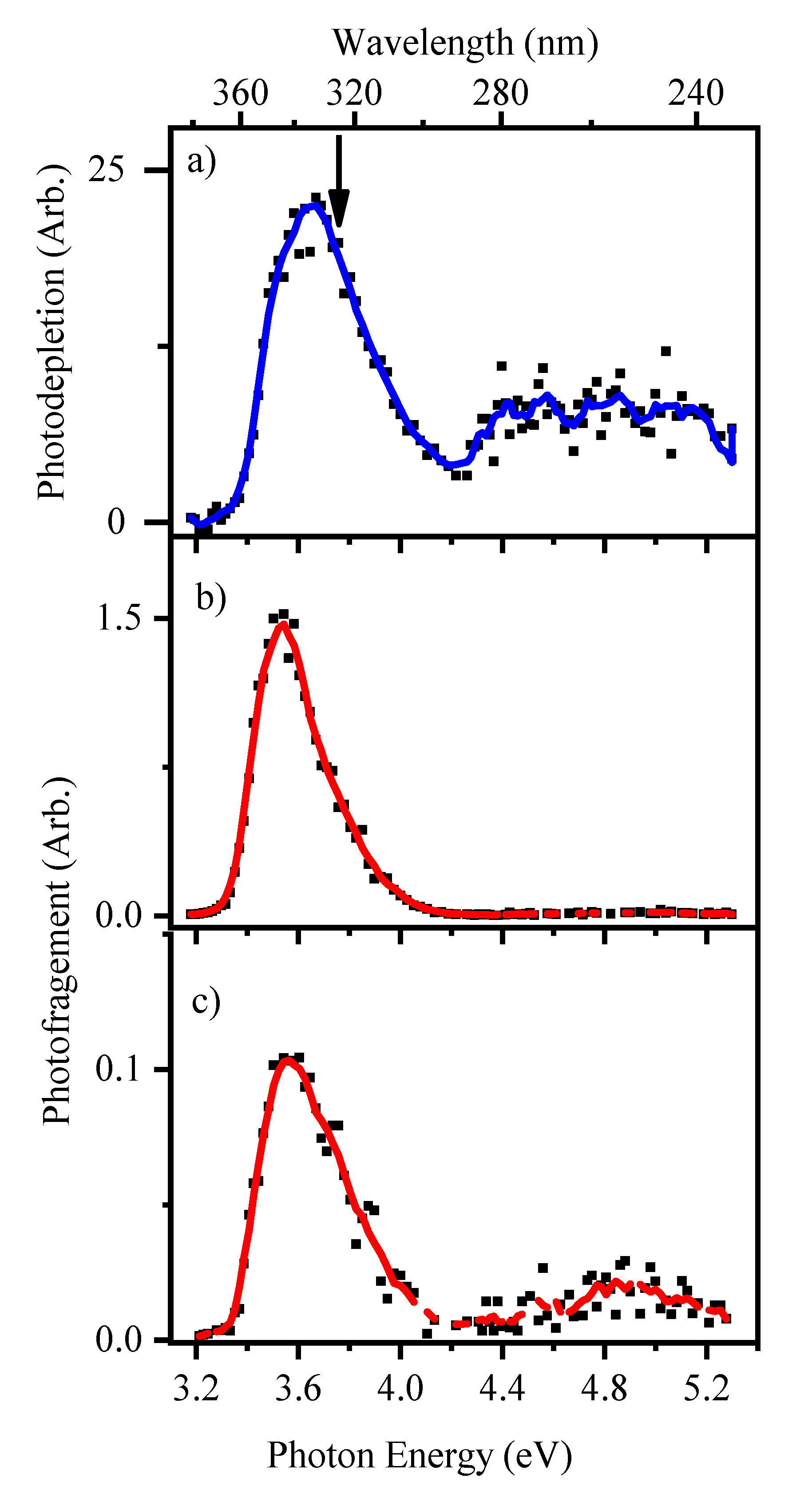
2.2.1. Gas-Phase Absorption Spectrum of Deprotonated 2-Thiouracil
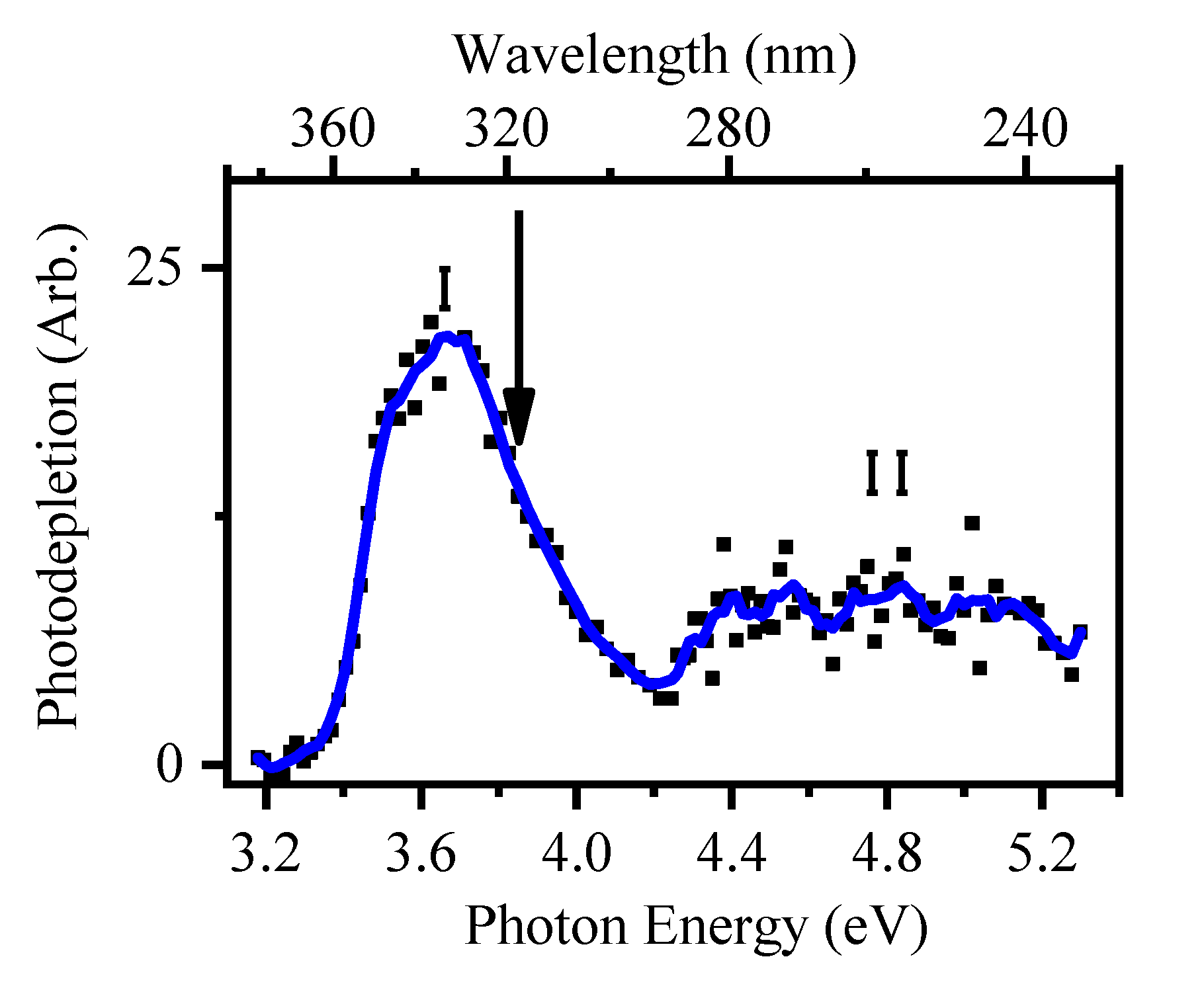
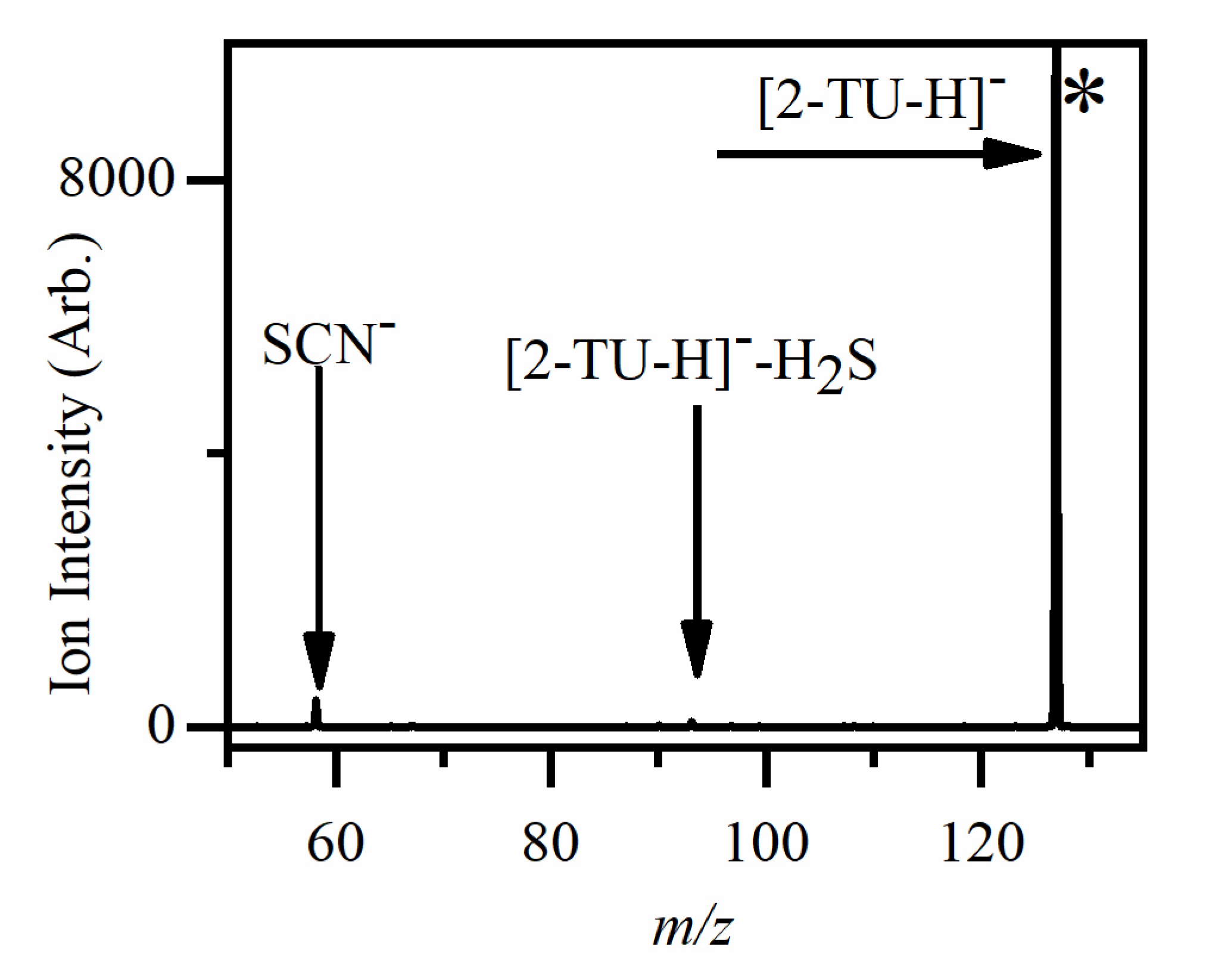
2.2.2. Photofragmentation of Deprotonated 2-Thiouracil
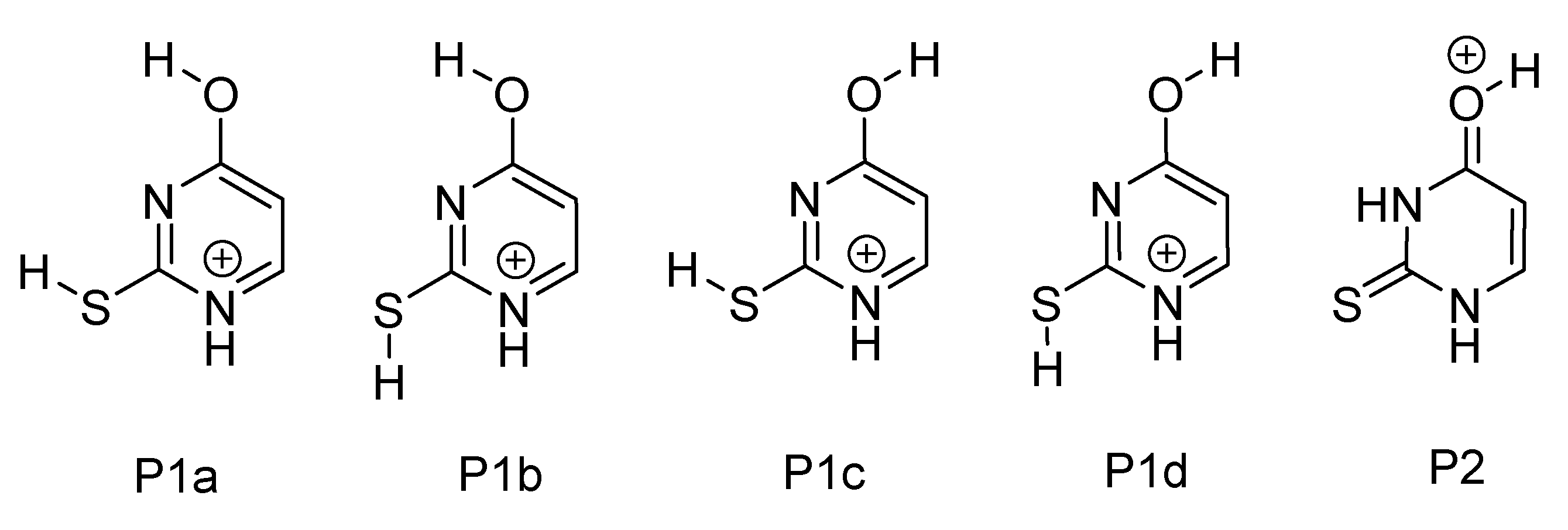
2.3. Protonated 2-Thiouracil [2-TU·H]+
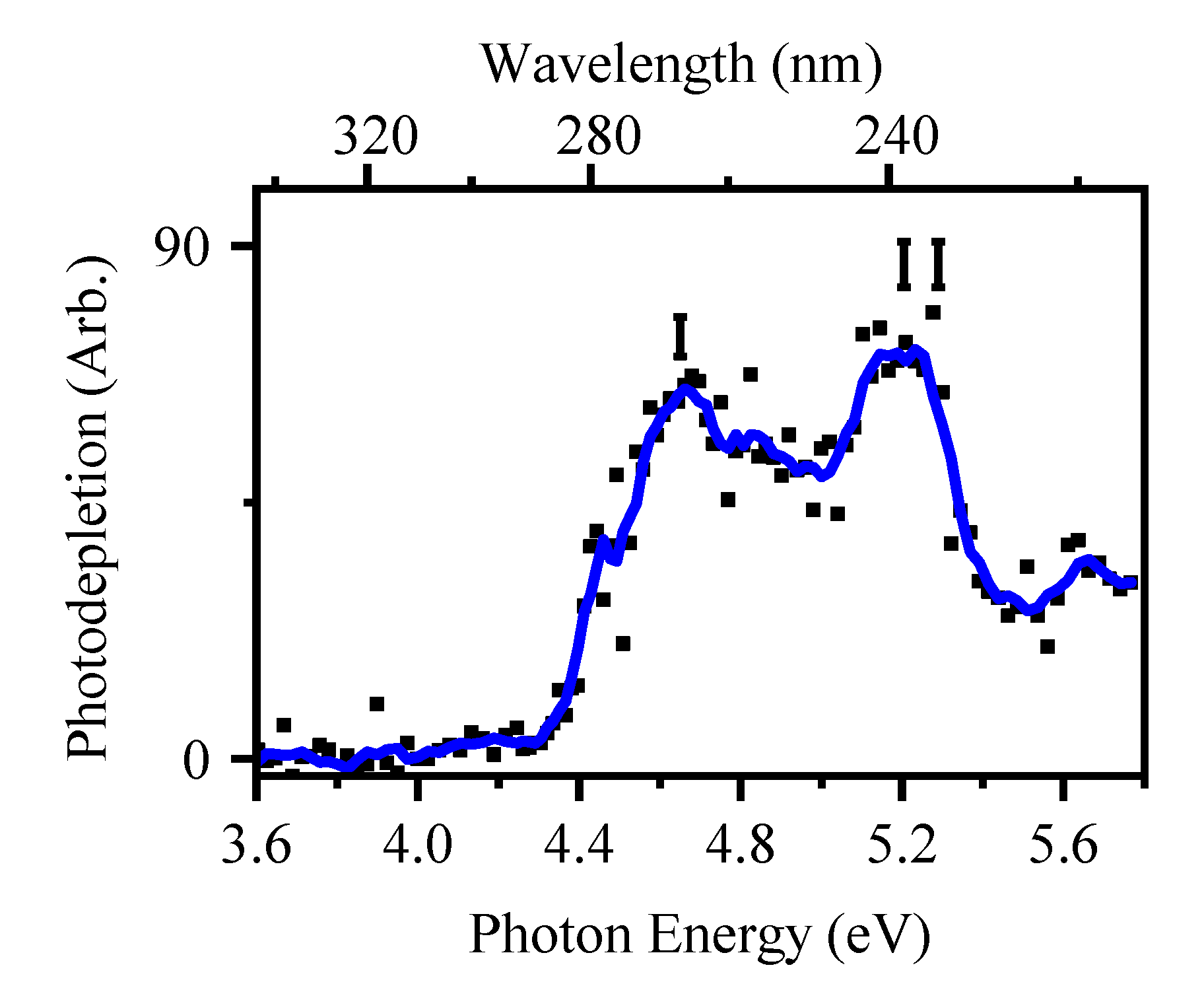
2.3.1. Gas-Phase and Solution-Phase Absorption Spectra of [2-TU·H]+
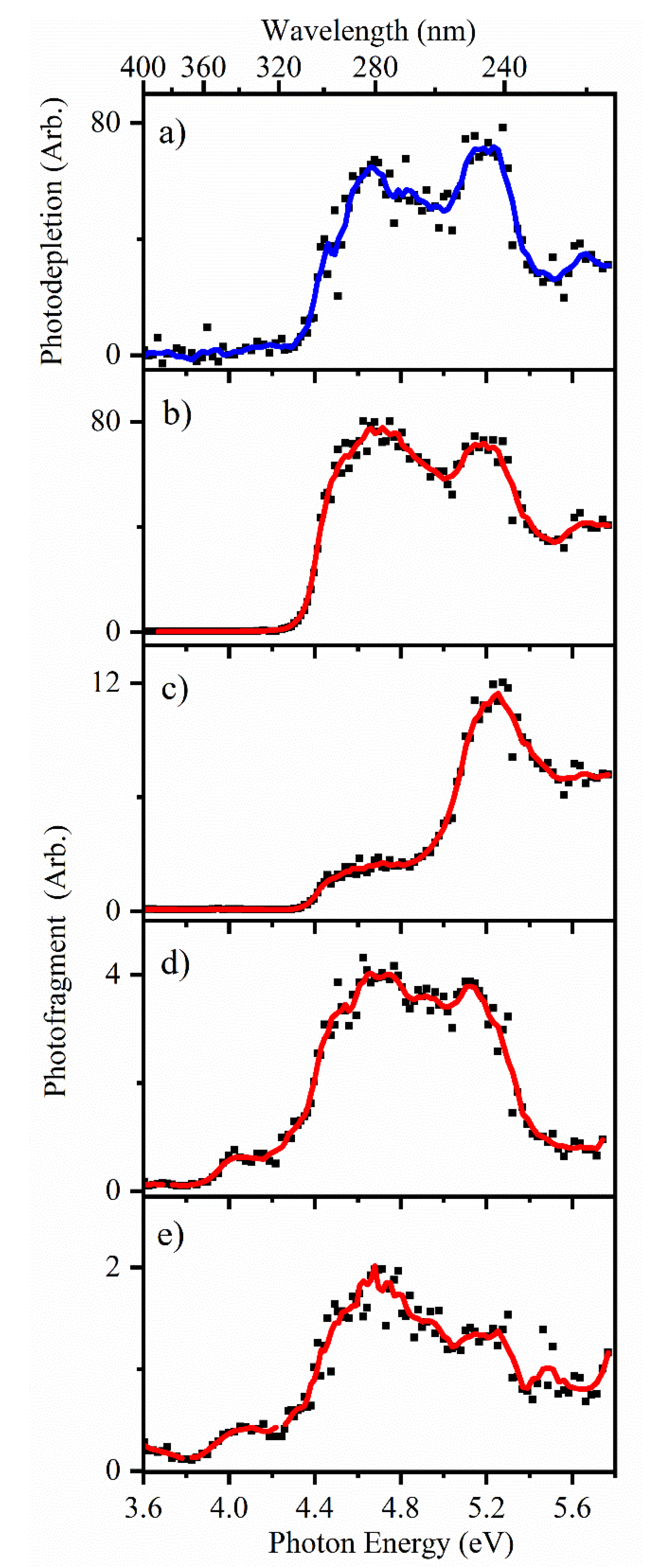
2.3.2. Photofragmentation of Protonated 2-Thiouracil
2.3.3. Comparison of Photofragmentation and HCD Fragmentation of [2-TU·H]+
3. Further Discussion
3.1. Deprotonated 2-Thiouracil
3.2. Protonated 2-Thiouracil [2-TU·H]+
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sobolewski, A.L.; Domcke, W. The chemical physics of the photostability of life. Europhys. News 2006, 37, 20–23. [Google Scholar] [CrossRef] [Green Version]
- Middleton, C.T.; de La Harpe, K.; Su, C.; Law, Y.K.; Crespo-Hernández, C.E.; Kohler, B. DNA Excited-State Dynamics: From Single Bases to the Double Helix. Annu. Rev. Phys. Chem. 2009, 60, 217–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbatti, M.; Aquino, A.J.A.; Szymczak, J.J.; Nachtigallova, D.; Hobza, P.; Lischka, H. Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases. Proc. Natl. Acad. Sci. USA 2010, 107, 21453–21458. [Google Scholar] [CrossRef] [Green Version]
- Kleinermanns, K.; Nachtigallová, D.; de Vries, M.S. Excited state dynamics of DNA bases. Int. Rev. Phys. Chem. 2013, 32, 308–342. [Google Scholar] [CrossRef]
- Spata, V.A.; Lee, W.; Matsika, S. Excimers and Exciplexes in Photoinitiated Processes of Oligonucleotides. J. Phys. Chem. Lett. 2016, 7, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum Mechanical Studies on the Photophysics and the Photochemistry of Nucleic Acids and Nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C.; Crespo-Hernández, C.E. Room-Temperature Phosphorescence of the DNA Monomer Analogue 4-Thiothymidine in Aqueous Solutions after UVA Excitation. J. Phys. Chem. Lett. 2010, 1, 2239–2243. [Google Scholar] [CrossRef]
- Harada, Y.; Okabe, C.; Kobayashi, T.; Suzuki, T.; Ichimura, T.; Nishi, N.; Xu, Y.-Z. Ultrafast Intersystem Crossing of 4-Thiothymidine in Aqueous Solution. J. Phys. Chem. Lett. 2010, 1, 480–484. [Google Scholar] [CrossRef]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. 2,4-Dithiothymine as a Potent UVA Chemotherapeutic Agent. J. Am. Chem. Soc. 2014, 136, 17930–17933. [Google Scholar] [CrossRef]
