
Continuous Bioinspired Oxidation of Sulfides

 and
and 
Abstract
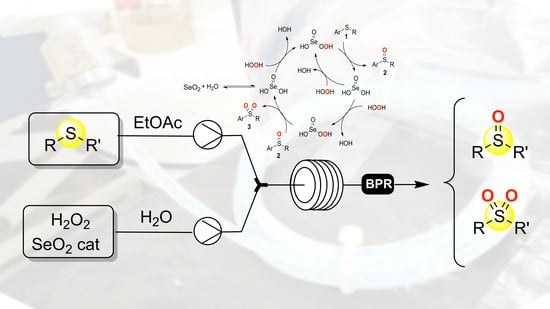
:
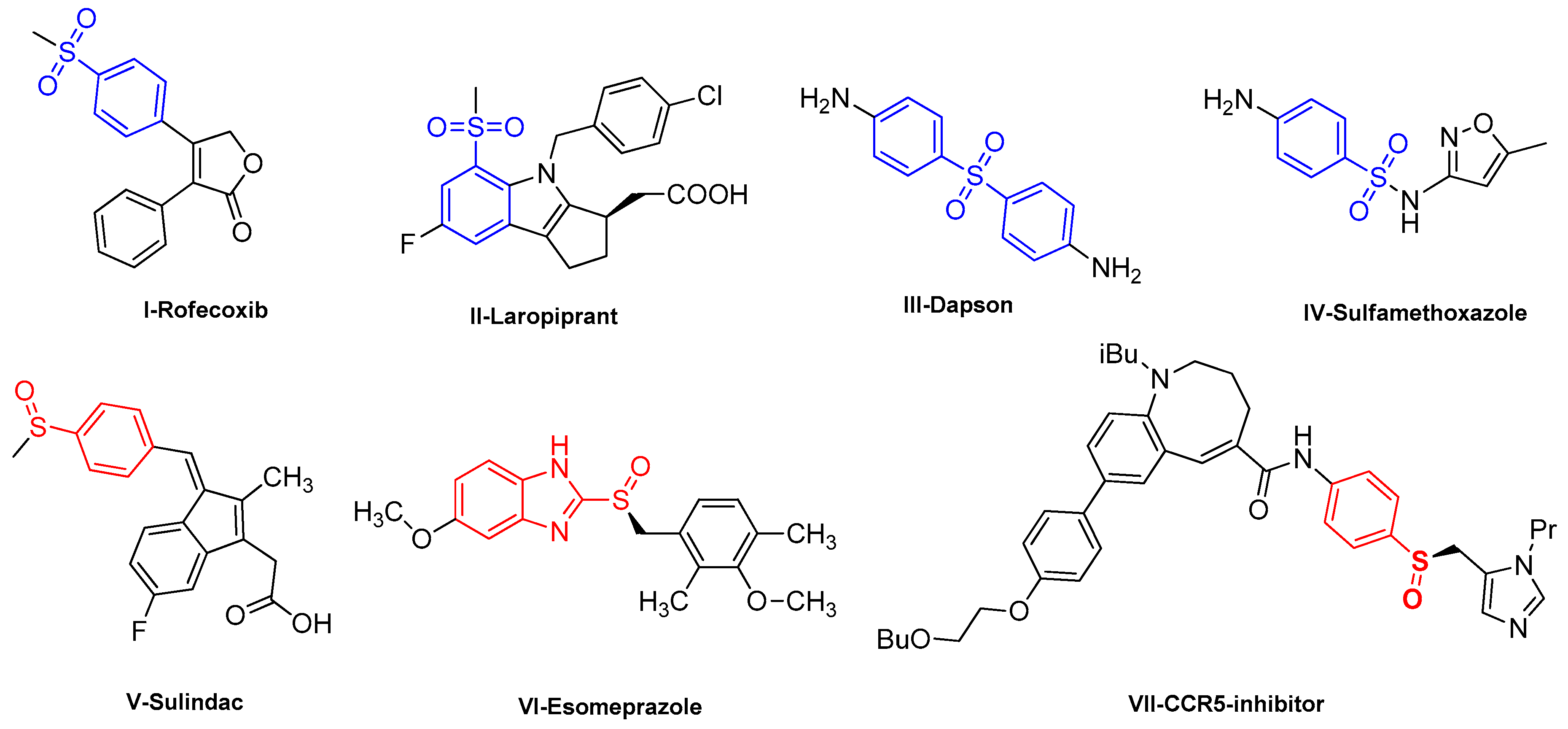
1. Introduction
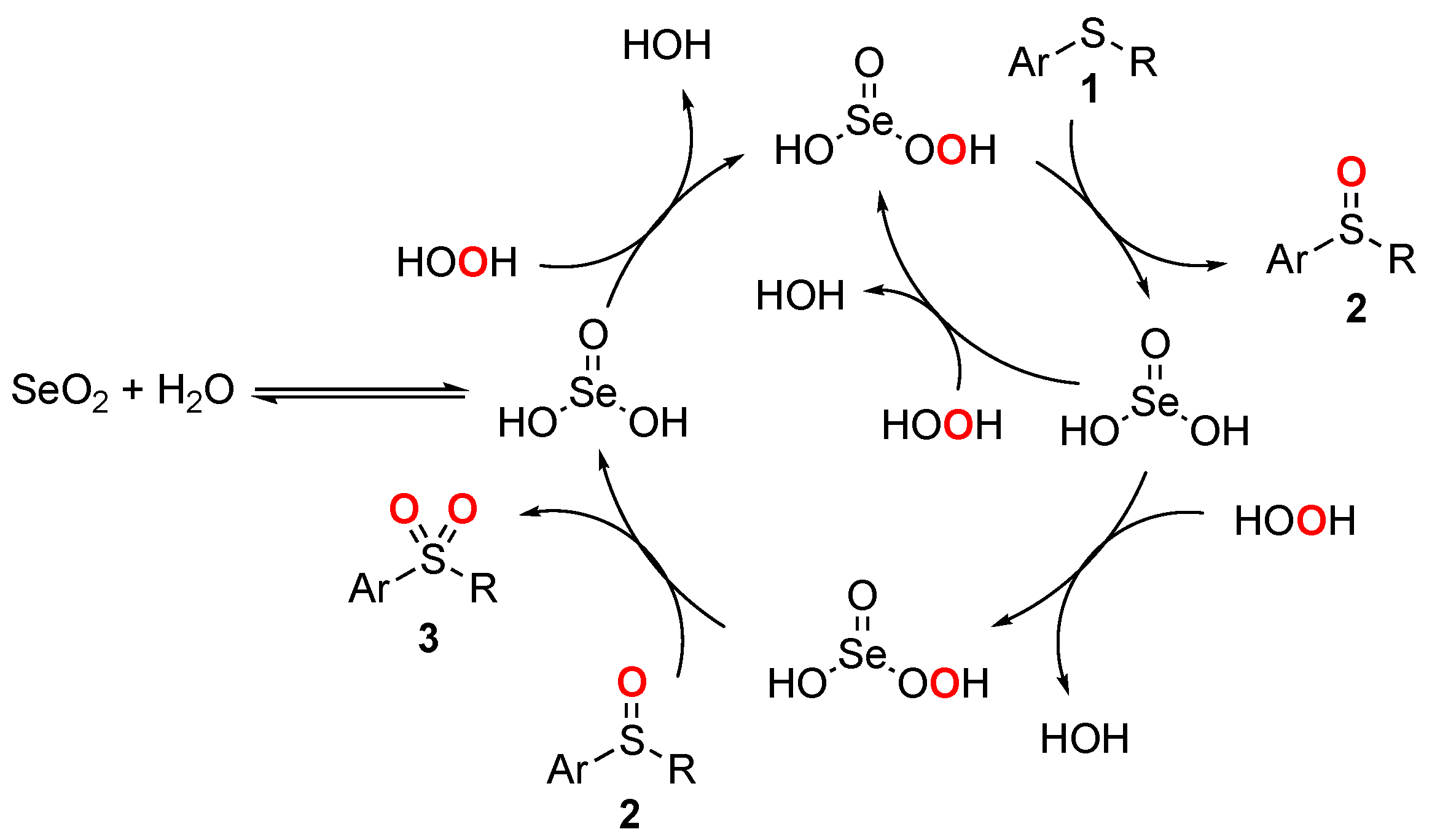
2. Results and Discussions
3. Materials and Methods
3.1. Synthesis of Sulfoxides 2a–f
3.2. Synthesis of Sulfones 2a–g
3.3. Spectral Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bäckvall, J.-E. Modern Oxidation Methods, 2nd ed.; Completely rev. and enlarged ed.; Bäckvall, J.-E., Ed.; Wiley-VCH: Weinheim, Germany, 2010; ISBN 978-3-527-32320-3. [Google Scholar]
- Surur, A.S.; Schulig, L.; Link, A. Interconnection of sulfides and sulfoxides in medicinal chemistry. Arch. Pharm. 2018, 1800248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Chen, Y.; Fan, E.; Sun, Z. Synthesis of 3-Substituted Aryl [4,5] isothiazoles through an All-Heteroatom Wittig-Equivalent Process. Org. Lett. 2016, 18, 2777–2779. [Google Scholar] [CrossRef] [PubMed]
- Pieters, L.; Van Dyck, S.; Gao, M.; Bai, R.; Hamel, E.; Vlietinck, A.; Lemière, G. Synthesis and Biological Evaluation of Dihydrobenzofuran Lignans and Related Compounds as Potential Antitumor Agents that Inhibit Tubulin Polymerization. J. Med. Chem. 1999, 42, 5475–5481. [Google Scholar] [CrossRef]
- Sturino, C.F.; O’Neill, G.; Lachance, N.; Boyd, M.; Berthelette, C.; Labelle, M.; Li, L.; Roy, B.; Scheigetz, J.; Tsou, N.; et al. Discovery of a Potent and Selective Prostaglandin D2 Receptor Antagonist, [(3R)-4-(4-Chloro- benzyl)-7-fluoro-5-(methylsulfonyl)-1,2,3,4-tetrahydrocyclopenta [b] indol-3-yl]-acetic Acid (MK-0524) †. J. Med. Chem. 2007, 50, 794–806. [Google Scholar] [CrossRef]
