
Sustainable Process for the Depolymerization/Oxidation of Softwood and Hardwood Kraft Lignins Using Hydrogen Peroxide under Ambient Conditions


Abstract
:1. Introduction
2. Results
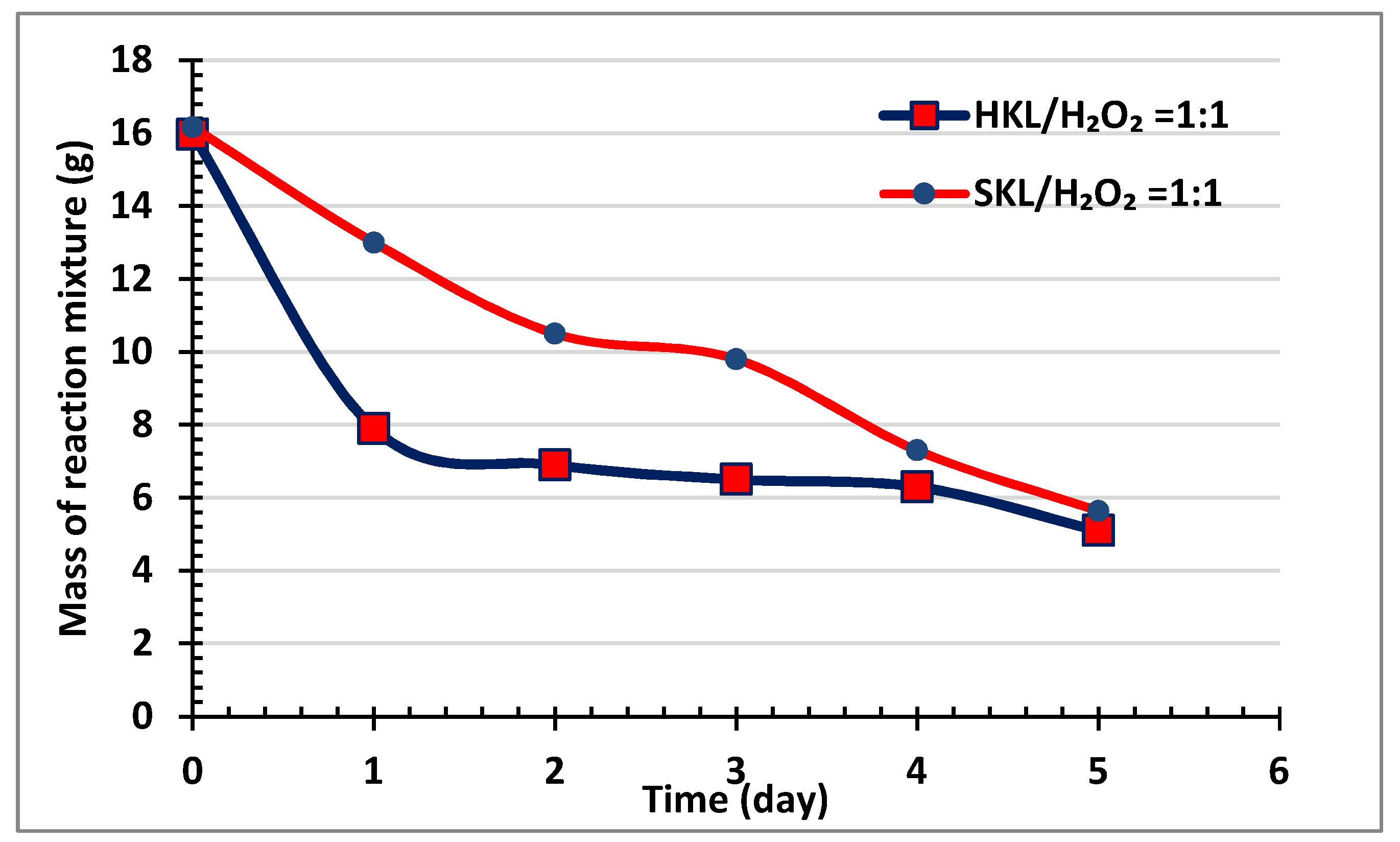
2.1. General Observations and Product Yields
2.2. Elemental Analysis
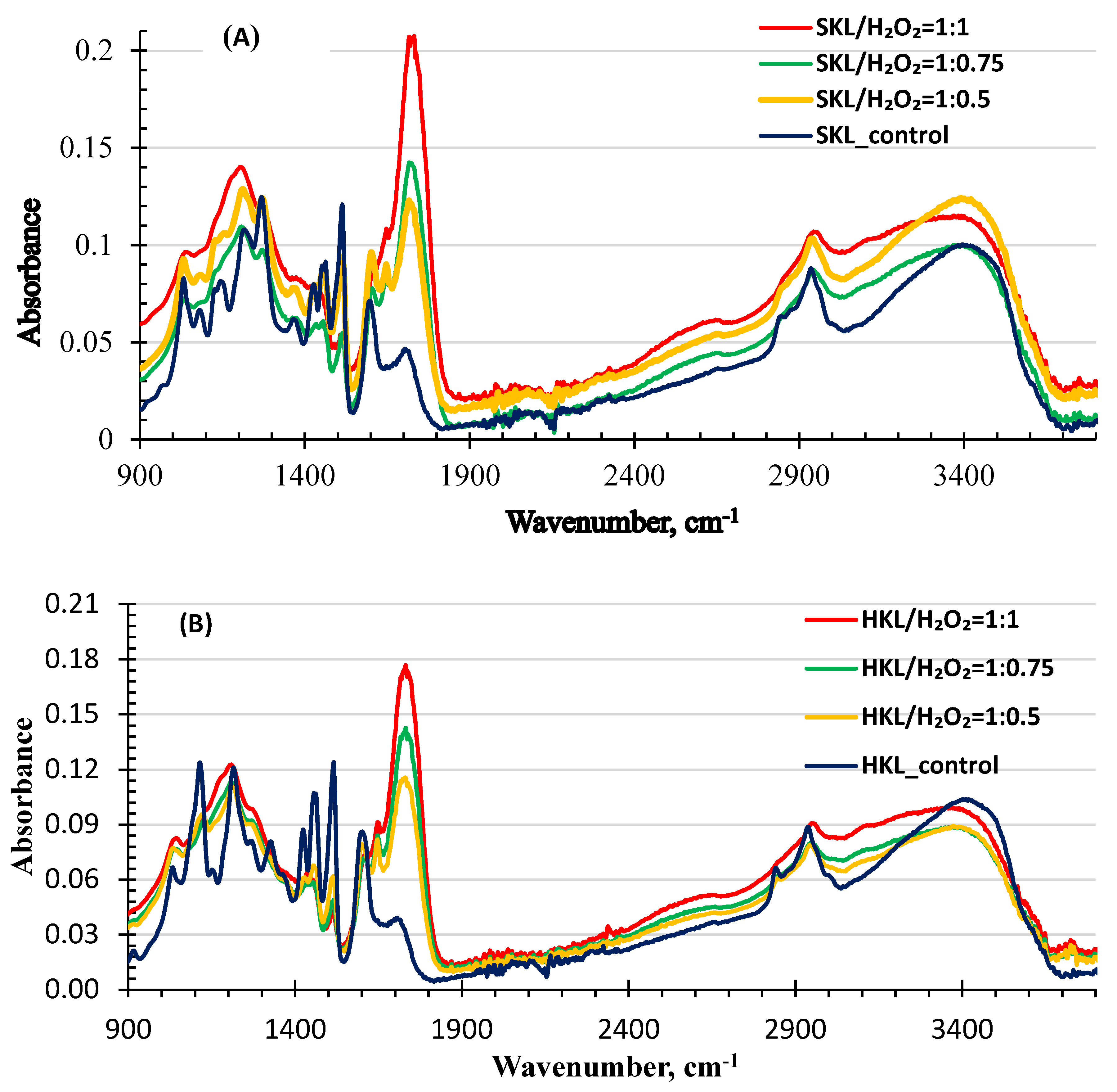
2.3. FTIR Analysis
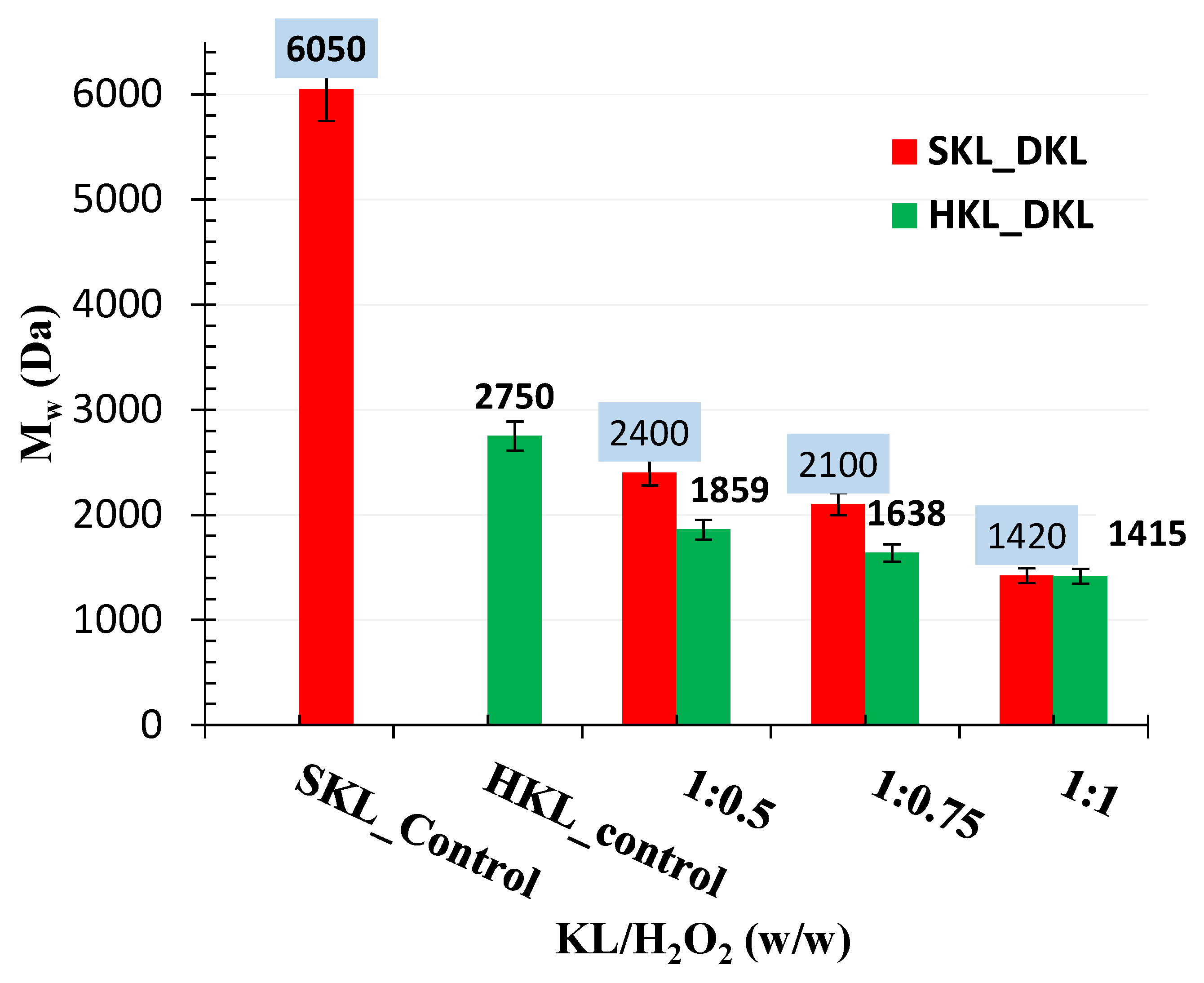
2.4. Molecular Weight Distribution by GPC
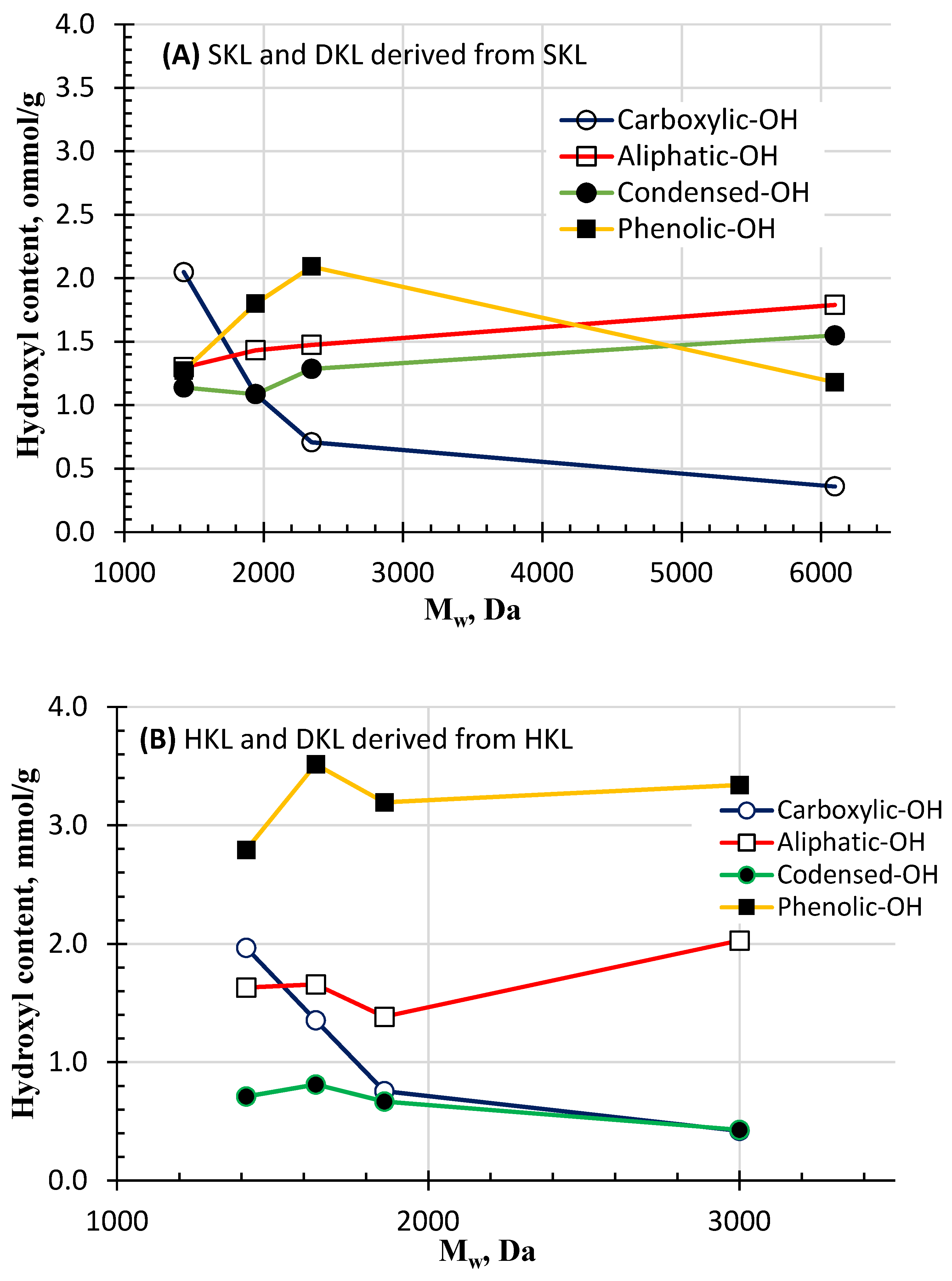
2.5. Functional Group Analysis Using 31P-NMR
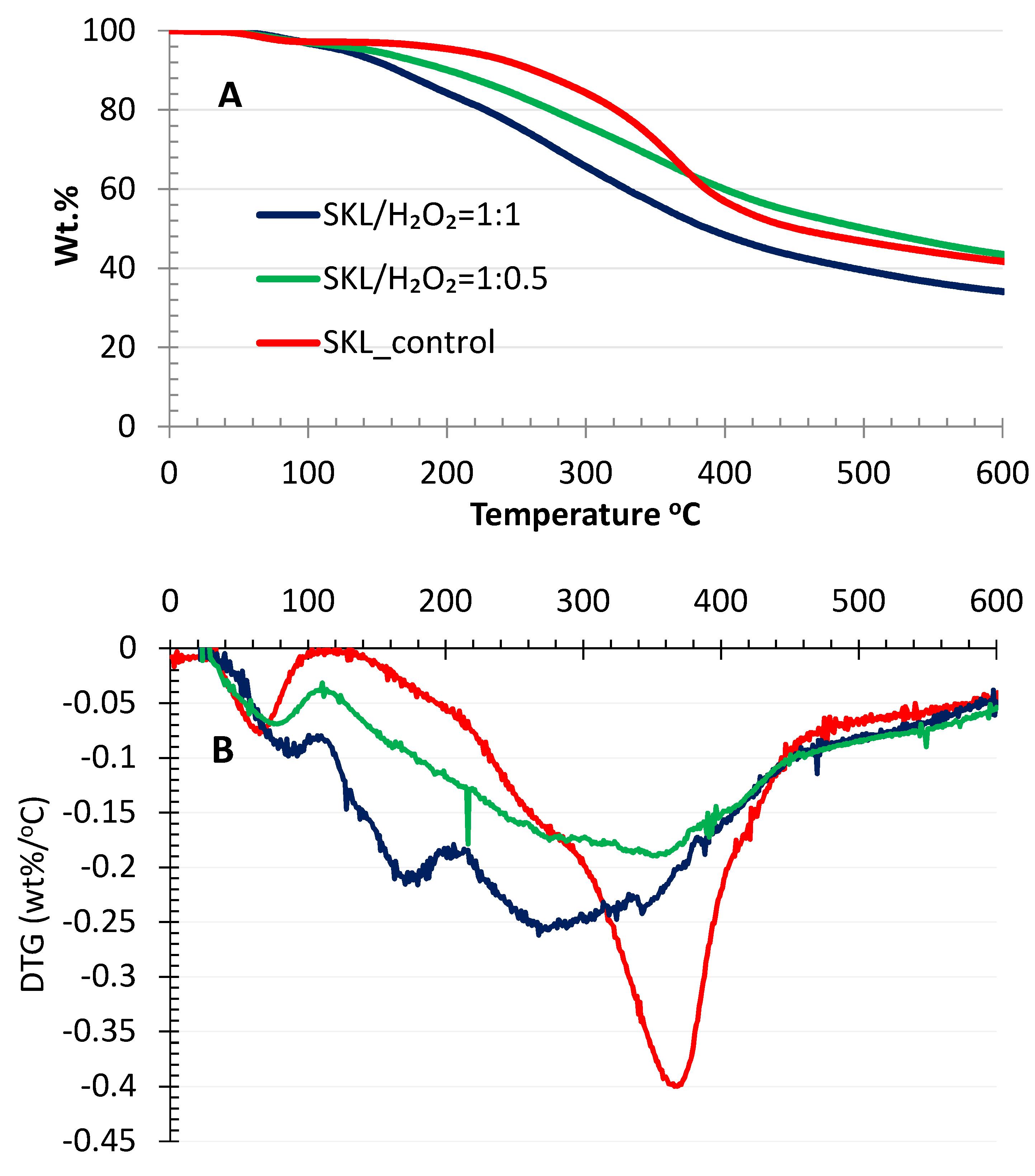
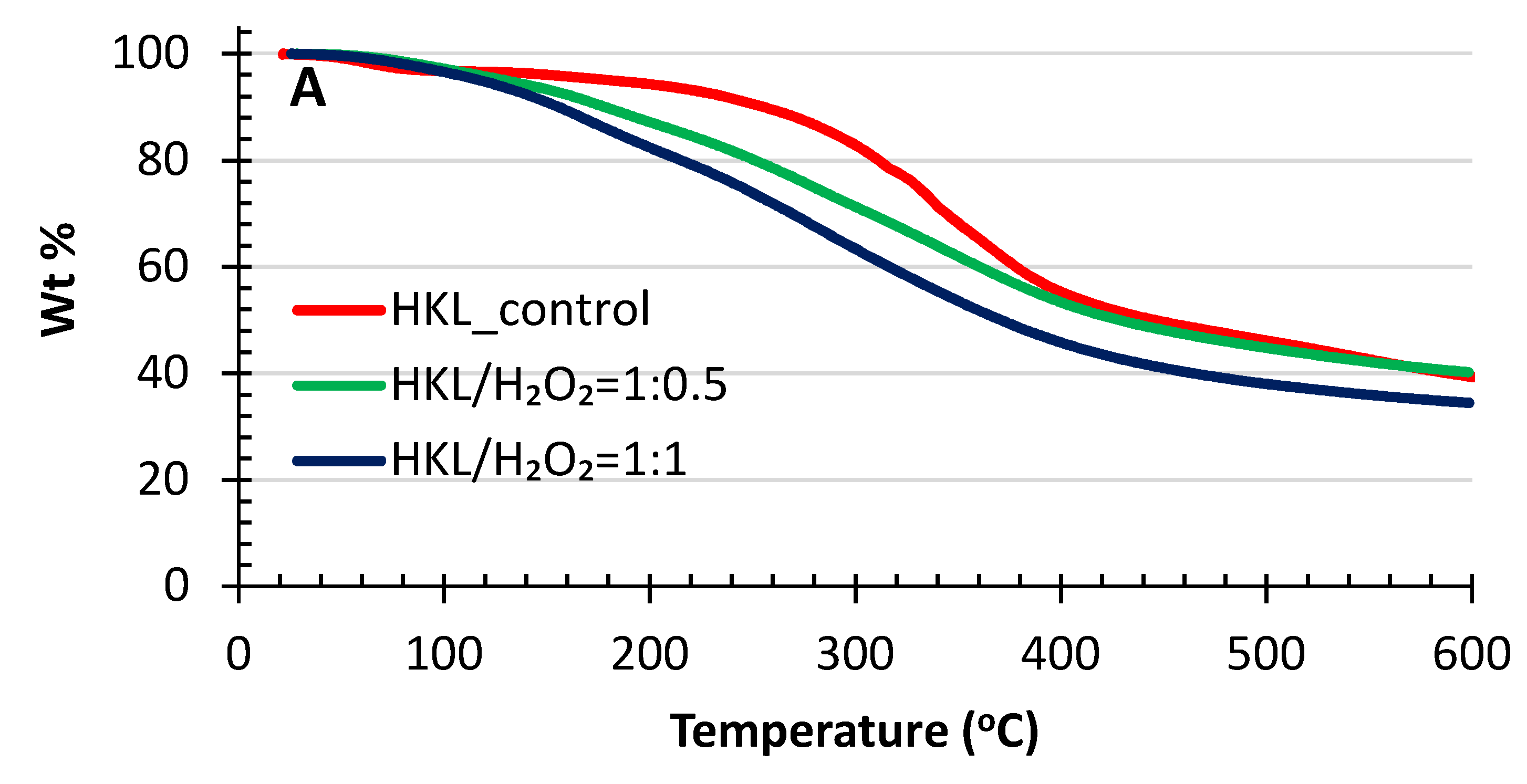
2.6. TGA Analysis
2.7. Py–GC/MS Analysis
