
Effects of Intra- and Interchain Interactions on Exciton Dynamics of PTB7 Revealed by Model Oligomers

 ,
,
Abstract
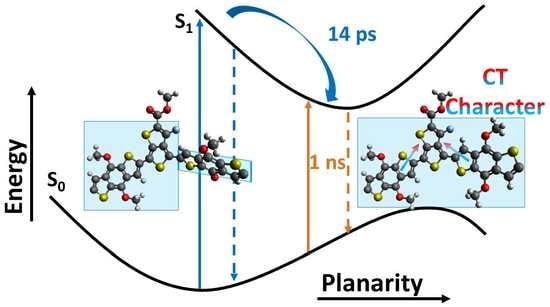
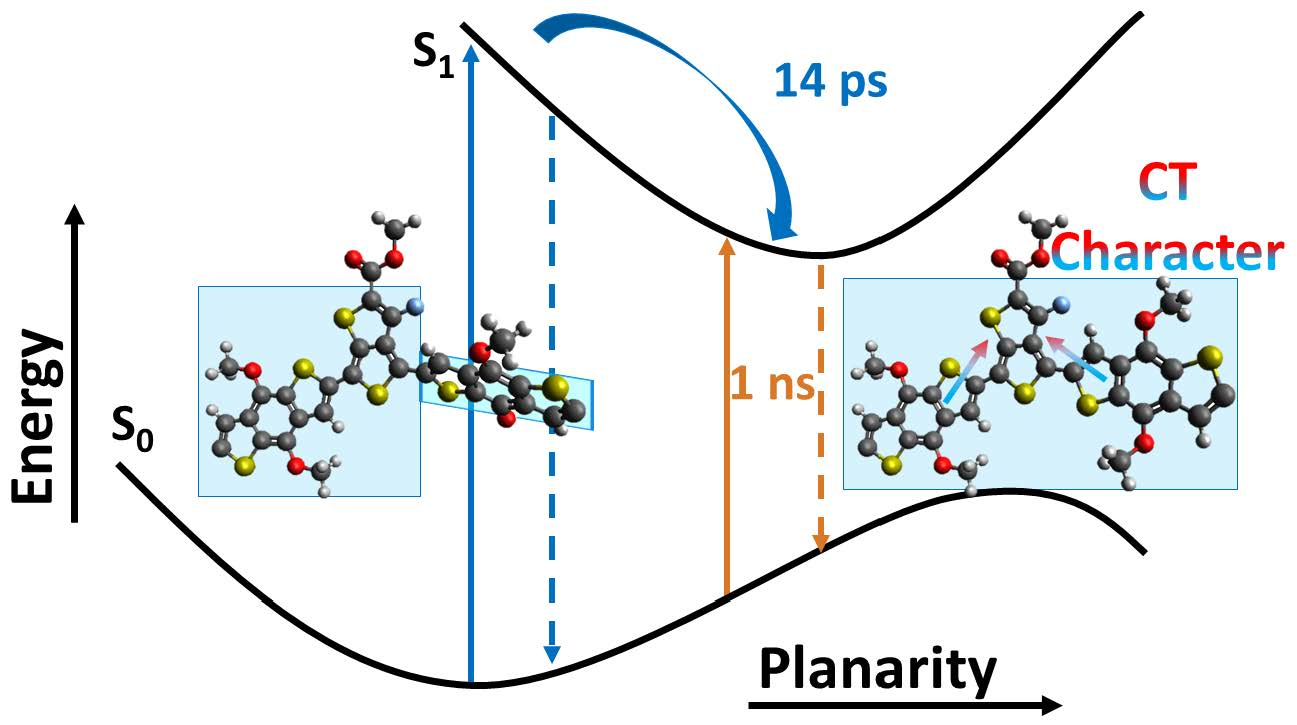
:
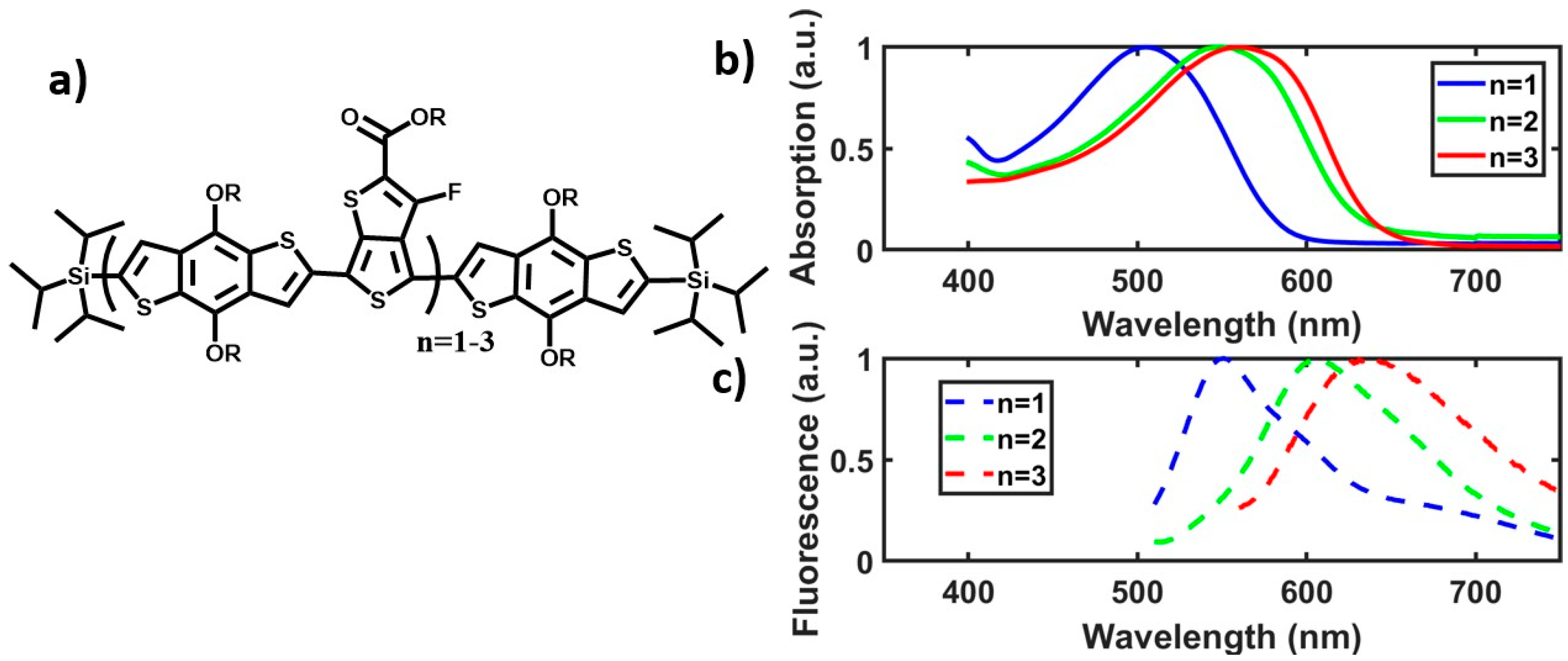
{kind=link}
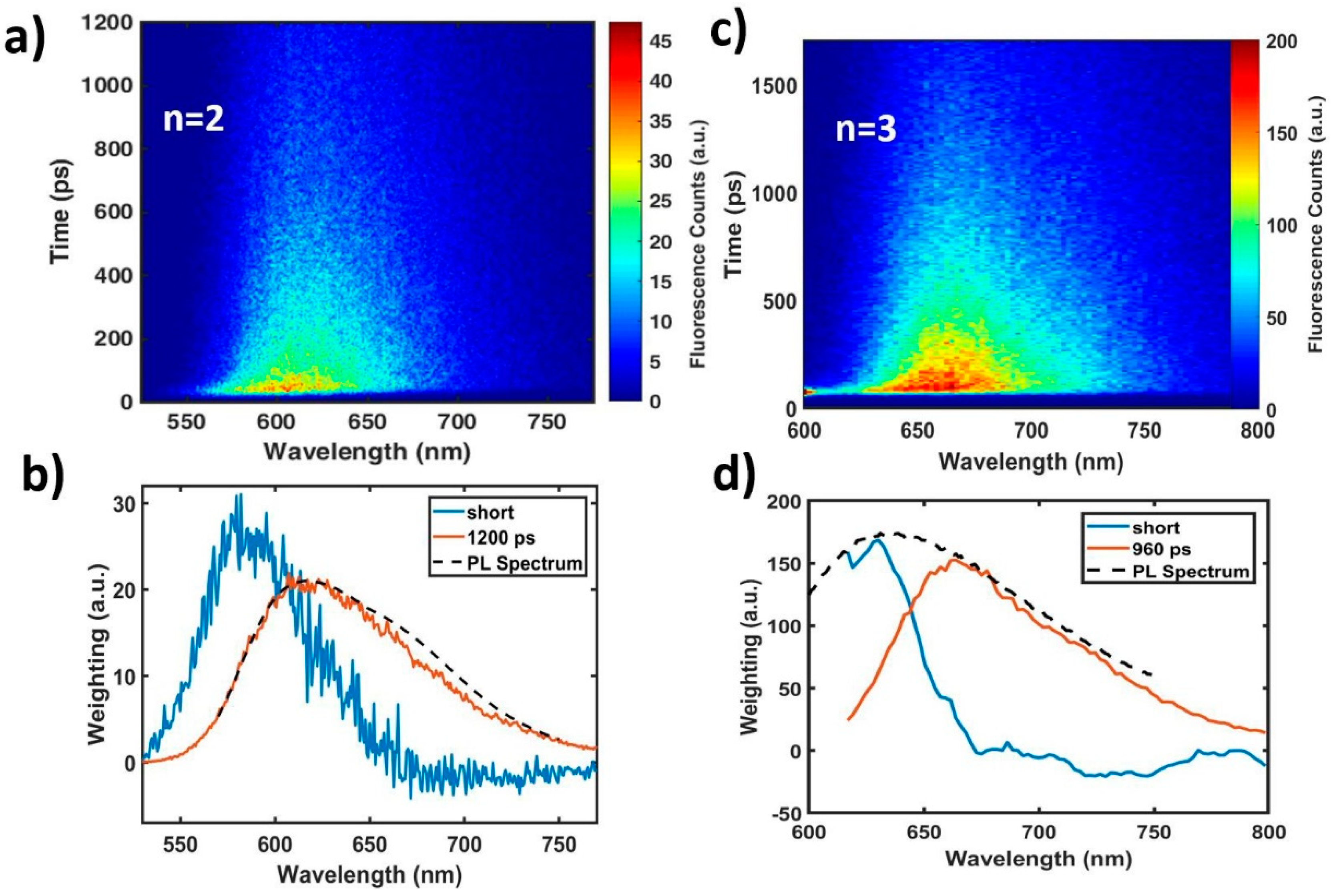
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussions
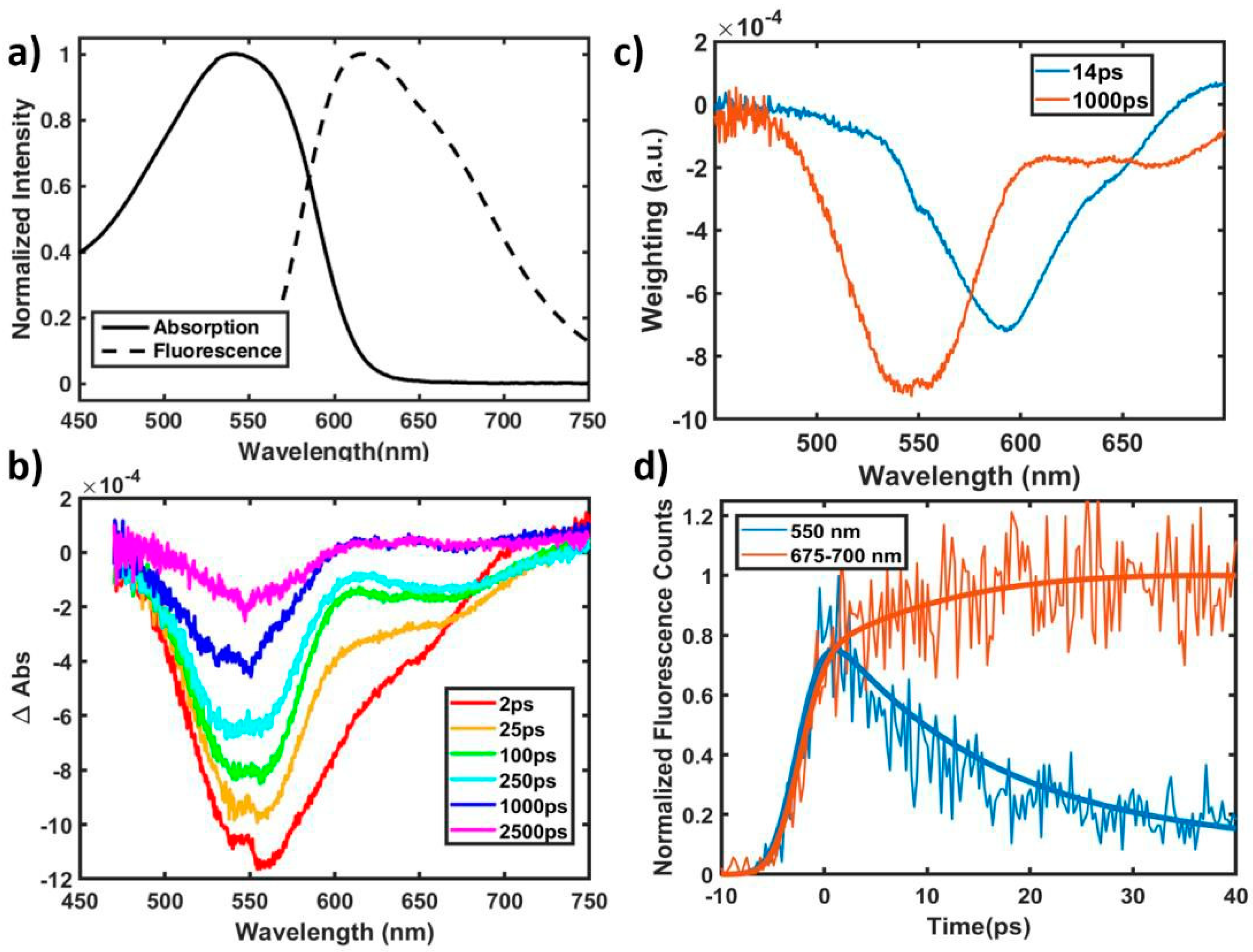
2.1. Excited State and Structural Dynamics in Oligomers
2.2. In-Chain Effects on CT Character.
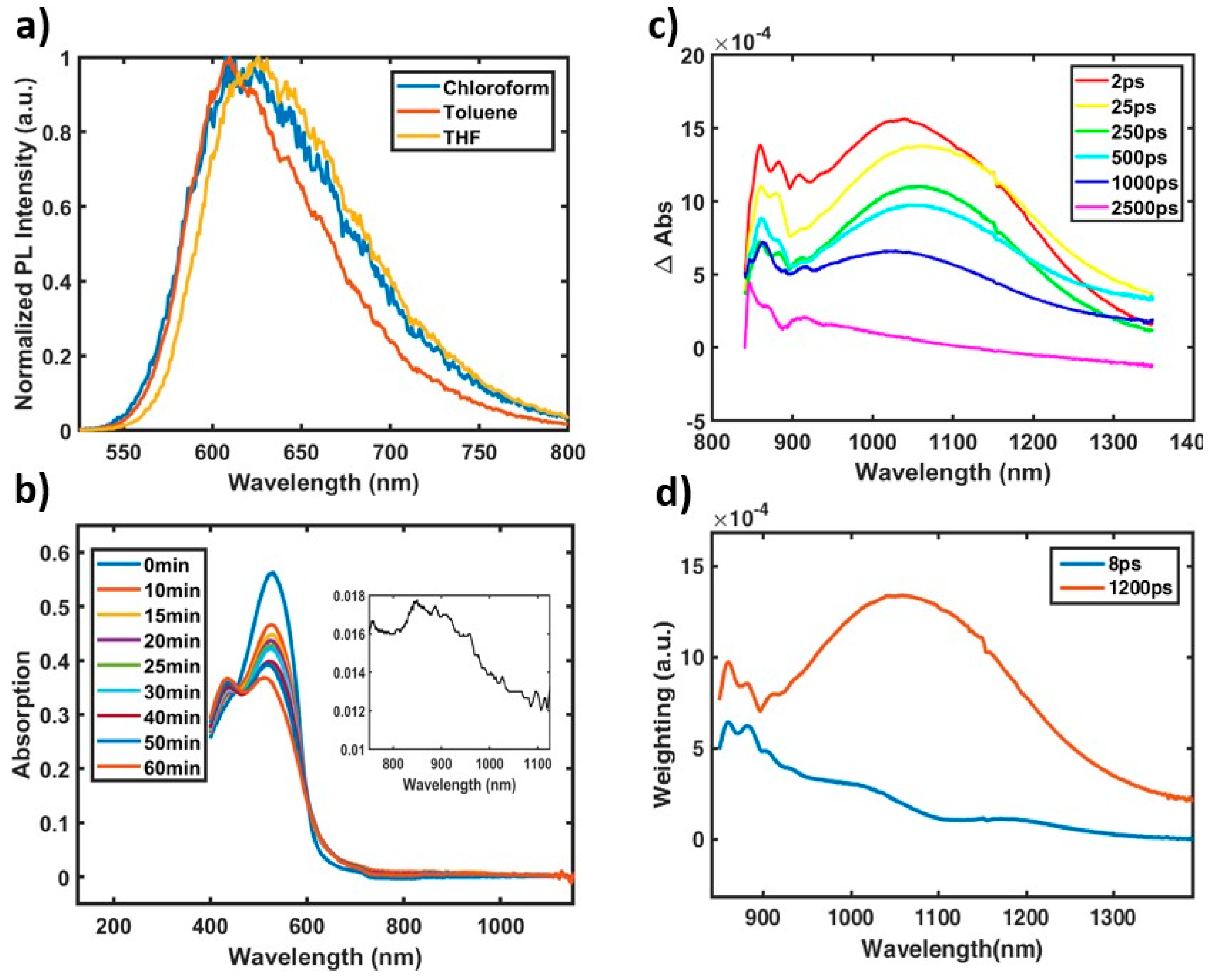
2.3. Aggregation Effects on CT Character.
3. Materials and Methods
3.1. Materials
3.2. Steady State Spectroscopy
3.3. Spectro-electrochemistry
3.4. Ultrafast Transient Absorption Spectroscopy
3.5. Streak Camera Fluorescence Measurements
3.6. Data Analyses.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yue, D.; Khatav, P.; You, F.; Darling, S.B. Deciphering the Uncertainties in Life Cycle Energy and Environmental Analysis of Organic Photovoltaics. Energy Environ. Sci. 2012, 5, 9163–9172. [Google Scholar] [CrossRef]
- Zhang, H.; Yao, H.; Hou, J.; Zhu, J.; Zhang, J.; Li, W.; Yu, R.; Gao, B.; Zhang, S.; Hou, J. Over 14% Efficiency in Organic Solar Cells Enabled by Chlorinated Nonfullerene Small-Molecule Acceptors. Adv. Mater. 2018, 30, 1800613. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ye, L.; Zhao, W.; Yan, H.; Yang, B.; Liu, D.; Li, W.; Ade, H.; Hou, J. A Wide Band-Gap Polymer with a Deep HOMO Level Enables 14.2% Efficiency in Polymer Solar Cells. J. Am. Chem. Soc. 2018, 140, 7159–7167. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhang, Y.; Wan, X.; Li, C.; Zhang, X.; Wang, Y.; Ke, X.; Xiao, Z.; Ding, L.; Xia, R.; et al. Organic and Solution-Processed Tandem Solar Cells with 17.3% Efficiency. Science. 2018, 361, 1094–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, N.E.; Savoie, B.M.; Marks, T.J.; Chen, L.X.; Ratner, M.A. The Next Breakthrough for Organic Photovoltaics? J. Phys. Chem. Lett. 2015, 6, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H.; et al. Aggregation and Morphology Control Enables Multiple Cases of High-Efficiency Polymer Solar Cells. Nat. Commun. 