- Mai, S.; Pollum, M.; Martínez-Fernández, L.; Dunn, N.; Marquetand, P.; Corral, I.; Crespo-Hernández, C.E.; González, L. The origin of efficient triplet state population in sulfur-substituted nucleobases. Nat. Commun. 2016, 7, 13077. [Google Scholar] [CrossRef]
- Ashwood, B.; Jockusch, S.; Crespo-Hernández, C. Excited-State Dynamics of the Thiopurine Prodrug 6-Thioguanine: Can N9-Glycosylation Affect Its Phototoxic Activity? Molecules 2017, 22, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwood, B.; Pollum, M.; Crespo-Hernández, C.E. Photochemical and Photodynamical Properties of Sulfur-Substituted Nucleic Acid Bases. Photochem. Photobiol. 2019, 95, 33–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karran, P.; Attard, N. Thiopurines in current medical practice: Molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 2008, 8, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.-N.; Qi, S.; Kim, S.; Kwon, N.; Kim, G.; Yim, Y.; Park, S.; Yoon, J. An Emerging Molecular Design Approach to Heavy-Atom-Free Photosensitizers for Enhanced Photodynamic Therapy under Hypoxia. J. Am. Chem. Soc. 2019, 141, 16243–16248. [Google Scholar] [CrossRef] [PubMed]
- Reelfs, O.; Karran, P.; Young, A.R. 4-thiothymidine sensitization of DNA to UVA offers potential for a novel photochemotherapy. Photochem. Photobiol. Sci. 2012, 11, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Trigiante, G.; Xu, Y.Z. Photodynamic Therapy: Fundamentals, Applications and Health Outcomes; Hugo, A.G., Ed.; Nova Science Publishers: Hauppauge, NJ, USA, 2015. [Google Scholar]
- Farrell, K.M.; Brister, M.M.; Pittelkow, M.; Sølling, T.I.; Crespo-Hernández, C.E. Heavy-Atom-Substituted Nucleobases in Photodynamic Applications: Substitution of Sulfur with Selenium in 6-Thioguanine Induces a Remarkable Increase in the Rate of Triplet Decay in 6-Selenoguanine. J. Am. Chem. Soc. 2018, 140, 11214–11218. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Varillas, R.; Teles-Ferreira, D.C.; Nenov, A.; Conti, I.; Ganzer, L.; Manzoni, C.; Garavelli, M.; Maria de Paula, A.; Cerullo, G. Observation of the Sub-100 Femtosecond Population of a Dark State in a Thiobase Mediating Intersystem Crossing. J. Am. Chem. Soc. 2018, 140, 16087–16093. [Google Scholar] [CrossRef]
