
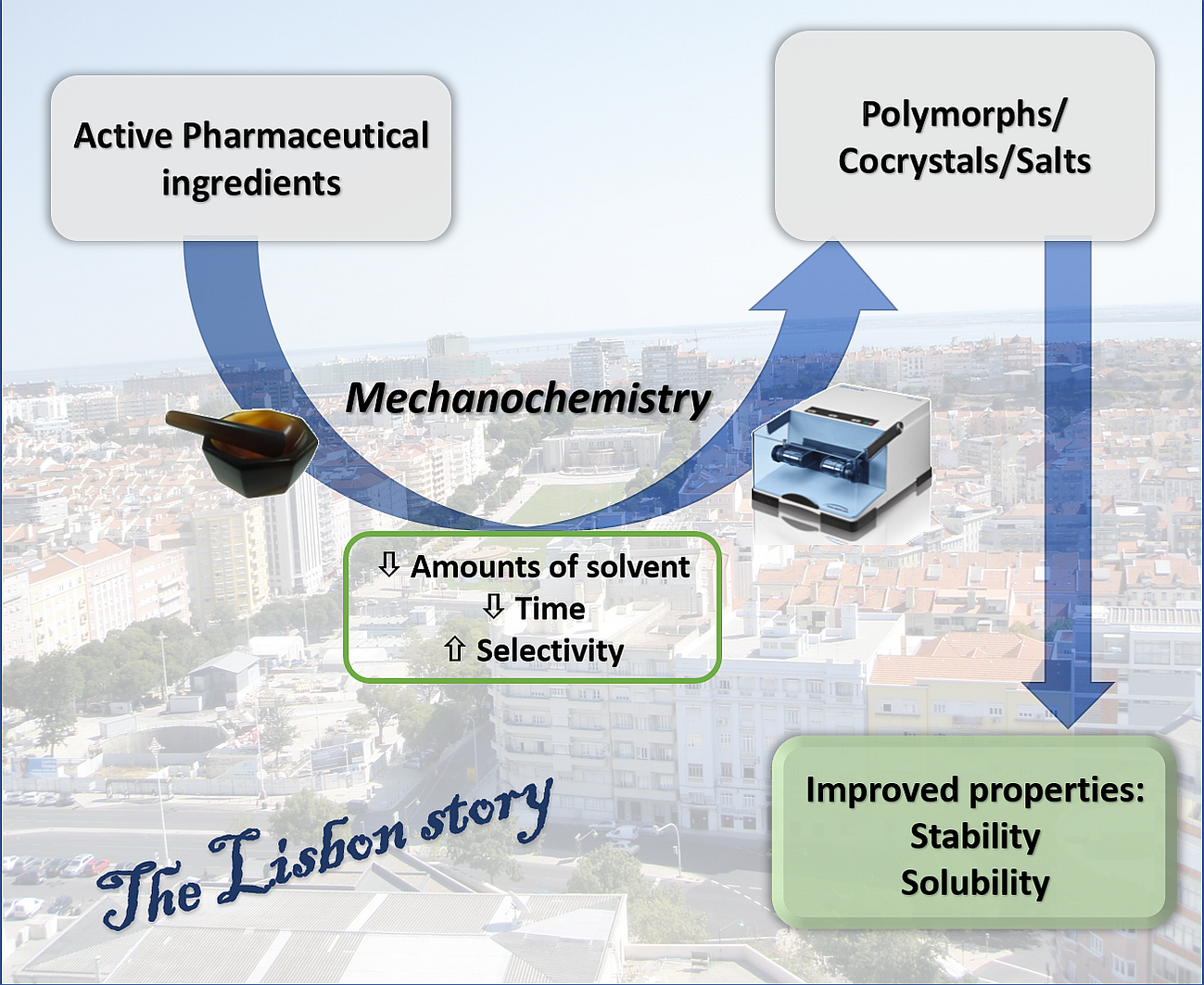
The Lisbon Supramolecular Green Story: Mechanochemistry towards New Forms of Pharmaceuticals

 , ,
, ,
Abstract
:
1. Introduction
2. Results
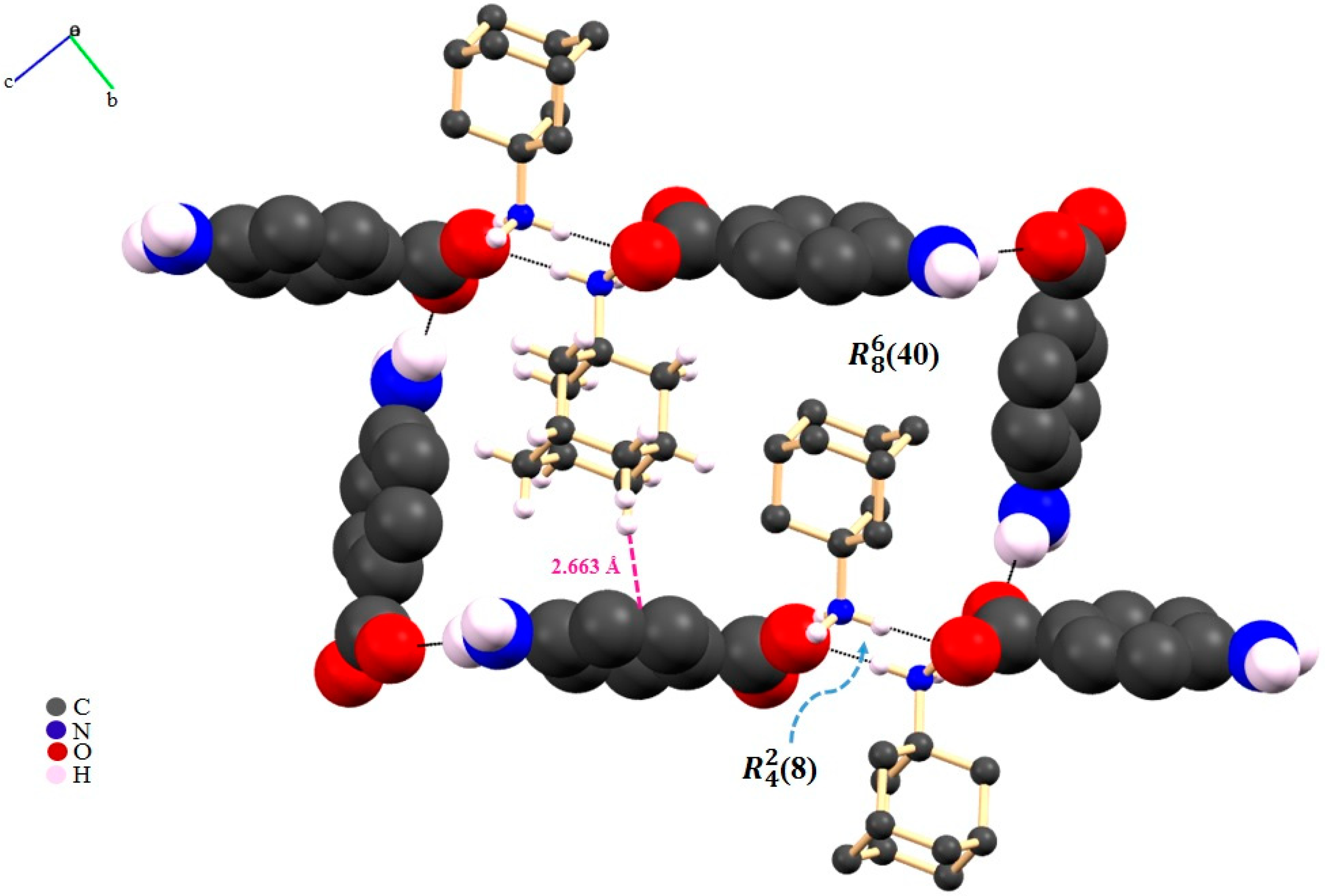
2.1. Gabapentin
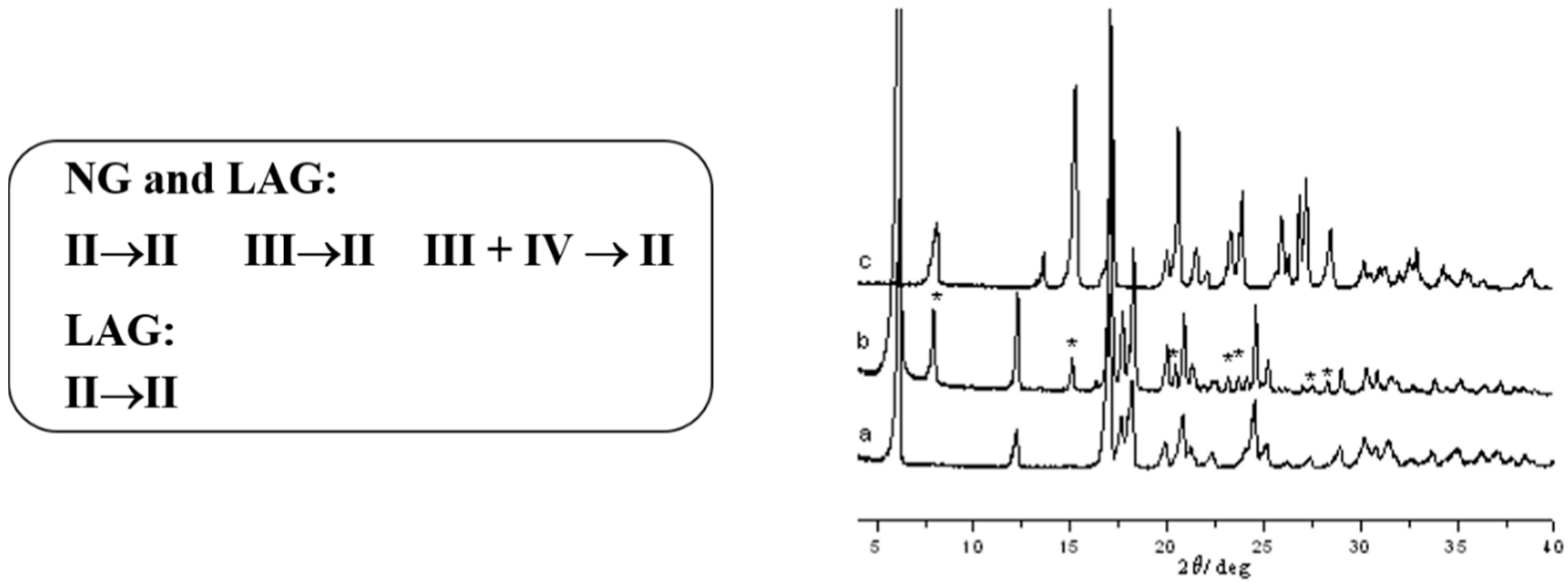
2.1.1. Polymorphic Screening and Control
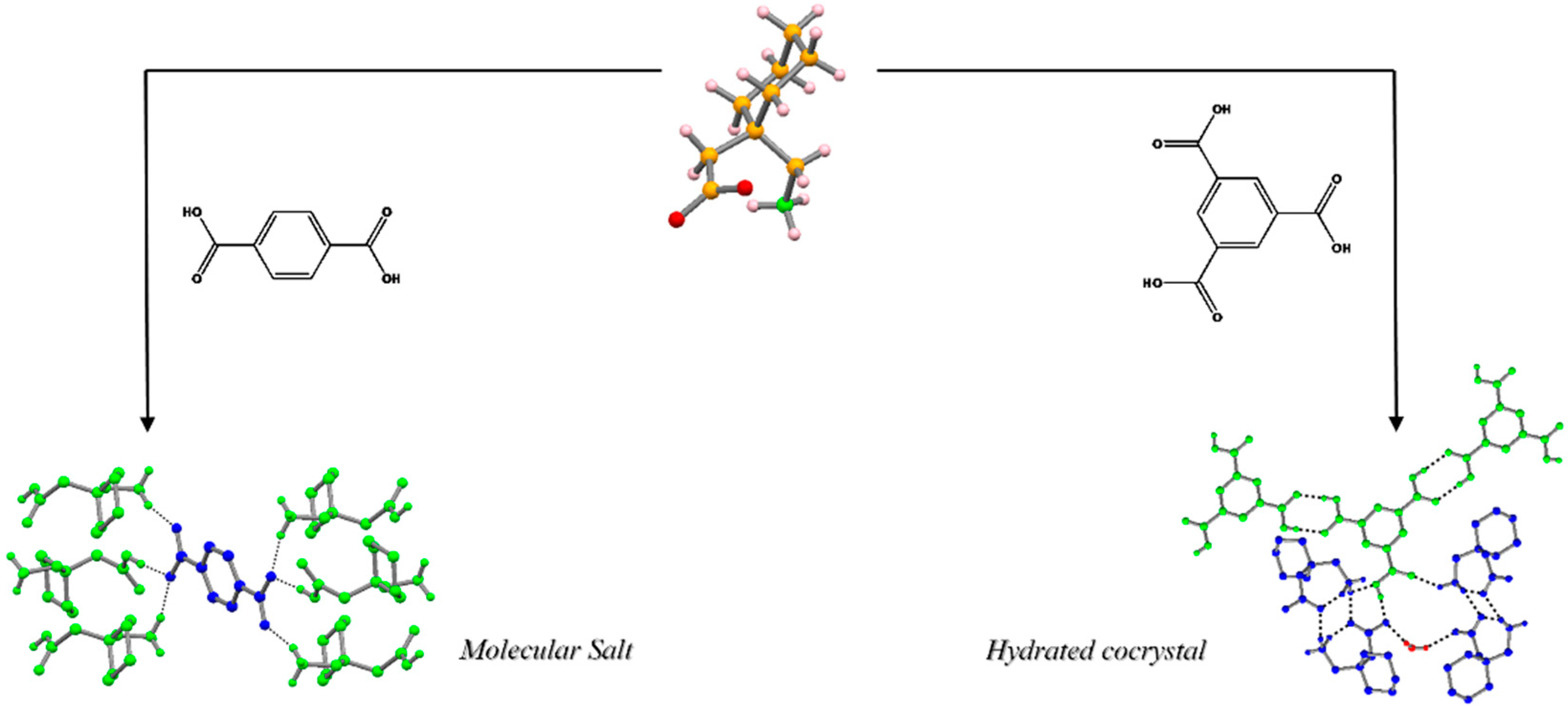
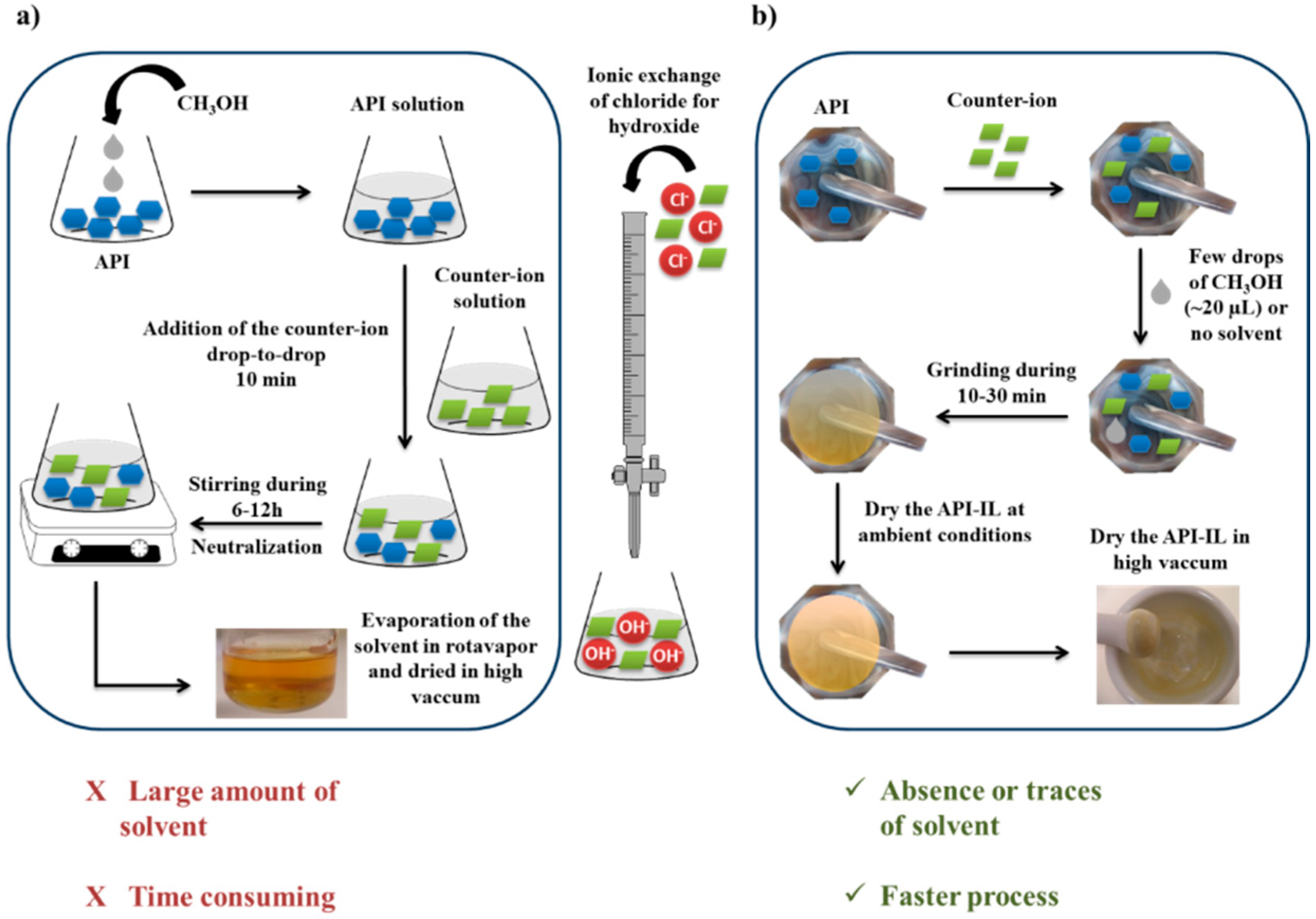
2.1.2. Novel Multicomponent Crystal Forms and Ionic Liquids
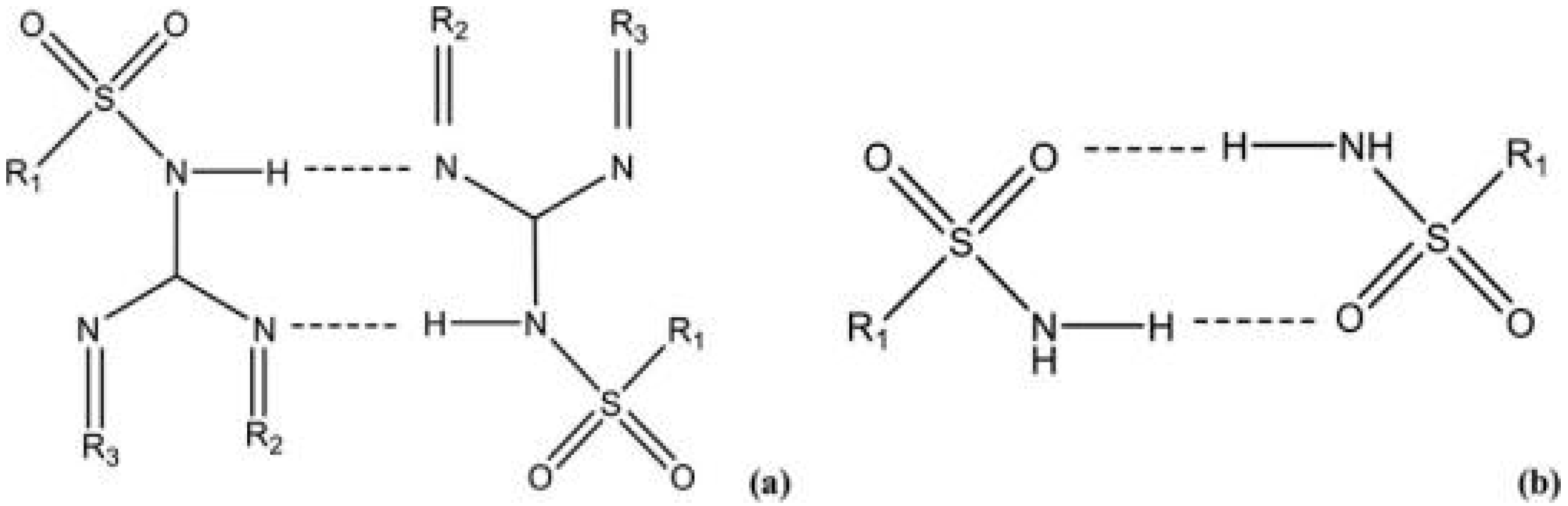
2.2. Sulfoxides: Robust Synthons in Co-Crystallization
2.2.1. Dapsone Co-Crystals
2.2.2. Sulfadimethoxine: Learning about Synthon Competition
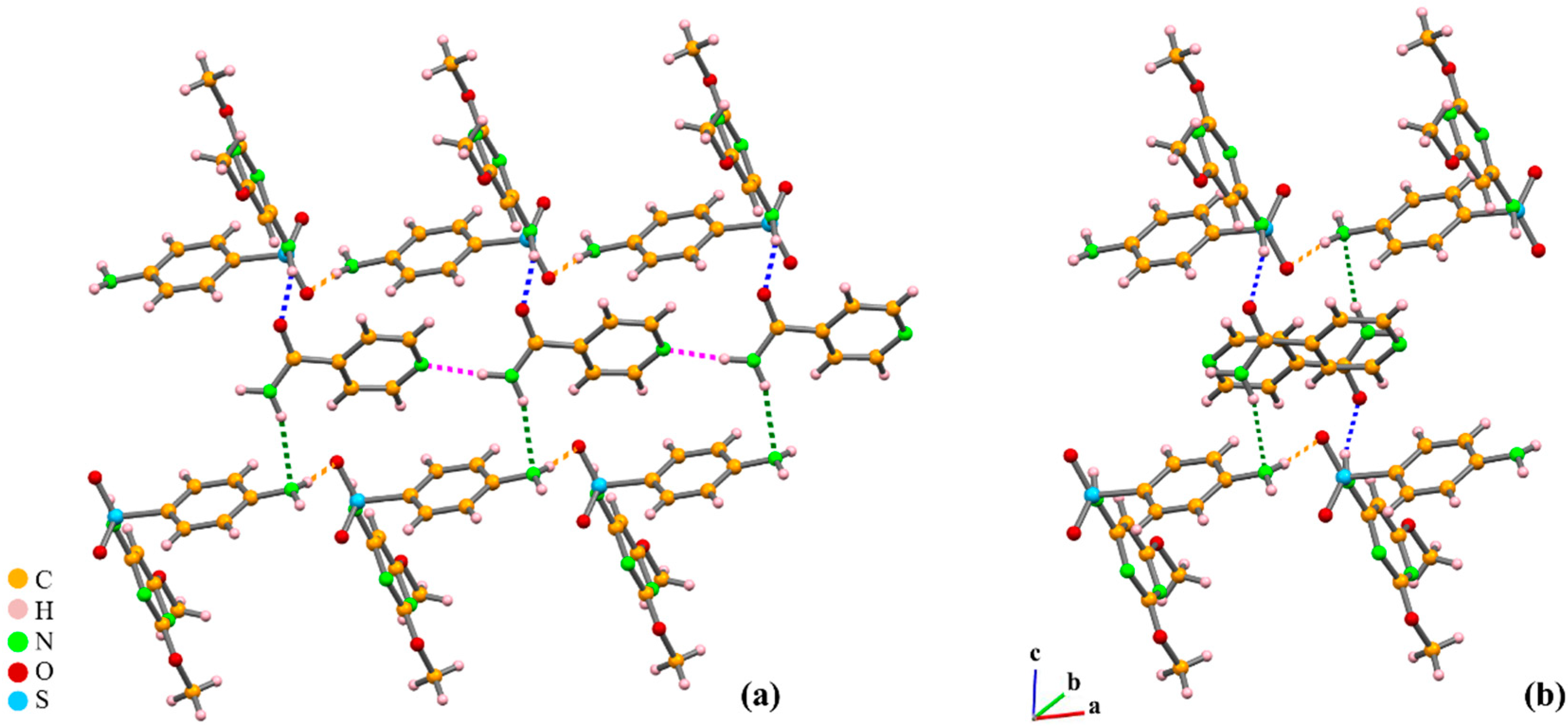
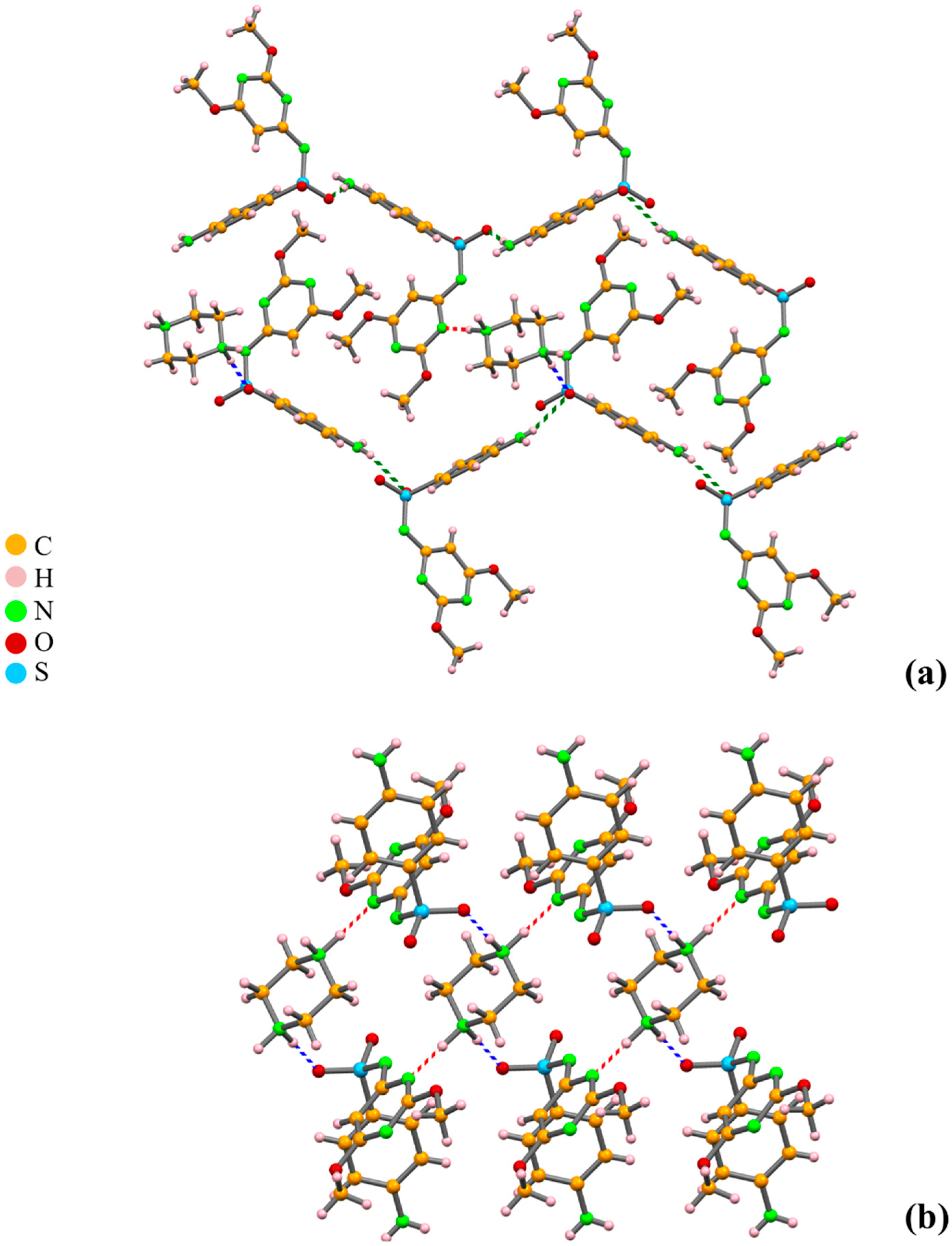
2.3. Discerning Co-Crystals from Salts by X-ray Diffraction and Solid-State NMR
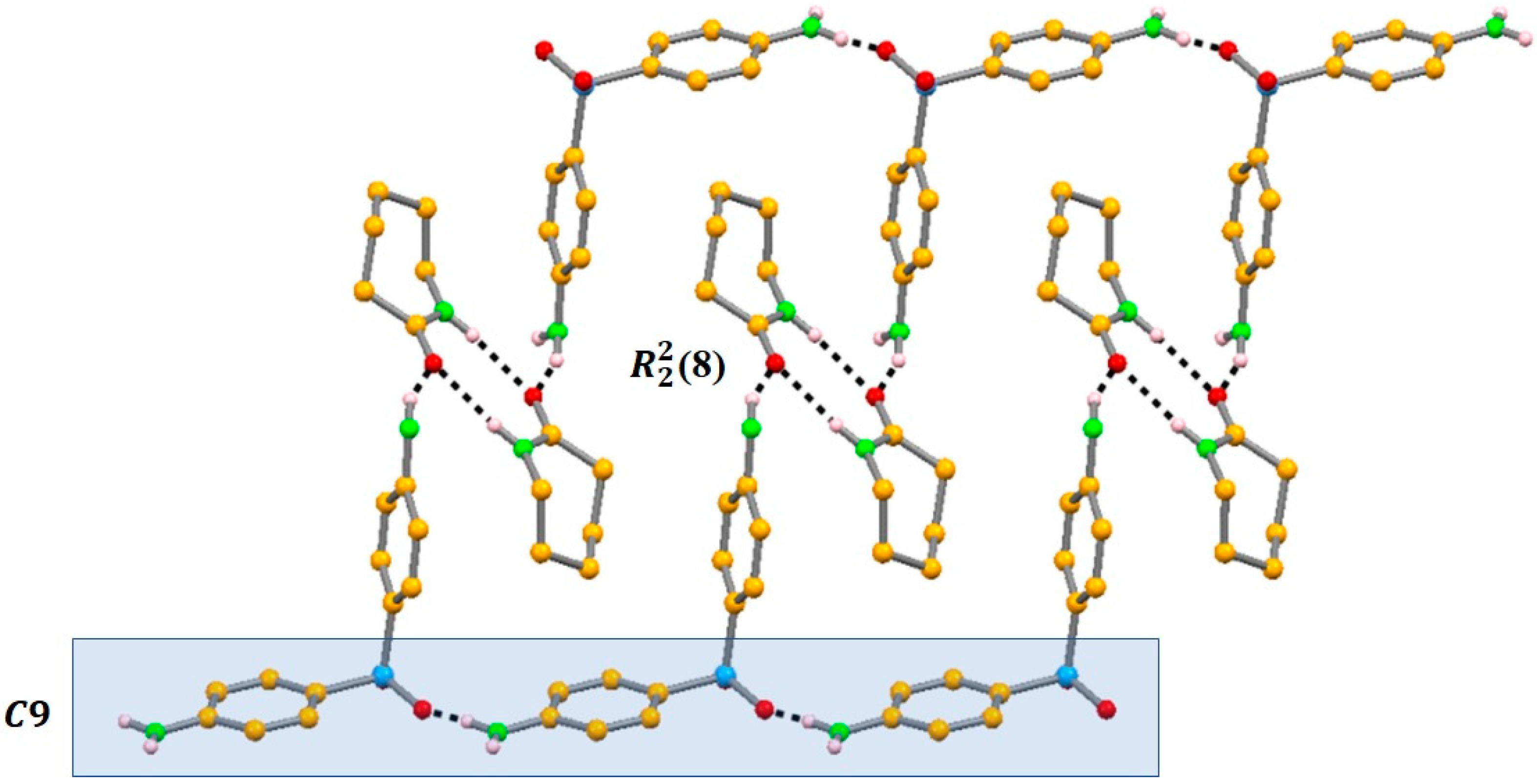
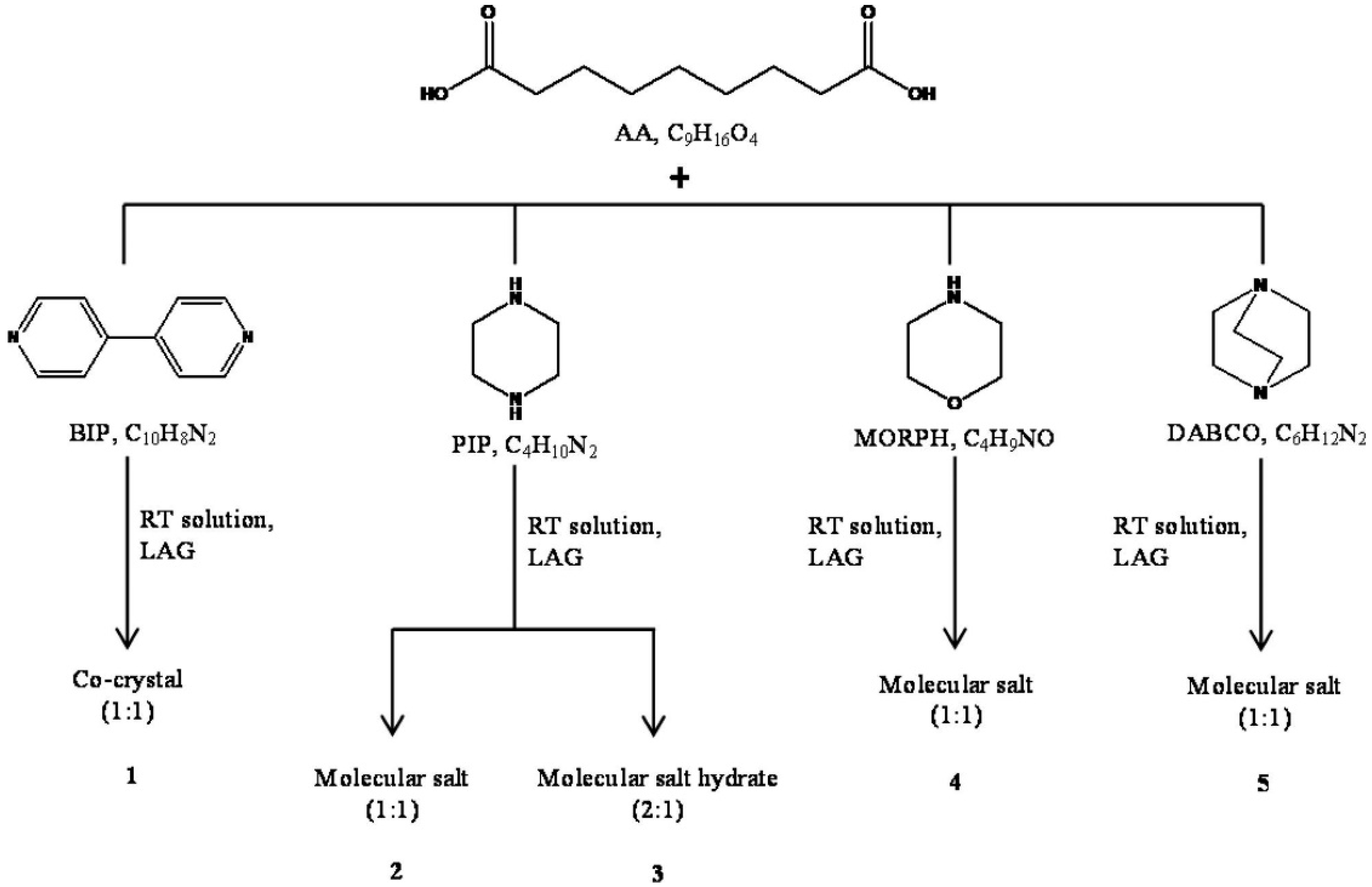
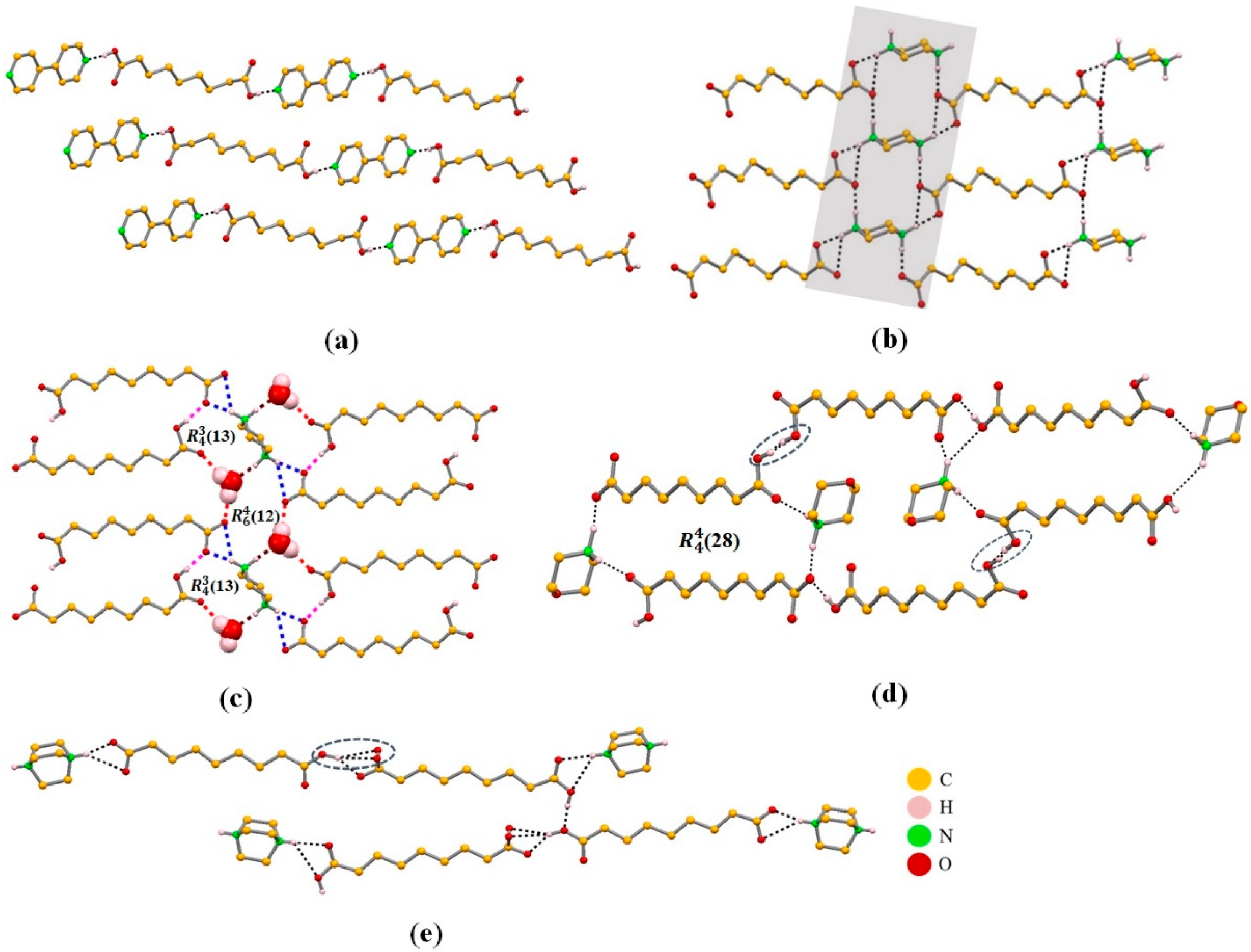
2.3.1. Azelaic Acid
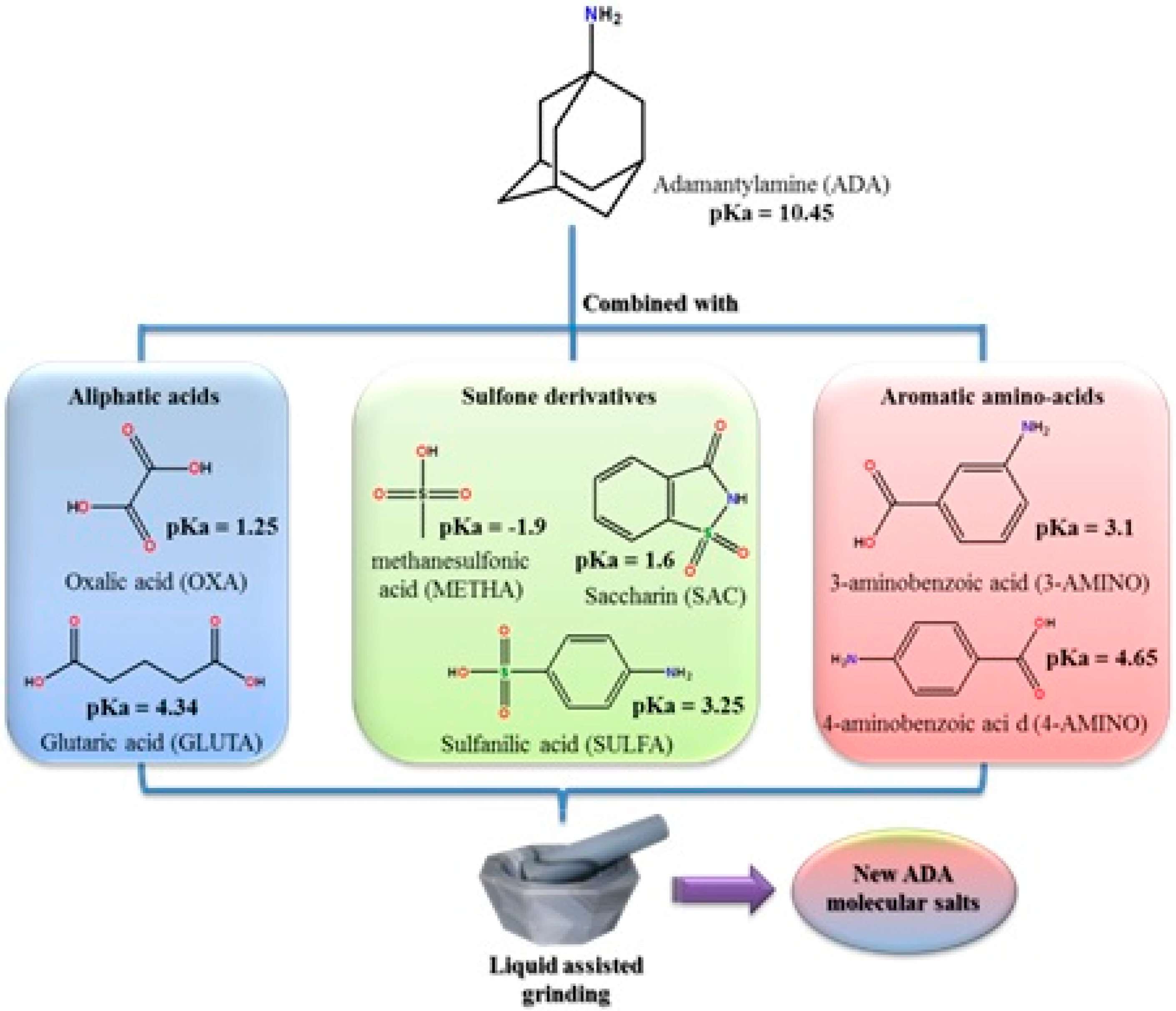
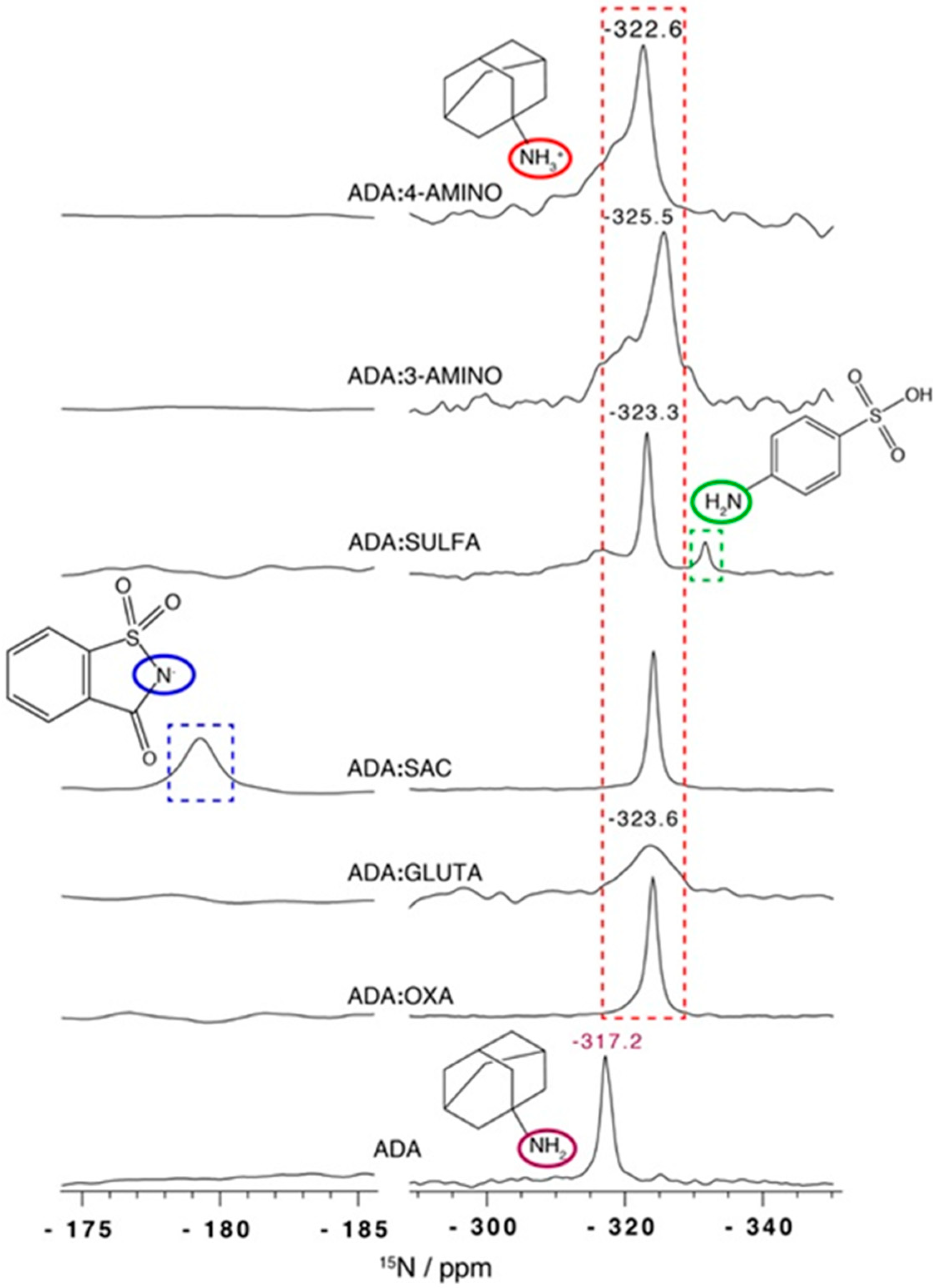
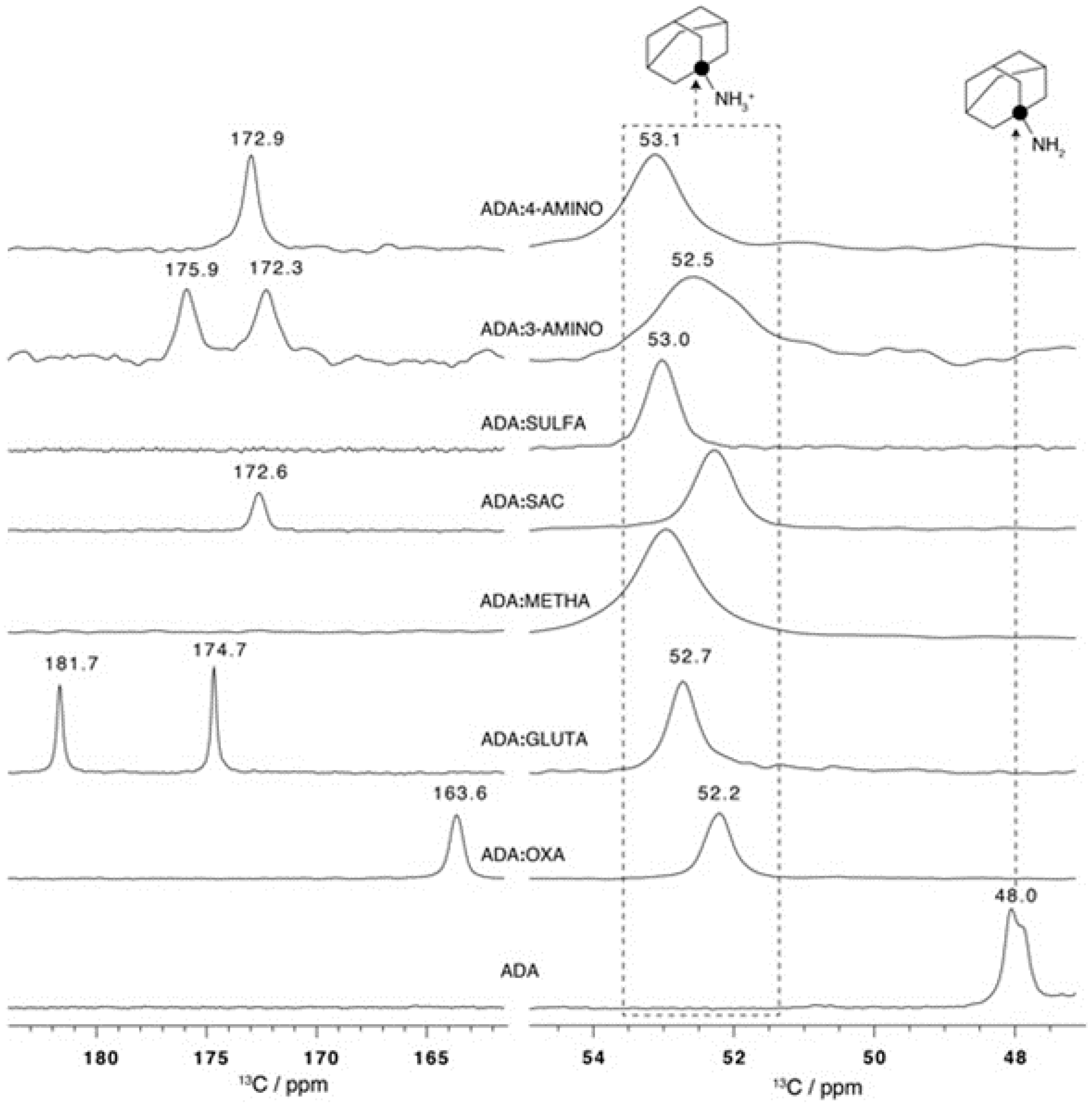
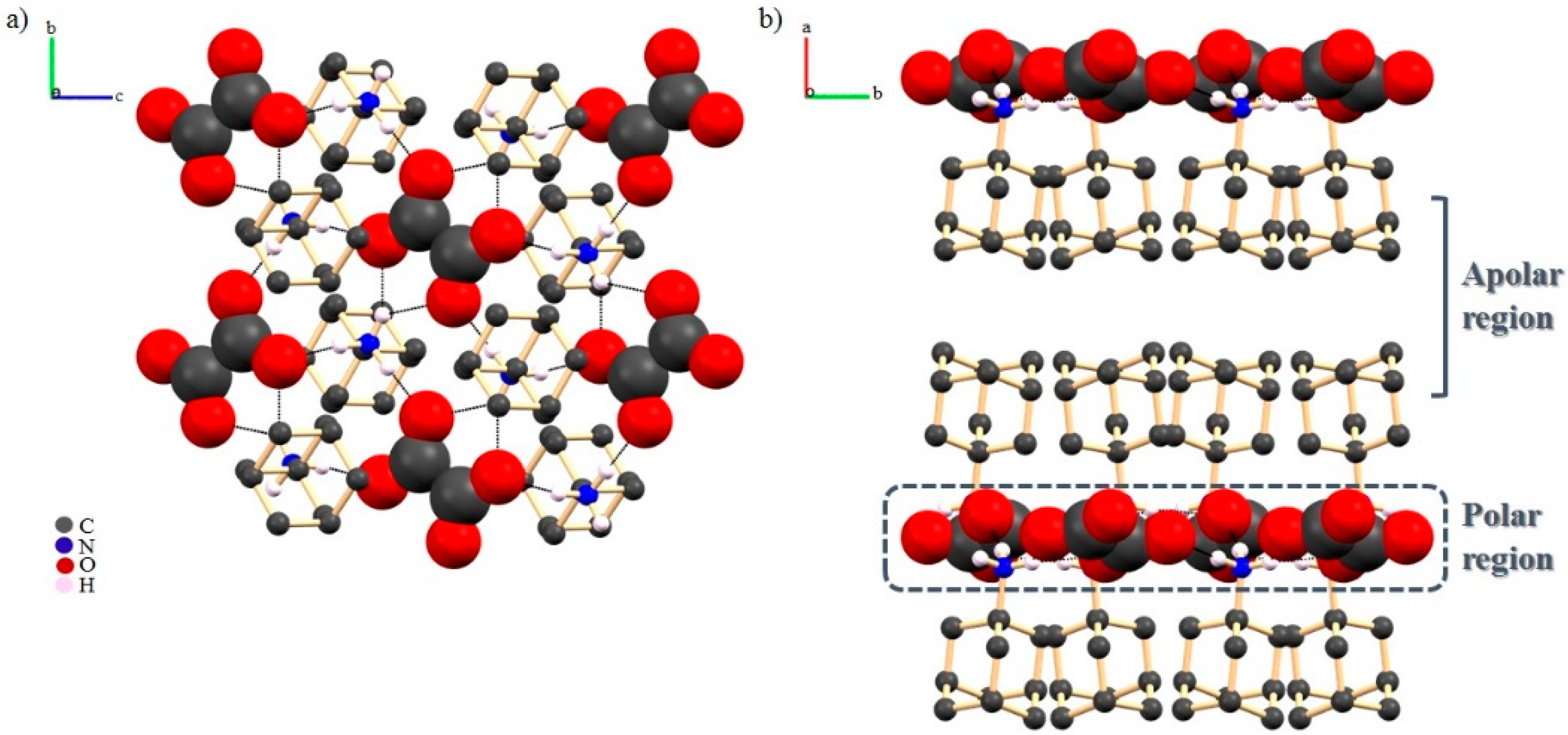
2.3.2. Adamantylamine
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brittain, H.G. Pharmaceutical cocrystals: The coming wave of new drug substances. J. Pharm. Sci. 2013, 102, 311–317. [Google Scholar] [CrossRef]
- Jones, W.; Motherwell, S.; Trask, A.V. Pharmaceutical cocrystals: An emerging approach to physical property enhancement. Mrs. Bull. 2006, 31, 875–879. [Google Scholar] [CrossRef] [Green Version]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [Green Version]
- Arenas-Garcia, J.I.; Herrera-Ruiz, D.; Morales-Rojas, H.; Hopfl, H. Interrelation of the dissolution behavior and solid-state features of acetazolamide cocrystals. Eur. J. Pharm. Sci. 2017, 96, 299–308. [Google Scholar] [CrossRef]
- Tan, D.; Loots, L.; Friscic, T. Towards medicinal mechanochemistry: Evolution of milling from pharmaceutical solid form screening to the synthesis of active pharmaceutical ingredients (APIs). Chem. Commun. 2016, 52, 7760–7781. [Google Scholar] [CrossRef]
- Desiraju, G. Supramolecular Synthons In Crystal Engineering-A New Organic-Synthesis. Angew. Chem. Int. Ed. Eng. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Desiraju, G. Crystal Engineering: The design of Organic Solids; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Bond, A.D. Fundamental Aspects of Salts and Co-crystals. In Pharmaceutical Salts and Co-crystals; Wouters, J., Queré, L., Eds.; RSC Publishing: Cambridge, UK, 2016; pp. 9–28. [Google Scholar]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef]