- Yazdanyar, S.; Boer, J.; Ingvarsson, G.; Szepietowski, J.C.; Jemec, G.B.E. Dapsone Therapy for Hidradenitis Suppurativa: A Series of 24 Patients. Dermatology 2011, 222, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Sunduru, N.; Salin, O.; Gylfe, Å.; Elofsson, M. Design, synthesis and evaluation of novel polypharmacological antichlamydial agents. Eur. J. Med. Chem. 2015, 101, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Aono, Y.; Horinaka, M.; Iizumi, Y.; Watanabe, M.; Taniguchi, T.; Yasuda, S.; Sakai, T. Sulindac sulfone inhibits the mTORC1 pathway in colon cancer cells by directly targeting voltage-dependent anion channel 1 and 2. Biochem. Biophys. Res. Commun. 2018, 505, 1203–1210. [Google Scholar] [CrossRef]
- Spencer, C.M.; Faulds, D. Esomeprazole. Drugs 2000, 60, 321–329. [Google Scholar] [CrossRef]
- Seto, M.; Aikawa, K.; Miyamoto, N.; Aramaki, Y.; Kanzaki, N.; Takashima, K.; Kuze, Y.; Iizawa, Y.; Baba, M.; Shiraishi, M. Highly Potent and Orally Active CCR5 Antagonists as Anti-HIV-1 Agents: Synthesis and Biological Activities of 1-Benzazocine Derivatives Containing a Sulfoxide Moiety. J. Med. Chem. 2006, 49, 2037–2048. [Google Scholar] [CrossRef]
- Santoro, S.; Azeredo, J.B.; Nascimento, V.; Sancineto, L.; Braga, A.L.; Santi, C. The green side of the moon: Ecofriendly aspects of organoselenium chemistry. RSC Adv. 2014, 4, 31521–31535. [Google Scholar] [CrossRef]
- Tidei, C.; Piroddi, M.; Galli, F.; Santi, C. Oxidation of thiols promoted by PhSeZnCl. Tetrahedron Lett. 2012, 53, 232–234. [Google Scholar] [CrossRef]
- Santoro, S.; Santi, C.; Sabatini, M.; Testaferri, L.; Tiecco, M. Eco-Friendly Olefin Dihydroxylation Catalyzed by Diphenyl Diselenide. Adv. Synth. Catal. 2008, 350, 2881–2884. [Google Scholar] [CrossRef]
- Santi, C.; Di Lorenzo, R.; Tidei, C.; Bagnoli, L.; Wirth, T. Stereoselective selenium catalyzed dihydroxylation and hydroxymethoxylation of alkenes. Tetrahedron 2012, 68, 10530–10535. [Google Scholar] [CrossRef]