- Yu, H.; Sanchez-Rodriguez, J.A.; Pollum, M.; Crespo-Hernández, C.E.; Mai, S.; Marquetand, P.; González, L.; Ullrich, S. Internal conversion and intersystem crossing pathways in UV excited, isolated uracils and their implications in prebiotic chemistry. Phys. Chem. Chem. Phys. 2016, 18, 20168–20176. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rodríguez, J.A.; Mohamadzade, A.; Mai, S.; Ashwood, B.; Pollum, M.; Marquetand, P.; González, L.; Crespo-Hernández, C.E.; Ullrich, S. 2-Thiouracil intersystem crossing photodynamics studied by wavelength-dependent photoelectron and transient absorption spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 19756–19766. [Google Scholar] [CrossRef] [Green Version]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. Increase in the photoreactivity of uracil derivatives by doubling thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef]
- Vendrell-Criado, V.; Sáez, J.A.; Lhiaubet-Vallet, V.; Cuquerella, M.C.; Miranda, M.A. Photophysical properties of 5-substituted 2-thiopyrimidines. Photochem. Photobiol. Sci. 2013, 12, 1460. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Suzuki, T.; Ichimura, T.; Xu, Y.-Z. Triplet Formation of 4-Thiothymidine and Its Photosensitization to Oxygen Studied by Time-Resolved Thermal Lensing Technique. J. Phys. Chem. B 2007, 111, 5518–5524. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Granucci, G.; Pollum, M.; Crespo-Hernández, C.E.; Persico, M.; Corral, I. Decoding the Molecular Basis for the Population Mechanism of the Triplet Phototoxic Precursors in UVA Light-Activated Pyrimidine Anticancer Drugs. Chem. A Eur. J. 2017, 23, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Koyama, D.; Milner, M.J.; Orr-Ewing, A.J. Evidence for a Double Well in the First Triplet Excited State of 2-Thiouracil. J. Phys. Chem. B 2017, 121, 9274–9280. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; González, L.; Corral, I. An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: The case of 6-thioguanine. Chem. Commun. 2012, 48, 2134. [Google Scholar] [CrossRef]
- Cui, G.; Fang, W. State-specific heavy-atom effect on intersystem crossing processes in 2-thiothymine: A potential photodynamic therapy photosensitizer. J. Chem. Phys. 2013, 138, 044315. [Google Scholar] [CrossRef]
- Gobbo, J.P.; Borin, A.C. 2-Thiouracil deactivation pathways and triplet states population. Comput. Theor. Chem. 2014, 1040–1041, 195–201. [Google Scholar] [CrossRef]
- Mai, S.; Marquetand, P.; González, L. A Static Picture of the Relaxation and Intersystem Crossing Mechanisms of Photoexcited 2-Thiouracil. J. Phys. Chem. A 2015, 119, 9524–9533. [Google Scholar] [CrossRef] [Green Version]