2014, 5, 5293. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Li, Y.; Yang, G.; Jiang, K.; Lin, H.; Ade, H.; Ma, W.; Yan, H. Efficient Organic Solar Cells Processed from Hydrocarbon Solvents. Nat. Energy 2016, 1, 15027. [Google Scholar] [CrossRef]
- Ma, W.; Yang, G.; Jiang, K.; Carpenter, J.H.; Wu, Y.; Meng, X.; McAfee, T.; Zhao, J.; Zhu, C.; Wang, C.; et al. Influence of Processing Parameters and Molecular Weight on the Morphology and Properties of High-Performance PffBT4T-2OD:PC71BM Organic Solar Cells. Adv. Energy Mater. 2015, 5, 1501400. [Google Scholar] [CrossRef]
- Lu, L.; Yu, L. Understanding Low Bandgap Polymer PTB7 and Optimizing Polymer Solar Cells Based on IT. Adv. Mater. 2014, 26, 4413–4430. [Google Scholar] [CrossRef]
- To, C.H.; Ng, A.; Dong, Q.; Djurišić, A.B.; Zapien, J.A.; Chan, W.K.; Surya, C. Effect of PTB7 Properties on the Performance of PTB7:PC 71 BM Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 13198–13207. [Google Scholar] [CrossRef]
- He, X.; Mukherjee, S.; Watkins, S.; Chen, M.; Qin, T.; Thomsen, L.; Ade, H.; McNeill, C.R. Influence of Fluorination and Molecular Weight on the Morphology and Performance of PTB7:PC71BM Solar Cells. J. Phys. Chem. C 2014, 118, 9918–9929. [Google Scholar] [CrossRef]
- Liu, F.; Zhao, W.; Tumbleston, J.R.; Wang, C.; Gu, Y.; Wang, D.; Briseno, A.L.; Ade, H.; Russell, T.P. Understanding the Morphology of PTB7:PCBM Blends in Organic Photovoltaics. Adv. Energy Mater. 2014, 4, 1–9. [Google Scholar] [CrossRef]
- Sharma, R.; Lee, H.; Gupta, V.; Kim, H.; Kumar, M.; Sharma, C.; Chand, S.; Yoo, S.; Gupta, D. Photo-Physics of PTB7, PCBM and ICBA Based Ternary Solar Cells. Org. Electron. 2016, 34, 111–117. [Google Scholar] [CrossRef]