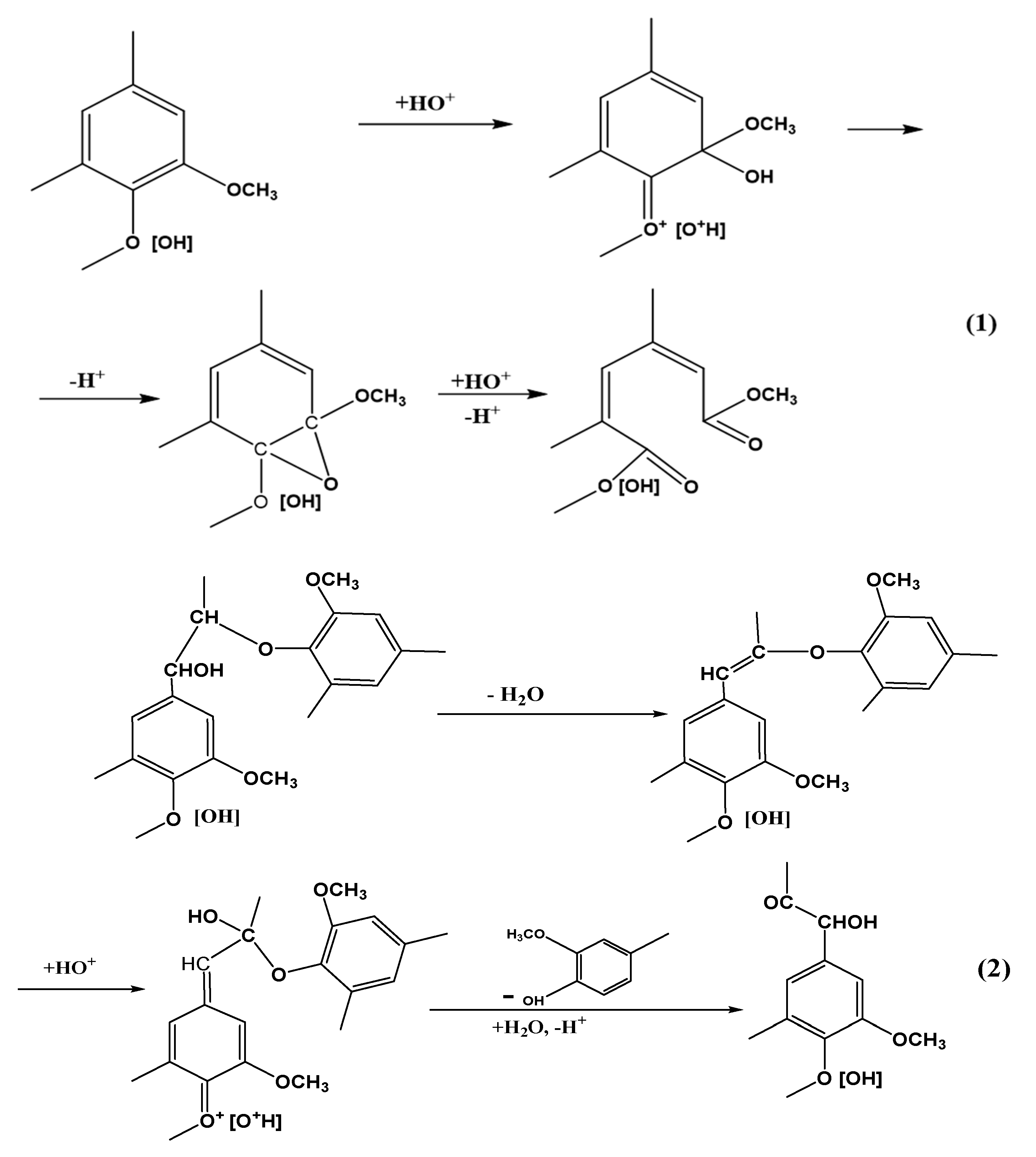
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Oxidative Treatment of SKL and HKL
4.3. Product Characterization
4.3.1. Elemental analysis
4.3.2. GPC Analysis
4.3.3. FTIR Analysis
4.3.4. 31P-NMR
4.3.5. TGA Analysis
4.3.6. Py–GC/MS
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef] [PubMed]
- McKendry, P. Energy production from biomass (part 1): Overview of biomass. Bioresour. Technol. 2002, 83, 37–46. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A.; Isac-García, J.; Martín-Martínez, F.J. Lignin and Lignans as Renewable Raw Materials: Chemistry, Technology and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 64–80. [Google Scholar]