- Mai, S.; Marquetand, P.; González, L. Intersystem Crossing Pathways in the Noncanonical Nucleobase 2-Thiouracil: A Time-Dependent Picture. J. Phys. Chem. Lett. 2016, 7, 1978–1983. [Google Scholar] [CrossRef] [Green Version]
- Teles-Ferreira, D.C.; Conti, I.; Borrego-Varillas, R.; Nenov, A.; Van Stokkum, I.H.M.; Ganzer, L.; Manzoni, C.; Paula, A.M.; Cerullo, G.; Garavelli, M. A Unified Experimental/Theoretical Description of the Ultrafast Photophysics of Single and Double Thionated Uracils. Chem. A Eur. J. 2020, 26, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.G.K.; Berenbeim, J.A.; Hawkridge, M.; Matthews, E.; Dessent, C.E.H. Mapping the intrinsic absorption properties and photodegradation pathways of the protonated and deprotonated forms of the sunscreen oxybenzone. Phys. Chem. Chem. Phys. 2019, 21, 14311–14321. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Dessent, C.E.H. Observation of Near-Threshold Resonances in the Flavin Chromophore Anions Alloxazine and Lumichrome. J. Phys. Chem. Lett. 2018, 9, 6124–6130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, E.; Cercola, R.; Dessent, C. Protomer-Dependent Electronic Spectroscopy and Photochemistry of the Model Flavin Chromophore Alloxazine. Molecules 2018, 23, 2036. [Google Scholar] [CrossRef] [Green Version]
- Matthews, E.; Dessent, C.E.H. Locating the proton in nicotinamide protomers via low-Resolution UV action spectroscopy of electrosprayed solutions. J. Phys. Chem. A 2016, 120, 9209–9216. [Google Scholar] [CrossRef]
- Pollum, M.; Crespo-Hernández, C.E. Communication: The dark singlet state as a doorway state in the ultrafast and efficient intersystem crossing dynamics in 2-thiothymine and 2-thiouracil. J. Chem. Phys. 2014, 140, 071101. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Fernandez, L.; Fahleson, T.; Norman, P.; Santoro, F.; Coriani, S.; Improta, R. Optical absorption and magnetic circular dichroism spectra of thiouracils: A quantum mechanical study in solution. Photochem. Photobiol. Sci. 2017, 16, 1415–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, S.; Mohamadzade, A.; Marquetand, P.; González, L.; Ullrich, S. Simulated and Experimental Time-Resolved Photoelectron Spectra of the Intersystem Crossing Dynamics in 2-Thiouracil. Molecules 2018, 23, 2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopyra, J.; Abdoul-Carime, H.; Kossoski, F.; Varella, M.T.N. Electron driven reactions in sulphur containing analogues of uracil: The case of 2-thiouracil. Phys. Chem. Chem. Phys. 2014, 16, 25054–25061. [Google Scholar] [CrossRef]