- Sancineto, L.; Mangiavacchi, F.; Tidei, C.; Bagnoli, L.; Marini, F.; Gioiello, A.; Scianowski, J.; Santi, C. Selenium-Catalyzed Oxacyclization of Alkenoic Acids and Alkenols. Asian J. Org. Chem. 2017, 6, 988–992. [Google Scholar] [CrossRef]
- Sancineto, L.; Tidei, C.; Bagnoli, L.; Marini, F.; Lenardão, E.; Santi, C. Selenium Catalyzed Oxidation of Aldehydes: Green Synthesis of Carboxylic Acids and Esters. Molecules 2015, 20, 10496–10510. [Google Scholar] [CrossRef]
- Santi, C.; Jacob, R.; Monti, B.; Bagnoli, L.; Sancineto, L.; Lenardão, E. Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry. Molecules 2016, 21, 1482. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.; Moody, T.S.; Smyth, M.; Wharry, S. A Perspective on Continuous Flow Chemistry in the Pharmaceutical Industry. Org. Process Res. Dev. 2020. [Google Scholar] [CrossRef]
- Valikhani, D.; Srivastava, P.L.; Allemann, R.K.; Wirth, T. Immobilised Enzymes for Sesquiterpene Synthesis in Batch and Flow Systems. ChemCatChem 2020, 12, 2194–2197. [Google Scholar] [CrossRef]
- Maljuric, S.; Jud, W.; Kappe, C.O.; Cantillo, D. Translating batch electrochemistry to single-pass continuous flow conditions: An organic chemist’s guide. J. Flow Chem. 2020, 10, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Cambié, D.; Noël, T. Solar Photochemistry in Flow. Top. Curr. Chem. 2018, 376. [Google Scholar] [CrossRef] [Green Version]
- Santi, M.; Seitz, J.; Cicala, R.; Hardwick, T.; Ahmed, N.; Wirth, T. Memory of Chirality in Flow Electrochemistry: Fast Optimisation with DoE and Online 2D-HPLC. Chem. A Eur. J. 2019, 25, 16230–16235. [Google Scholar] [CrossRef]
- Di Schino, L.; Incipini, L.; Dragone, V.; Tidei, C.; Scalera, C.; Santi, C. Green Chemistry for Enviromental Sustainability: An Example of “Bio-Logic” Approach. In Proceedings of the 1st World Sustainability Forum, Basel, Switzerland, 1–30 November 2011; MDPI: Basel, Switzerland, 2011; p. 675. [Google Scholar]