- Xu, C.; Arancon, R.A.D.; Labidi, J.; Luque, R. Lignin depolymerization strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef] [PubMed]
- Heitner, C.; Dimmel, D.; Schmidt, J. Lignin and Lignans: Advances in Chemistry; CRC press: Boca Raton, FL, USA, 2016; pp. 5–7. [Google Scholar]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C.C. Depolymerization of lignins and their applications for the preparation of polyols and rigid polyurethane foams: A review. Renew. Sustain. Energy Rev. 2016, 60, 317–329. [Google Scholar] [CrossRef]
- An, L.; Si, C.; Wang, G.; Sui, W.; Tao, Z. Enhancing the solubility and antioxidant activity of high-molecular-weight lignin by moderate depolymerization via in situ ethanol/acid catalysis. Ind. Crop. Prod. 2019, 128, 177–185. [Google Scholar] [CrossRef]
- Morohoshi, N.; Glasser, W.G. The structure of lignins in pulps. 4. Comparative evaluation of five lignin depolymerization techniques. Wood Sci. Technol. 1979, 13, 165–178. [Google Scholar] [CrossRef]
- Freudenberg, K.; Chen, C.L.; Harkin, J.M.; Nimz, H.; Renner, H. Observations on lignin. Chem. Commun. (London) 1965, 224–225. [Google Scholar] [CrossRef]
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright side of lignin depolymerization: Toward new platform chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [Green Version]