- Friscic, T. Supramolecular concepts and new techniques in mechanochemistry: Cocrystals, cages, rotaxanes, open metal-organic frameworks. Chem. Soc. Rev. 2012, 41, 3493–3510. [Google Scholar] [CrossRef]
- Shamshina, J.; Berton, P.; Wang, H.; Zhou, X.; Gurau, G.; Rodgers, R. Ionic Liquids in Pharmaceutical Industry. In Green Techniques for Organic Synthesis and Medicinal Chemistry, 2nd ed.; Zhang, W., Cue, B., Cue, B.W., Cue, B.W., Eds.; John Wiley & Sons, Ltd: Oxford, UK, 2018; pp. 539–577. [Google Scholar]
- Nangia, A.; Desiraju, G.R. Pseudopolymorphism: Occurrences of hydrogen bonding organic solvents in molecular crystals. Chem. Commun. 1999, 7, 605–606. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friscic, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, D.; Maini, L.; Grepioni, F. Mechanochemical preparation of co-crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. [Google Scholar] [CrossRef]
- Delori, A.; Friscic, T.; Jones, W. The role of mechanochemistry and supramolecular design in the development of pharmaceutical materials. CrystEngComm 2012, 14, 2350–2362. [Google Scholar] [CrossRef]
- Friscic, T.; Jones, W. Application of Mechanochemistry in the Synthesis and Discovery of New Pharmaceutical Forms: Co-crystals, Salts and Coordination Compounds. In Pharmaceutical Salts and Co-Crystals; Wouters, J., Queré, L., Eds.; RSC Publishing: Cambridge, UK, 2016; pp. 154–187. [Google Scholar]
- Ventura, B.; Bertocco, A.; Braga, D.; Catalano, L.; d’Agostino, S.; Grepioni, F.; Taddei, P. Luminescence Properties of 1,8-Naphthalimide Derivatives in Sol;ution, in Their Crystals, and in Co-crystals: Toward Room-Temperature Phosphorescence from Organic Materials. J. Phys. Chem. C 2014, 118, 18646–18658. [Google Scholar] [CrossRef]
- Weyna, D.R.; Shattock, T.; Vishweshwar, P.; Zaworotko, M.J. Synthesis and Structural Characterization of Cocrystals and Pharmaceutical Cocrystals: Mechanochemistry vs Slow Evaporation from Solution. Cryst. Growth Des. 2009, 9, 1106–1123. [Google Scholar] [CrossRef]
- Friscic, T. New opportunities for materials synthesis using mechanochemistry. J. Mater. Chem. 2010, 20, 7599–7605. [Google Scholar] [CrossRef]