- Kopyra, J.; Abdoul-Carime, H. Unusual temperature dependence of the dissociative electron attachment cross section of 2-thiouracil. J. Chem. Phys. 2016, 144, 034306. [Google Scholar] [CrossRef]
- Zheng, Y.; Sanche, L. Clustered DNA Damages induced by 0.5 to 30 eV Electrons. Int. J. Mol. Sci. 2015, 20, 3749. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, E.; Orlando, T.M.; Sanche, L. Biomolecular Damage Induced by Ionizing Radiation: The Direct and Indirect Effects of Low-Energy Electrons on DNA. Annu. Rev. Phys. Chem. 2015, 66, 379–398. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Dessent, C.E.H. Experiment and theory confirm that UV laser photodissociation spectroscopy can distinguish protomers formed via electrospray. Phys. Chem. Chem. Phys. 2017, 19, 17434–17440. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-L.; Kunin, A.; Matthews, E.; Yoshikawa, N.; Dessent, C.E.H.; Neumark, D.M. Photodissociation dynamics of the iodide-uracil (I–U) complex. J. Chem. Phys. 2016, 145, 044319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, Y.-W.; Akinyemi, T.E.; Steill, J.D.; Oomens, J.; Rodgers, M.T. Infrared multiple photon dissociation action spectroscopy of protonated uracil and thiouracils: Effects of thioketo-substitution on gas-phase conformation. Int. J. Mass Spectrom. 2010, 297, 139–151. [Google Scholar] [CrossRef]
- Morgan, W.J.; Fortenberry, R.C. Additional diffuse functions in basis sets for dipole-bound excited states of anions. Theor. Chem. Acc. 2015, 134, 47. [Google Scholar] [CrossRef]
- Gutowski, M.; Jordan, K.D.; Skurski, P. Electronic Structure of Dipole-Bound Anions. J. Phys. Chem. A 1998, 102, 2624–2633. [Google Scholar] [CrossRef]
- Skurski, P.; Gutowski, M.; Simons, J. How to choose a one-electron basis set to reliably describe a dipole-bound anion. Int. J. Quantum Chem. 2000, 80, 1024–1038. [Google Scholar] [CrossRef]
- Simons, J. Theoretical Study of Negative Molecular Ions. Annu. Rev. Phys. Chem. 2011, 62, 107–128. [Google Scholar] [CrossRef] [Green Version]
- Simons, J. Molecular Anions. J. Phys. Chem. A 2008, 112, 6401–6511. [Google Scholar] [CrossRef]
- Stein, T.; Kronik, L.; Baer, R. Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. [Google Scholar] [CrossRef]