- Manley, E.F.; Strzalka, J.; Fauvell, T.J.; Jackson, N.E.; Leonardi, M.J.; Eastham, N.D.; Marks, T.J.; Chen, L.X. In Situ GIWAXS Analysis of Solvent and Additive Effects on PTB7 Thin Film Microstructure Evolution during Spin Coating. Adv. Mater. 2017, 29, 1703933. [Google Scholar] [CrossRef]
- Dang, M.T.; Hirsch, L.; Wantz, G. P3HT:PCBM, Best Seller in Polymer Photovoltaic Research. Adv. Mater. 2011, 23, 3597–3602. [Google Scholar] [CrossRef]
- Dante, M.; Peet, J.; Nguyen, T.Q. Nanoscale Charge Transport and Internal Structure of Bulk Heterojunction Conjugated Polymer/Fullerene Solar Cells by Scanning Probe Microscopy. J. Phys. Chem. C 2008, 112, 7241–7249. [Google Scholar] [CrossRef]
- Montanari, I.; Nogueira, A.F.; Nelson, J.; Durrant, J.R.; Winder, C.; Loi, M.A.; Sariciftci, N.S.; Brabec, C. Transient Optical Studies of Charge Recombination Dynamics in a Polymer/Fullerene Composite at Room Temperature. Appl. Phys. Lett. 2002, 81, 3001–3003. [Google Scholar] [CrossRef] [Green Version]
- Spano, F.C.; Silva, C. H- and J-Aggregate Behavior in Polymeric Semiconductors. Annu. Rev. Phys. Chem. 2014, 65, 477–500. [Google Scholar] [CrossRef]
- Yamagata, H.; Spano, F.C. Interplay between Intrachain and Interchain Interactions in Semiconducting Polymer Assemblies: The HJ-Aggregate Model. J. Chem. Phys. 2012, 136, 1–14. [Google Scholar] [CrossRef]
- Fauvell, T.J.; Zheng, T.; Jackson, N.E.; Ratner, M.A.; Yu, L.; Chen, L.X. Photophysical and Morphological Implications of Single-Strand Conjugated Polymer Folding in Solution. Chem. Mater. 2016, 28, 2814–2822. [Google Scholar] [CrossRef]