- Frankenbach, G.M.; Etter, M.C. Relationship between Symmetry in Hydrogen-Bonded Benzoic-Acids and the Formation of Acentric Crystal-Structures. Chem. Mater. 1992, 4, 272–278. [Google Scholar] [CrossRef]
- Do, J.L.; Friscic, T. Mechanochemistry: A Force of Synthesis. ACS Cent. Sci. 2017, 3, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Hernández, J.G.; Bolm, C. Altering Product Selectivity by Mechanochemistry. J. Org. Chem. 2017, 82, 4007–4019. [Google Scholar] [CrossRef]
- Available online: https://www.lithmachine.com/planetary-ball-mill-machine_p163.html (accessed on 25 May 2020).
- Shan, N.; Toda, F.; Jones, W. Mechanochemistry and co-crystal formation: Effect of solvent on reaction kinetics. Chem. Commun. 2002, 20, 2372–2373. [Google Scholar] [CrossRef]
- Andre, V.; Braga, D.; Grepioni, F.; Duarte, M.T. Crystal Forms of the Antibiotic 4-Aminosalicylic Acid: Solvates and Molecular Salts with Dioxane, Morpholine, and Piperazine. Cryst. Growth Des. 2009, 9, 5108–5116. [Google Scholar] [CrossRef]
- Andre, V.; Duarte, M.T.; Braga, D.; Grepioni, F. Polymorphic Ammonium Salts of the Antibiotic 4-Aminosalicylic Acid. Cryst. Growth Des. 2012, 12, 3082–3090. [Google Scholar] [CrossRef]
- Andre, V.; da Piedade, M.F.M.; Duarte, M.T. Revisiting paracetamol in a quest for new co-crystals. CrystEngComm 2012, 14, 5005–5014. [Google Scholar] [CrossRef]
- Maheshwari, C.; Andre, V.; Reddy, S.; Roy, L.; Duarte, T.; Rodrigez-Hornedo, N. Tailoring aqueous solubility of a highly soluble compound via cocrystallization: effect of co-former ionization, pH(max) and solute-solvent interactions. CrystEngComm 2012, 14, 4801–4811. [Google Scholar] [CrossRef]
- Andre, V.; Cunha-Silva, L.; Duarte, M.T.; Santos, P.P. First Crystal Structures of the Antihypertensive Drug Perindopril Erbumine: A Novel Hydrated Form and Polymorphs alpha and beta. Cryst. Growth Des. 2011, 11, 3703–3706. [Google Scholar] [CrossRef]
- Andre, V.; Hardeman, A.; Halasz, I.; Stein, R.S.; Jackson, G.J.; Reid, D.G.; Duer, M.J.; Curfs, C.; Duarte, M.T.; Friscic, T. Mechanosynthesis of the Metallodrug Bismuth Subsalicylate from Bi2O3 and Structure of Bismuth Salicylate without Auxiliary Organic Ligands. Angew. Chem. Int. Ed. 2011, 50, 7858–7861. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, S.; Andre, V.; Antunes, A.M.M.; Vilela, S.M.F.; Amariei, G.; Arenas-Vivo, A.; Rosal, R.; Horcajada, P.; Duarte, M.T. Novel Antibacterial Azelaic Acid BioMOFs. Cryst. Growth Des. 2020, 20, 370–382. [Google Scholar] [CrossRef]