- Rößiger, B.; Röver, R.; Unkelbach, G.; Pufky-Heinrich, D. Production of bio-phenols for industrial application: Scale-up of the base-catalyzed depolymerization of lignin. Green Sustain. Chem. 2017, 7, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Hewson, W.B.; Hibbert, H. Studies on Lignin and Related Compounds. LXV. Re-ethanolysis of Isolated Lignins. J. Am. Chem. Soc. 1943, 65, 1173–1176. [Google Scholar] [CrossRef]
- Gasson, J.R.; Forchheim, D.; Sutter, T.; Hornung, U.; Kruse, A.; Barth, T. Modeling the lignin degradation kinetics in an ethanol/formic acid solvolysis approach. Part 1. Kinetic model development. Ind. Eng. Chem. Res. 2012, 51, 10595–10606. [Google Scholar] [CrossRef]
- Forchheim, D.; Gasson, J.R.; Hornung, U.; Kruse, A.; Barth, T. Modeling the lignin degradation kinetics in a ethanol/formic acid solvolysis approach. Part 2. Validation and transfer to variable conditions. Ind. Eng. Chem. Res. 2012, 51, 15053–15063. [Google Scholar] [CrossRef]
- Kristianto, I.; Limarta, S.O.; Lee, H.; Ha, J.M.; Suh, D.J.; Jae, J. Effective depolymerization of concentrated acid hydrolysis lignin using a carbon-supported ruthenium catalyst in ethanol/formic acid media. Bioresour. Technol. 