- Noguchi, Y.; Hiyama, M.; Akiyama, H.; Koga, N. First-principles investigation on Rydberg and resonance excitations: A case study of the firefly luciferin anion. J. Chem. Phys. 2014, 141, 044309. [Google Scholar] [CrossRef] [PubMed]
- Støchkel, K.; Milne, B.F.; Nielsen, S.B. Absorption Spectrum of the Firefly Luciferin Anion Isolated in Vacuo. J. Phys. Chem. A 2011, 115, 2155–2159. [Google Scholar] [CrossRef] [PubMed]
- Cercola, R.; Matthews, E.; Dessent, C.E.H. Photoexcitation of Adenosine 5’-Triphosphate Anions in Vacuo: Probing the Influence of Charge State on the UV Photophysics of Adenine. J. Phys. Chem. B 2017, 121, 5553–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cercola, R.; Matthews, E.; Dessent, C.E.H. Near-threshold electron transfer in anion-nucleobase clusters: Does the identity of the anion matter? Mol. Phys. 2019, 117, 3001–3010. [Google Scholar] [CrossRef] [Green Version]
- Cercola, R.; Uleanya, K.O.; Dessent, C.E.H. Electron detachment dynamics of the iodide-guanine cluster: Does ionization occur from the iodide or from guanine? Mol. Phys. 2019. [Google Scholar] [CrossRef]
- Spencer, P.S.; Schaumburg, H.H. Nervous system degeneration produced by acrylamide monomer. Environ. Health Perspect. 1975, 11, 129–133. [Google Scholar] [CrossRef]
- Dessent, C.E.H.; Bailey, C.G.; Johnson, M.A. On the vibrational fine structure in the near-threshold photofragmentation spectrum of the I−⋅CH3 I complex: Spectroscopic observation of nonadiabatic effects in electron-molecule scattering. J. Chem. Phys. 1996, 105, 10416–10423. [Google Scholar] [CrossRef]
- Pedersen, Ø.S.; Byskov, S.C.; Turecek, F.; Nielsen, B.S. Structures of Protonated Thymine and Uracil and Their Monohydrated Gas-Phase Ions from Ultraviolet Action Spectroscopy and Theory. J. Phys. Chem. A 2014, 118, 4256–4265. [Google Scholar] [CrossRef]
- Berdakin, M.; Féraud, G.; Dedonder-Lardeux, C.; Jouvet, C.; Pino, G.A. Excited states of protonated DNA/RNA bases. Phys. Chem. Chem. Phys. 2014, 16, 10643–10650. [Google Scholar] [CrossRef] [Green Version]
- Molina, E.R.; Ortiz, D.; Salpin, J.Y.; Spezia, R. Elucidating collision induced dissociation products and reaction mechanisms of protonated uracil by coupling chemical dynamics simulations with tandem mass spectrometry experiments. J. Mass Spectrom. 2015, 50, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Lucas, B.; Barat, M.; Fayeton, J.A.; Jouvet, C.; Çarçabal, P.; Grégoire, G. Statistical versus non-statistical photo-fragmentation of protonated GWG tri-peptide induced by UV excitation. Chem. Phys. 2008, 347, 324–330. [Google Scholar] [CrossRef]