- Jensen, A.A.; Mosbacher, J.; Elg, S.; Lingenhoehl, K.; Lohmann, T.; Johansen, T.N.; Abrahamsen, B.; Mattsson, J.P.; Lehmann, A.; Bettler, B.; et al. The anticonvulsant gabapentin (Neurontin) does not act through gamma-aminobutyric acid-B receptors. Mol. Pharmacol. 2002, 61, 1377–1384. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.P.; Gee, N.S.; Su, T.Z.; Kocsis, J.D.; Welty, D.F.; Brown, J.P.; Dooley, D.J.; Boden, P.; Singh, L. A summary of mechanistic hypotheses of gabapentin pharmacology. Epilepsy Res. 1998, 29, 233–249. [Google Scholar] [CrossRef]
- Ettinger, A.B.; Argoff, C.E. Use of Antiepileptic drugs for nonepileptic conditions: Psychiatric disorders and chronic pain. Neurotherapeutics 2007, 4, 75–83. [Google Scholar] [CrossRef]
- Reece, H.A.; Levendis, D.C. Polymorphs of gabapenitin. Acta Crystallogr. C 2008, 64, CO105–CO108. [Google Scholar] [CrossRef]
- Ibers, J.A. Gabapentin and gabapentin monohydrate. Acta Crystallogr. C 2001, 57, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Grepioni, F.; Maini, L.; Rubini, K.; Polito, M.; Brescello, R.; Cotarca, L.; Duarte, M.T.; Andre, V.; Piedade, M.F.M. Polymorphic gabapentin: Thermal behaviour, reactivity and interconversion of forms in solution and solid-state. New J. Chem. 2008, 32, 1788–1795. [Google Scholar] [CrossRef]
- Vasudev, P.G.; Aravinda, S.; Ananda, K.; Veena, S.D.; Nagarajan, K.; Shamala, N.; Balaram, P. Crystal Structures of a New Polymorphic Form of Gabapentin Monohydrate and the E and Z Isomers of 4-Tertiarybutylgabapentin. Chem. Biol. Drug Des. 2009, 73, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.E.; Greenman, B.J. Gabapentin monohydrate and a process for producing the same. US4894476-A, 16 January 1990. [Google Scholar]
- Braga, D.; Grepioni, F.; Maini, L.; Polito, M. Crystal Polymorphism and Multiple Crystal Forms. Mol. Networks 2009, 132, 25–50. [Google Scholar]
- Domingos, S.; Andre, V.; Quaresma, S.; Martins, I.C.B.; da Piedade, M.F.M.; Duarte, M.T. New forms of old drugs: Improving without changing. J. Pharm. Pharmacol. 2015, 67, 830–846. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and fictions about polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef] [PubMed]
- Brittain, H.G. Polymorphism and solvatomorphism 2006. J. Pharm. Sci. 2008, 97, 3611–3636. [Google Scholar] [CrossRef]
- Martins, I.C.B.; Gomes, J.R.B.; Duarte, M.T.; Mafra, L. Understanding Polymorphic Control of Pharmaceuticals Using lmidazolium-Based Ionic Liquid Mixtures as Crystallization Directing Agents. Cryst. Growth Des. 2017, 17, 428–432. [Google Scholar] [CrossRef]