- Scharber, M.C.; Sariciftci, N.S. Efficiency of Bulk-Heterojunction Organic Solar Cells. Prog. Polym. Sci. 2013, 38, 1929–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolczynski, B.S.; Szarko, J.M.; Son, H.J.; Liang, Y.; Yu, L.; Chen, L.X. Ultrafast Intramolecular Exciton Splitting Dynamics in Isolated Low-Band-Gap Polymers and Their Implications in Photovoltaic Materials Design. J. Am. Chem. Soc. 2012, 134, 4142–4152. [Google Scholar] [CrossRef] [PubMed]
- Szarko, J.M.; Guo, J.; Liang, Y.; Lee, B.; Rolczynski, B.S.; Strzalka, J.; Xu, T.; Loser, S.; Marks, T.J.; Yu, L.; et al. When Function Follows Form: Effects of Donor Copolymer Side Chains on Film Morphology and BHJ Solar Cell Performance. Adv. Mater. 2010, 22, 5468–5472. [Google Scholar] [CrossRef] [PubMed]
- Rolczynski, B.S.; Szarko, J.M.; Son, H.J.; Yu, L.; Chen, L.X. Effects of Exciton Polarity in Charge-Transfer Polymer/PCBM Bulk Heterojunction Films. J. Phys. Chem. Lett. 2014, 5, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Szarko, J.M.; Rolczynski, B.S.; Lou, S.J.; Xu, T.; Strzalka, J.; Marks, T.J.; Yu, L.; Chen, L.X. Photovoltaic Function and Exciton/Charge Transfer Dynamics in a Highly Efficient Semiconducting Copolymer. Adv. Funct. Mater. 2014, 24, 10–26. [Google Scholar] [CrossRef]
- Chosrovian, H.; Rentsch, S.; Grebner, D.; Dahm, D.U.; Birckner, E.; Naarmann, H. Time-Resolved Fluorescence Studies on Thiophene Oligomers in Solution. Synth. Met. 1993, 60, 23–26. [Google Scholar] [CrossRef]
- Jackson, N.E.; Savoie, B.M.; Kohlstedt, K.L.; Olvera de la Cruz, M.; Schatz, G.C.; Chen, L.X.; Ratner, M.A. Controlling Conformations of Conjugated Polymers and Small Molecules: The Role of Nonbonding Interactions. J. Am. Chem. Soc. 2013, 135, 10475–10483. [Google Scholar] [CrossRef]
- Lanzani, G.; Nisoli, M.; De Silvestri, S.; Tubino, R. Femtosecond Optical Dynamics of α-Conjugated Hexamethylsexithiophene in Solution. Synth. Met. 1996, 76, 39–41. [Google Scholar] [CrossRef]