- Dessent, C.E.H.; Kim, J.; Johnson, M.A. Spectroscopic observation of vibrational Feshbach resonances in near-threshold photoexcitation of X-center dot CH3NO2 (X− = I− and Br−). Faraday Discuss. 2000, 115, 395–406. [Google Scholar] [CrossRef]
- Matthews, E.; Cercola, R.; Mensa-Bonsu, G.; Neumark, D.M.; Dessent, C.E.H. Photoexcitation of iodide ion-pyrimidine clusters above the electron detachment threshold: Intracluster electron transfer versus nucleobase-centred excitations. J. Chem. Phys. 2018, 148, 084304. [Google Scholar] [CrossRef]
- Sen, A.; Hou, G.-L.; Wang, X.-B.; Dessent, C.E.H. Electron Detachment as a Probe of Intrinsic Nucleobase Dynamics in Dianion-Nucleobase Clusters: Photoelectron Spectroscopy of the Platinum II Cyanide Dianion Bound to Uracil, Thymine, Cytosine, and Adenine. J. Phys. Chem. B 2015, 119, 11626–11631. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Matthews, E.M.; Hou, G.-L.; Wang, X.-B.; Dessent, C.E.H. Photoelectron spectroscopy of hexachloroplatinate-nucleobase complexes: Nucleobase excited state decay observed via delayed electron emission. J. Chem. Phys. 2015, 143, 184307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, E.; Sen, A.; Yoshikawa, N.; Bergström, E.; Dessent, C.E.H. UV laser photoactivation of hexachloroplatinate bound to individual nucleobases: In vacuo as molecular level probes of a model photopharmaceutical. Phys. Chem. Chem. Phys. 2016, 18, 15143–15152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, A.; Luxford, T.F.M.; Yoshikawa, N.; Dessent, C.E.H. Solvent evaporation versus proton transfer in nucleobase–Pt(CN)4,62− dianion clusters: A collisional excitation and electronic laser photodissociation spectroscopy study. Phys. Chem. Chem. Phys. 2014, 16, 15490–15500. [Google Scholar] [CrossRef] [PubMed]
- Antoine, R.; Dugourd, P. Visible and ultraviolet spectroscopy of gas phase protein ions. Phys. Chem. Chem. Phys. 2011, 13, 16494–16509. [Google Scholar] [CrossRef] [PubMed]
- Wellman, S.M.; Jockusch, R.A. Moving in on the Action: An Experimental Comparison of Fluorescence Excitation and Photodissociation Action Spectroscopy. J. Phys. Chem. A 2015, 119, 6333–6338. [Google Scholar] [CrossRef]