- Andre, V.; Fernandes, A.; Santos, P.P.; Duarte, M.T. On the Track of New Multicomponent Gabapentin Crystal Forms: Synthon Competition and pH Stability. Cryst. Growth Des. 2011, 11, 2325–2334. [Google Scholar] [CrossRef]
- Martins, I.C.B.; Oliveira, M.C.; Diogo, H.P.; Branco, L.C.; Duarte, M.T. MechanoAPI-ILs: Pharmaceutical Ionic Liquids Obtained through Mechanochemical Synthesis. Chemsuschem 2017, 10, 1360–1363. [Google Scholar] [CrossRef]
- Rafilovich, M.; Bernstein, J.; Hickey, M.B.; Tauber, M. Benzidine: A co-crystallization agent for proton acceptors. Cryst. Growth Des. 2007, 7, 1777–1782. [Google Scholar] [CrossRef]
- Eccles, K.S.; Elcoate, C.J.; Stokes, S.P.; Maguire, A.R.; Lawrence, S.E. Sulfoxides: Potent Co-Crystal Formers. Cryst. Growth Des. 2010, 10, 4243–4245. [Google Scholar] [CrossRef]
- Martins, I.; Martins, M.; Fernandes, A.; Andre, V.; Duarte, M.T. An insight into dapsone co-crystals: Sulfones as participants in supramolecular interactions. CrystEngComm 2013, 15, 8173–8179. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. B 2002, 58, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, H.; Stieger, N.; Liebenberg, W.; Caira, M.R. Solvatomorphism of the Antibacterial Dapsone: X-ray Structures and Thermal Desolvation Kinetics. Cryst. Growth Des. 2012, 12, 1683–1692. [Google Scholar] [CrossRef]
- Smith, G.; Wermuth, U.D. Hydrogen bonding in cyclic imides and amide carboxylic acid derivatives from the facile reaction of cis-cyclohexane-1,2-carboxylic anhydride with o- and p-anisidine and m- and p-aminobenzoic acids. Acta Crystallogr. C 2012, 68. [Google Scholar]
- Skold, O. Sulfonamide resistance: Mechanisms and trends. Drug Resist Update 2000, 3, 155–160. [Google Scholar] [CrossRef]
- Caira, M.R. Sulfa drugs as model cocrystal formers. Mol. Pharmaceut. 2007, 4, 310–316. [Google Scholar] [CrossRef]
- Hunter, C.A. Quantifying intermolecular interactions: Guidelines for the molecular recognition toolbox. Angew. Chem. Int. Ed. 2004, 43, 5310–5324. [Google Scholar] [CrossRef]
- Adsmond, D.A.; Grant, D.J.W. Hydrogen bonding in sulfonamides. J. Pharm. Sci. 2001, 90, 2058–2077. [Google Scholar] [CrossRef] [PubMed]
- Goud, N.R.; Khan, R.A.; Nangia, A. Modulating the solubility of sulfacetamide by means of cocrystals. CrystEngComm 2014, 16, 5859–5869. [Google Scholar] [CrossRef]