- Cerra, B.; Mangiavacchi, F.; Santi, C.; Lozza, A.M.; Gioiello, A. Selective continuous flow synthesis of hydroxy lactones from alkenoic acids. React. Chem. Eng. 2017, 2, 467–471. [Google Scholar] [CrossRef]
- Mello, R.; Olmos, A.; Alcalde-Aragonés, A.; Díaz-Rodríguez, A.; González Núñez, M.E.; Asensio, G. Oxidation of Sulfides with a Silica-Supported Peracid in Supercritical Carbon Dioxide under Flow Conditions: Tuning Chemoselectivity with Pressure. Eur. J. Org. Chem. 2010, 2010, 6200–6206. [Google Scholar] [CrossRef]
- Maggi, R.; Chitsaz, S.; Loebbecke, S.; Piscopo, C.G.; Sartori, G.; Schwarzer, M. Highly chemoselective metal-free oxidation of sulfides with diluted H2O2 in a continuous flow reactor. Green Chem. 2011, 13, 1121. [Google Scholar] [CrossRef]
- Laudadio, G.; Straathof, N.J.W.; Lanting, M.D.; Knoops, B.; Hessel, V.; Noël, T. An environmentally benign and selective electrochemical oxidation of sulfides and thiols in a continuous-flow microreactor. Green Chem. 2017, 19, 4061–4066. [Google Scholar] [CrossRef] [Green Version]
- Silva, F.; Baker, A.; Stansall, J.; Michalska, W.; Yusubov, M.S.; Graz, M.; Saunders, R.; Evans, G.J.S.; Wirth, T. Selective Oxidation of Sulfides in Flow Chemistry: Selective Oxidation of Sulfides in Flow Chemistry. Eur. J. Org. Chem. 2018, 2018, 2134–2137. [Google Scholar] [CrossRef]
- Drabowicz, J.; Mikołajczyk, M. A Facile and Selective Oxidation of Organic Sulphides to Sulphoxides with Hydrogen Peroxide/Selenium Dioxide System. Synthesis 1978, 1978, 758–759. [Google Scholar] [CrossRef]
- Drabowicz, J.; łyzwa, P.; Mikołajczyk, M. A New Procedure for Oxidation of Sulfides to Sulfones. Phosphorus Sulfur Relat. Elem. 1983, 17, 169–172. [Google Scholar] [CrossRef]