- Jedrychowski, M.P.; Huttlin, E.L.; Haas, W.; Sowa, M.E.; Rad, R.; Gygi, S.P. Evaluation of HCD- and CID-type Fragmentation Within Their Respective Detection Platforms for Murine Phosphoproteomics. Mol. Cell. Proteomics 2011, 10, M111.009910. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Macek, B.; Lange, O.; Makarov, A.; Horning, S.; Mann, M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | Tautomer | Relative Energy (kJ mol−1) a,b | % Boltzmann Population b,c | VDE |
|---|---|---|---|---|
| [2-TU-H]− | D1 | 0.0 (0.0) | 99.9 (99.8) | 3.82 |
| D2 | 45.9 (15.3) | 0 (<0.1) | 3.66 | |
| D3a | 101 (58.9) | 0 (<0.1) | 2.91 | |
| D3b | 74.6 (49.5) | 0 (<0.1) | 3.00 | |
| D4a | 58.2 (51.7) | 0 (<0.1) | 3.93 | |
| D4b | 56.5 (51.5) | 0 (<0.1) | 2.54 | |
| [2-TU·H]+ | P1a | 0.0 (0.0) | 93.9 (35.9) | |
| P1b | 6.81 (1.32) | 6.01 (21.1) | ||
| P1c | 17.6 (8.10) | 0.077 (1.37) | ||
| P1d | 26.8 (9.55) | 0.003 (0.76) | ||
| P2 | 20.2 (-0.32) | 0.026 (40.9) |
| Photofragment m/z | Proposed Ion Structures | Associated Neutral | Present in HCD a | Photofragment Intensity at 3.6 eV b |
|---|---|---|---|---|
| 58 | SCN− | C3H3NO | √ | √ (w) |
| 93 |  | H2S | X | √ (vw) |
| Photofragment m/z | Proposed Structure | Neutral or Radical Loss | HCD a | Photofragment Intensity at 4.6 eV b,c | Photofragment Intensity at 5.2 eV b,c |
|---|---|---|---|---|---|
| 128 |  | H· | √ | √ (w) | √ (m) |
| 112 | C4H2NO+ | NH3 | √ | √ (w) | √ (w) |
| 96 |  | HS· | X | √ (vs) | √ (vs) |
| 79 | C4HNO+ | HS· + NH3 | X | X | √ (vw) |
| 70 |  | HNCS | √ | √ (m) | √ (m) |
| 68 | C3H4N2+ | HS· + CO | X | X | √ (vw) |
| 60 | CNSH2+ | NH3 + C3O | √ | √ (vw) | √ (vw) |
| 53 |  | NH3 + HNCS | √ | √ (vw) | √ (vw) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uleanya, K.O.; Cercola, R.; Nikolova, M.; Matthews, E.; Wong, N.G.K.; Dessent, C.E.H. Observation of Enhanced Dissociative Photochemistry in the Non-Native Nucleobase 2-Thiouracil. Molecules 2020, 25, 3157. https://doi.org/10.3390/molecules25143157
Uleanya KO, Cercola R, Nikolova M, Matthews E, Wong NGK, Dessent CEH. Observation of Enhanced Dissociative Photochemistry in the Non-Native Nucleobase 2-Thiouracil. Molecules. 2020; 25(14):3157. https://doi.org/10.3390/molecules25143157
Chicago/Turabian StyleUleanya, Kelechi O., Rosaria Cercola, Maria Nikolova, Edward Matthews, Natalie G. K. Wong, and Caroline E. H. Dessent. 2020. "Observation of Enhanced Dissociative Photochemistry in the Non-Native Nucleobase 2-Thiouracil" Molecules 25, no. 14: 3157. https://doi.org/10.3390/molecules25143157