- Domingos, S.; Fernandes, A.; Duarte, M.T.; Piedade, M.F.M. New Multicomponent Sulfadimethoxine Crystal Forms: Sulfonamides as Participants in Supramolecular Interactions. Cryst. Growth Des. 2016, 16, 1879–1892. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The salt-cocrystal continuum: The influence of crystal structure on ionization state. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, C.; Tapmeyer, L.; Gumbert, S.D.; Prill, D.; Bolte, M.; Schmidt, M.U.; Saal, C. Absolute Configuration of Pharmaceutical Research Compounds Determined by X-ray Powder Diffraction. Angew. Chem. Int. Ed. 2018, 57, 9150–9153. [Google Scholar] [CrossRef]
- Dempah, K.E.; Barich, D.H.; Kaushal, A.M.; Zong, Z.X.; Desai, S.D.; Suryanarayanan, R.; Kirsch, L.; Munson, E.J. Investigating Gabapentin Polymorphism Using Solid-State NMR Spectroscopy. Aaps. Pharmscitech. 2013, 14, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, J.S.; Byard, S.J.; Seaton, C.C.; Sadiq, G.; Davey, R.J.; Schroeder, S.L.M. Proton transfer and hydrogen bonding in the organic solid state: A combined XRD/XPS/ssNMR study of 17 organic acid-base complexes. Phys. Chem. Chem. Phys. 2014, 16, 1150–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mafra, L.; Santos, S.M.; Siegel, R.; Alves, I.; Paz, F.A.A.; Dudenko, D.; Spiess, H.W. Packing Interactions in Hydrated and Anhydrous Forms of the Antibiotic Ciprofloxacin: A Solid-State NMR, X-ray Diffraction, and Computer Simulation Study. J. Am. Chem. Soc. 2012, 134, 71–74. [Google Scholar] [CrossRef]
- Martins, I.C.B.; Sardo, M.; Santos, S.M.; Femandes, A.; Antunes, A.; Andre, V.; Mafra, L.; Duarte, M.T. Packing Interactions and Physicochemical Properties of Novel Multicomponent Crystal Forms of the Anti-Inflammatory Azelaic Acid Studied by X-ray and Solid-State NMR. Cryst. Growth Des. 2016, 16, 154–166. [Google Scholar] [CrossRef]
- Liu, R.H.; Smith, M.K.; Basta, S.A.; Farmer, E.R. Azelaic acid in the treatment of papulopustular rosacea—A systematic review of randomized controlled trials. Arch. Dermatol. 2006, 142, 1047–1052. [Google Scholar] [CrossRef]
- Thompson, L.J.; Voguri, R.S.; Male, L.; Tremayne, M. The crystal structures and melting point properties of isonicotinamide cocrystals with alkanediacids HO2C(CH2)(n-2)CO2H n=7-9. CrystEngComm 2011, 13, 4188–4195. [Google Scholar] [CrossRef]