- Młochowski, J.; Wójtowicz-Młochowska, H. Developments in Synthetic Application of Selenium(IV) Oxide and Organoselenium Compounds as Oxygen Donors and Oxygen-Transfer Agents. Molecules 2015, 20, 10205–10243. [Google Scholar] [CrossRef] [Green Version]
- Chuo, T.H.; Boobalan, R.; Chen, C. Camphor-based schiff base of 3-endo-aminoborneol (SBAB): Novel ligand for vanadium-catalyzed asymmetric sulfoxidation and subsequent kinetic resolution. ChemistrySelect 2016, 1, 2174–2180. [Google Scholar] [CrossRef]
- Yuan, Y.; Shi, X.; Liu, W. Transition-Metal-Free, Chemoselective Aerobic Oxidations of Sulfides and Alcohols with Potassium Nitrate and Pyridinium Tribromide or Bromine. Synlett 2011, 2011, 559–564. [Google Scholar] [CrossRef]
- Gogoi, S.R.; Boruah, J.J.; Sengupta, G.; Saikia, G.; Ahmed, K.; Bania, K.K.; Islam, N.S. Peroxoniobium (V)-catalyzed selective oxidation of sulfides with hydrogen peroxide in water: A sustainable approach. Catal. Sci. Technol. 2015, 5, 595–610. [Google Scholar] [CrossRef]
- Fukuda, N.; Ikemoto, T. Imide-Catalyzed Oxidation System: Sulfides to Sulfoxides and Sulfones. J. Org. Chem. 2010, 75, 4629–4631. [Google Scholar] [CrossRef]
- Russell, G.A.; Pecoraro, J.M. Pummerer reaction of para-substituted benzylic sulfoxides. J. Org. Chem. 1979, 44, 3990–3991. [Google Scholar] [CrossRef]
- Yang, C.; Jin, Q.; Zhang, H.; Liao, J.; Zhu, J.; Yu, B.; Deng, J. Tetra-(tetraalkylammonium)octamolybdate catalysts for selective oxidation of sulfides to sulfoxides with hydrogen peroxide. Green Chem. 2009, 11, 1401. [Google Scholar] [CrossRef]
- Jain, S.L.; Sain, B. Perfluorinated resinsulphonic acid (Nafion-H®®) catalyzed highly efficient oxidations of organic compounds with hydrogen peroxide. Appl. Catal. A Gen. 2006, 301, 259–264. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | H2O2 eq [conc] | Total Flow RatemL/min | Residence Time (min) | NMR Conversion % | Selectivity 2/3 |