- Busby, E.; Carroll, E.C.; Chinn, E.M.; Chang, L.; Moulé, A.J.; Larsen, D.S. Excited-State Self-Trapping and Ground-State Relaxation Dynamics in Poly3-Hexylthiophene. Resolved with Broadband Pump- Dump-Probe Spectroscopy. J. Phys. Chem. Lett. 2011, 2, 2764–2769. [Google Scholar] [CrossRef]
- Zheng, T.; Yu, L. Roles of Quinoidal Character, Regioregularity, and Polydispersity in Determining the Photovoltaic Performance of Conjugated Copolymers. Macromolecules 2014, 47, 6252–6259. [Google Scholar] [CrossRef]
- Liang, Y.; Yu, L. Development of Semiconducting Polymers for Solar Energy Harvesting. Polym. Rev. 2010, 50, 454–473. [Google Scholar] [CrossRef]
- Risko, C.; McGehee, M.D.; Brédas, J.-L. A Quantum-Chemical Perspective into Low Optical-Gap Polymers for Highly-Efficient Organic Solar Cells. Chem. Sci. 2011, 2, 1200. [Google Scholar] [CrossRef]
- Berezin, M.M.Y.; Achilefu, S. Fluorescence Lifetime Measurements and Biological Imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickler, S.J.; Berg, R.A. Relationship between Absorption Intensity and Fluorescence Lifetime of Molecules. J. Chem. Phys. 1962, 37, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Tamai, Y.; Fan, Y.; Kim, V.O.; Ziabrev, K.; Rao, A.; Barlow, S.; Marder, S.R.; Friend, R.H.; Menke, S.M. Ultrafast Long-Range Charge Separation in Nonfullerene Organic Solar Cells. ACS Nano 2017, 11, 12473–12481. [Google Scholar] [CrossRef]
- Hwang, I.; Beaupré, S.; Leclerc, M.; Scholes, G.D. Ultrafast Relaxation of Charge-Transfer Excitons in Low-Bandgap Conjugated Copolymers. Chem. Sci. 2012, 3, 2270–2277. [Google Scholar] [CrossRef]
- Szarko, J.M.; Guo, J.; Rolczynski, B.S.; Chen, L.X. Nanoscale Structure, Dynamics and Power Conversion Efficiency Correlations in Small Molecule and Oligomer-Based Photovoltaic Devices. Nano Rev. 2011, 2, 7249. [Google Scholar] [CrossRef]