- Braga, D.; Dichiarante, E.; Palladino, G.; Grepioni, F.; Chierotti, M.R.; Gobetto, R.; Pellegrino, L. Remarkable reversal of melting point alternation by co-crystallization. CrystEngComm 2010, 12, 3534–3536. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Panikkattu, S.V.; DeHaven, B.; Desper, J. Establishing Supramolecular Control over Solid-State Architectures: A Simple Mix and Match Strategy. Cryst. Growth Des. 2012, 12, 2579–2587. [Google Scholar] [CrossRef]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 11. [Google Scholar] [CrossRef]
- Stanicova, J.; Miskovsky, P.; Sutiak, V. Amantadine: An antiviral and antiparkinsonian agent. Vet. Med.-Czech 2001, 46, 244–256. [Google Scholar] [CrossRef] [Green Version]
- Hubsher, G.; Haider, M.; Okun, M.S. Amantadine The journey from fighting flu to treating Parkinson disease. Neurology 2012, 78, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Sood, R.; Chanu, K.V.; Bhatia, S.; Khandia, R.; Pateriya, A.K.; Nagarajan, S.; Dimri, U.; Kulkarni, D.D. Amantadine resistance among highly pathogenic avian influenza viruses (H5N1) isolated from India. Microb. Pathog. 2016, 91, 35–40. [Google Scholar] [CrossRef]
- Rodnitzky, R.L.; Narayanan, N.S. Amantadine’s role in the treatment of levodopa-induced dyskinesia. Neurology 2014, 82, 288–289. [Google Scholar] [CrossRef] [Green Version]
- Dolin, R.; Reichman, R.C.; Madore, H.P.; Maynard, R.; Linton, P.N.; Webberjones, J. A CONTROLLED TRIAL OF AMANTADINE AND RIMANTADINE IN THE PROPHYLAXIS OF INFLUENZA-A INFECTION. N. Eng. J. Med. 1982, 307, 580–584. [Google Scholar] [CrossRef]
- Heim, C.; Sontag, K.H. Memantine prevents progressive functional neurodegeneration in rats. J. Neural Transm. Suppl. 1995, 46, 117–130. [Google Scholar]
- Martins, I.C.B.; Sardo, M.; Alig, E.; Fink, L.; Schmidt, M.U.; Mafra, L.; Duarte, M.T. Enhancing Adamantylamine Solubility through Salt Formation: Novel Products Studied by X-ray Diffraction and Solid-State NMR. Cryst. Growth Des. 2019, 19, 1860–1873. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
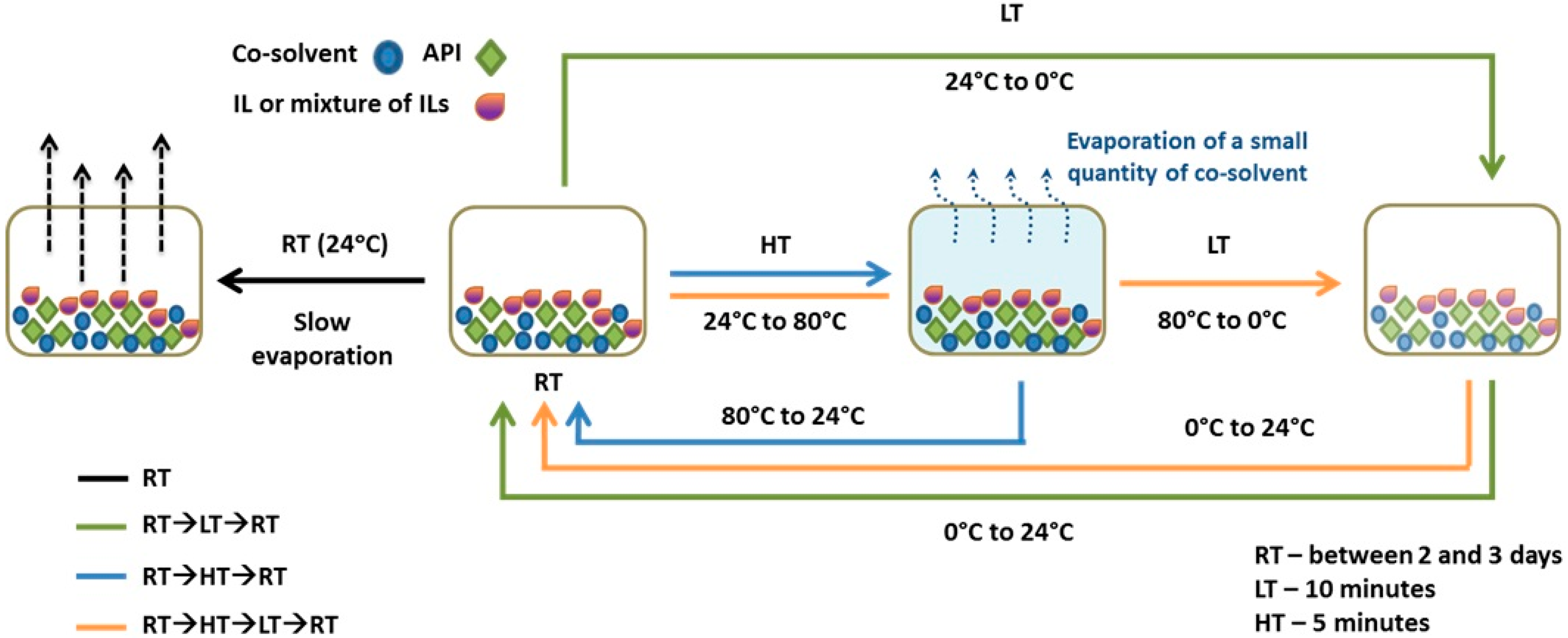
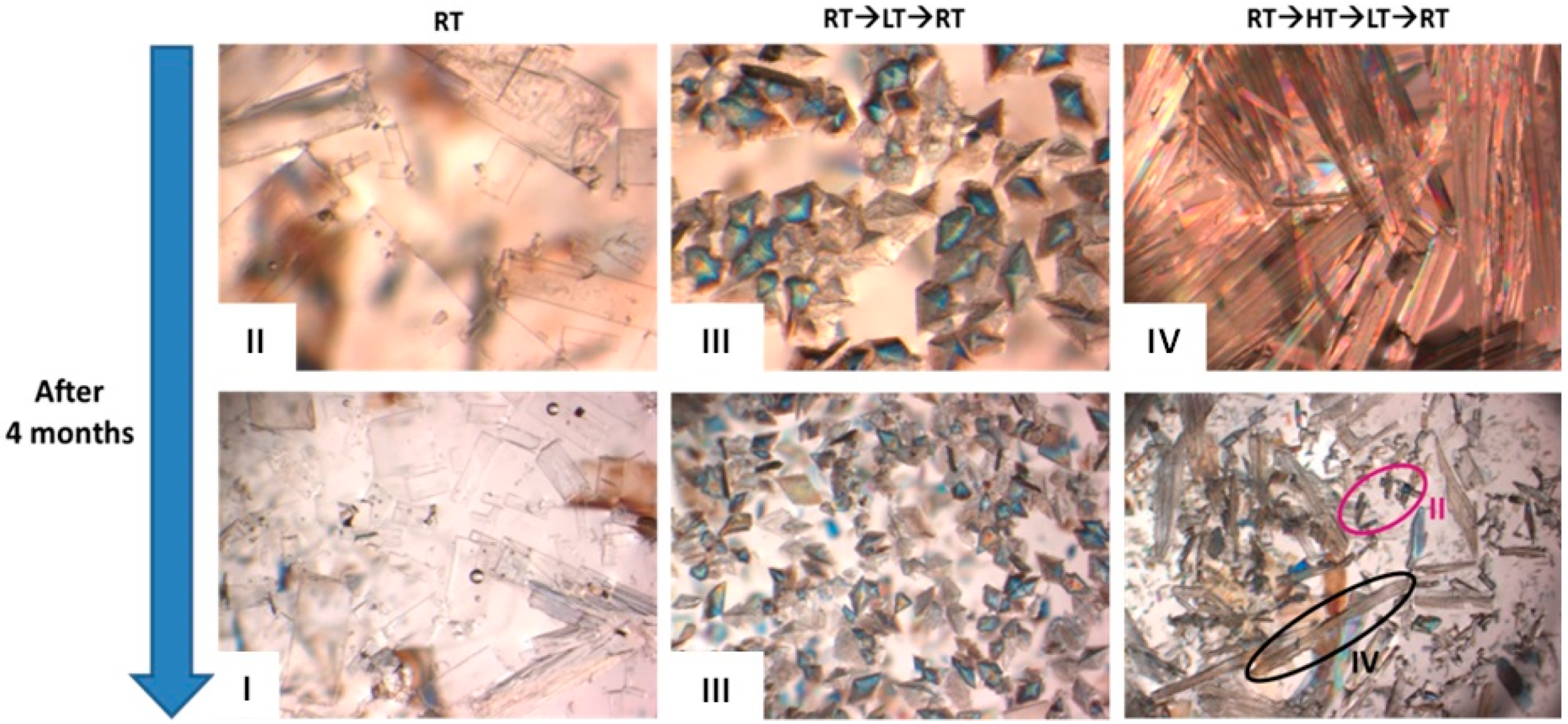
| Entry | RTIL in Methanol | RT | RT→LT→RT | RT→HT→RT | RT→HT→LT→RT |
|---|---|---|---|---|---|
| 1 | Control Samplea | Form II | Form II | Form II | Form II |
| 2 | C4mimN(CF3SO2)2 | Form II | Form IV | Form III | Form III |
| 3 | C4mimBF4 | Form II | Form II + III | Form II | Form II + IV |
| 4 | C6mimBF4 | Form II | Form III | Form II + IV | Form IV |