|---|---|---|---|---|---|
| 1 | 1 [0.5 M] | 0.3 | 6.5 | 59% | 95:5 |
| 2 | 1 [0.5 M] | 0.02 | 100 | 61% | 100:0 |
| 3 | 2 [1.0 M] | 0.3 | 6.5 | 60% | 100:0 |
| 4 | 2 [1.0 M] | 0.2 | 10 | 78% | 100:0 |
| 5 | 2 [1.0 M] | 0.1 | 20 | 85% | 100:0 |
| 6 | 5 [2.5 M] | 0.1 | 20 | >99% | 71:29 |
| 7 | 10 [5.0 M] | 0.1 | 20 | >99% | 0:100 |
| 8 | 10 [5.0 M] | 0.2 | 10 | >99% | 0:100 |

| Substrate | Product | NMR Conversion (Isolated Yield) 2 1 | Selectivity 2/3 1 |
|---|---|---|---|
 |  | 85% (85%) | 100:0 |
| 1a | 2a | ||
 |  | 91% (91%) | 100:0 |
| 1b | 2b | ||
 |  | 93% (89%) | 100:0 |
| 1c | 2c | ||
 |  | 20% 99% 2(75%) | 100:0 80:20 |
| 1d | 2d | ||
 |  | 80% (74%) | 100:0 |
| 1e | 2e | ||
 |  | 99% (99%) | 100:0 |
| 1f | 2f | ||
 |  | n.d. 3 | n.d. 3 |
| 1g | 2g |

| Substrate | Product | Yield of 3 |
|---|---|---|
 |  | > 99% |
| 1a | 3a | |
 |  | > 99% |
| 1b | 3b | |
 |  | > 99% |
| 1c | 3c | |
 |  | > 99% 1 |
| 1d | 3d | |
 |  | > 99% |
| 1e | 3e | |
 |  | > 99% |
| 1f | 3f | |
 |  | > 99% |
| 1g | 3g |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangiavacchi, F.; Crociani, L.; Sancineto, L.; Marini, F.; Santi, C. Continuous Bioinspired Oxidation of Sulfides. Molecules 2020, 25, 2711. https://doi.org/10.3390/molecules25112711
Mangiavacchi F, Crociani L, Sancineto L, Marini F, Santi C. Continuous Bioinspired Oxidation of Sulfides. Molecules. 2020; 25(11):2711. https://doi.org/10.3390/molecules25112711
Chicago/Turabian StyleMangiavacchi, Francesca, Letizia Crociani, Luca Sancineto, Francesca Marini, and Claudio Santi. 2020. "Continuous Bioinspired Oxidation of Sulfides" Molecules 25, no. 11: 2711. https://doi.org/10.3390/molecules25112711