- Guo, J.; Liang, Y.; Szarko, J.; Lee, B.; Son, H.J.; Rolczynski, B.S.; Yu, L.; Chen, L.X.; Son, H.J.; Rolczynski, B.S.; et al. Structure, Dynamics, and Power Conversion Efficiency Correlations in a New Low Bandgap Polymer: PCBM Solar Cell. J. Phys. Chem. B 2010, 114, 742–748. [Google Scholar] [CrossRef]
- Reid, O.G.; Pensack, R.D.; Song, Y.; Scholes, G.D.; Rumbles, G. Charge Photogeneration in Neat Conjugated Polymers. Chem. Mater. 2014, 26, 561–575. [Google Scholar] [CrossRef]
- Mosquera, M.A.; Jackson, N.E.; Fauvell, T.J.; Kelley, M.S.; Chen, L.X.; Schatz, G.C.; Ratner, M.A. Exciton Absorption Spectra by Linear Response Methods: Application to Conjugated Polymers. J. Am. Chem. Soc. 2017, 139, 3728–3735. [Google Scholar] [CrossRef]
- Soon, Y.W.; Cho, H.; Low, J.; Bronstein, H.; McCulloch, I.; Durrant, J.R. Correlating Triplet Yield, Singlet Oxygen Generation and Photochemical Stability in Polymer/Fullerene Blend Films. Chem. Commun. 2013, 49, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Huynh, U.N.V.; Basel, T.P.; Ehrenfreund, E.; Vardeny, Z.V. Transient Magnetic Field Effect of Photoexcitations in Donor-Acceptor Organic Semiconductors. J. Phys. Chem. Lett. 2018, 9, 4544–4549. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, R.S.; Tsige, M. Chain Length and Torsional Dependence of Exciton Binding Energies in P3HT and PTB7 Conjugated Polymers: A First-Principles Study. Polymer 2014, 55, 2667–2672. [Google Scholar] [CrossRef]
- Gerhard, M.; Arndt, A.P.; Bilal, M.; Lemmer, U.; Koch, M.; Howard, I.A. Field-Induced Exciton Dissociation in PTB7-Based Organic Solar Cells. Phys. Rev. B 2017, 95, 1–12. [Google Scholar] [CrossRef]