| 5 | C4mimBF4 + C6mimBF4 | Form II | Form III | Form IV | Form IV |
| 6 | C4mimN(CF3SO2)2 + C6mimN(CF3SO2)2 | Form II + III | Form II + III + IV | Form III | Form IV |
| 7 | C6mimN(CF3SO2)2 + C6mimBF4 | Form II | Form II | Form II | Form II |
| Acid-base Neutralization (Traditional Method) | Mechanochemistry (New Method) | |||||||
|---|---|---|---|---|---|---|---|---|
| Compound Name a | Physical State at RT | Yield (%) | Tg (°C) | Reaction Time (h) | Physical State at RT | Yield (%) | Tg (°C) | Reaction Time (min) |
| (EMIM)(GBP) | Transparent liquid | 96.5 | −70.9 | 15 | Transparent liquid | 97.7 | −72.2 | 180b |
| (choline)(GBP) | Transparent viscous liquid | 98.5 | −59.2 | 15 | Transparent viscous liquid | 99.0 | −59.0 | 180b |
| (GBP)(D-glu) | Orange viscous liquid | 92.5 | −48.6 | 10 | Orange viscous liquid | 95.2 | −44.0 | 30 |
| (GBP)(gly) | Transparent liquid | 97.2 | −56.2 | 10 | Transparent liquid | 99.3 | −49.1 | 15 |
| (TMG)(L-glut) | Light yellow liquid | 93.7 | −36.2 | 6 | Light yellow liquid | 97.0 | −38.9 | 10 |
| (DBU)(L-glut) | Yellow liquid | 98.4 | −39.5 | 6 | Yellow liquid | 99.5 | −44.5 | 10 |
| Compound | Solubility (mg/mL) | Melting Point (°C) | Melting Point Conformers (°C) |
|---|---|---|---|
| ADA | 1.03 | 206–208 | - |
| ADA∙HCl | 50 | 300 | - |
| ADA:OXA (I) | 0.81 | 223 | 189–191 |
| ADA:GLUTA (II) | 58.8 | 183 | 95–98 |
| ADA:METHA (V) | 200 | 153.9 a | 17–19 |
| ADA:SAC (VI) | 14.3 | 242.6 | 228 |
| ADA:SULFA (VII) | 5.6 | 347.3 a | 288 |
| ADA:3-AMINO (VIII) | 9.1 | 244.2 | 178–180 |
| ADA:4-AMINO (IX) | 0.96 | 245.9 | 187–189 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira da Silva, J.L.; Minas da Piedade, M.F.; André, V.; Domingos, S.; Martins, I.C.B.; Duarte, M.T. The Lisbon Supramolecular Green Story: Mechanochemistry towards New Forms of Pharmaceuticals. Molecules 2020, 25, 2705. https://doi.org/10.3390/molecules25112705
Ferreira da Silva JL, Minas da Piedade MF, André V, Domingos S, Martins ICB, Duarte MT. The Lisbon Supramolecular Green Story: Mechanochemistry towards New Forms of Pharmaceuticals. Molecules. 2020; 25(11):2705. https://doi.org/10.3390/molecules25112705
Chicago/Turabian StyleFerreira da Silva, João Luís, M. Fátima Minas da Piedade, Vânia André, Sofia Domingos, Inês C. B. Martins, and M. Teresa Duarte. 2020. "The Lisbon Supramolecular Green Story: Mechanochemistry towards New Forms of Pharmaceuticals" Molecules 25, no. 11: 2705. https://doi.org/10.3390/molecules25112705