- Pandit, B.; Jackson, N.E.; Zheng, T.; Fauvell, T.J.; Manley, E.F.; Orr, M.; Brown-Xu, S.; Yu, L.; Chen, L.X. Molecular Structure Controlled Transitions between Free-Charge Generation and Trap Formation in a Conjugated Copolymer Series. J. Phys. Chem. C 2016, 120, 4189–4198. [Google Scholar] [CrossRef]
- Hannah, D.C.; Yang, J.; Kramer, N.J.; Schatz, G.C.; Kortshagen, U.R.; Schaller, R.D. Ultrafast Photoluminescence in Quantum-Confined Silicon Nanocrystals Arises from an Amorphous Surface Layer. ACS Photonics 2014, 1, 960–967. [Google Scholar] [CrossRef]
- MATLAB; The Mathworks Inc.: Natick, MA, USA, 2017.
- Henry, E.R.; Hofrichter, J. Singular Value Decomposition: Application to Analysis of Experimental Data. Methods Enzymol. 1992, 210, 129–192. [Google Scholar]
- Van Stokkum, I.H.M.; Larsen, D.S.; Van Grondelle, R. Global and Target Analysis of Time-Resolved Spectra. Biochim. Biophys. Acta - Bioenerg. 2004, 1657, 82–104. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fauvell, T.J.; Cai, Z.; Kirschner, M.S.; Helweh, W.; Kim, P.; Zheng, T.; Schaller, R.D.; Yu, L.; Chen, L.X. Effects of Intra- and Interchain Interactions on Exciton Dynamics of PTB7 Revealed by Model Oligomers. Molecules 2020, 25, 2441. https://doi.org/10.3390/molecules25102441
Fauvell TJ, Cai Z, Kirschner MS, Helweh W, Kim P, Zheng T, Schaller RD, Yu L, Chen LX. Effects of Intra- and Interchain Interactions on Exciton Dynamics of PTB7 Revealed by Model Oligomers. Molecules. 2020; 25(10):2441. https://doi.org/10.3390/molecules25102441
Chicago/Turabian StyleFauvell, Thomas J., Zhengxu Cai, Matthew S. Kirschner, Waleed Helweh, Pyosang Kim, Tianyue Zheng, Richard D. Schaller, Luping Yu, and Lin X. Chen. 2020. "Effects of Intra- and Interchain Interactions on Exciton Dynamics of PTB7 Revealed by Model Oligomers" Molecules 25, no. 10: 2441. https://doi.org/10.3390/molecules25102441