2017, 234, 424–431. [Google Scholar] [CrossRef]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C.C. Production of polyols via direct hydrolysis of kraft lignin: Effect of process parameters. Bioresour. Technol. 2013, 139, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Xu, Y.; Zhang, X. Catalytic oxidation of biorefinery lignin to value-added chemicals to support sustainable biofuel production. ChemSusChem 2015, 8, 24–51. [Google Scholar] [CrossRef]
- Das, L.; Kolar, P.; Sharma-Shivappa, R.; Classen, J.; Osborne, J. Oxidative depolymerization of lignin using supported niobium catalysts. Chem. Eng. 2017, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Irmak, S.; Wilkins, M. Conversion of lignin into renewable carboxylic acid compounds by advanced oxidation processes. Renew. Energy. 2019, 135, 951–962. [Google Scholar] [CrossRef]
- Crestini, C.; Caponi, M.C.; Argyropoulos, D.S.; Saladino, R. Immobilized methyltrioxo rhenium (MTO)/H2O2 systems for the oxidation of lignin and lignin model compounds. Bioorganic Med. Chem. 2006, 14, 5292–5302. [Google Scholar] [CrossRef]
- Sun, Y.; Argyropoulos, D.S. A comparison of the reactivity and efficiency of ozone, chlorine dioxide, dimethyldioxirane and hydrogen peroxide with residual kraft lignin. Holzforschung-Int. J. Biol. Chem. Phys. Technol. Wood 1996, 50, 175–182. [Google Scholar] [CrossRef]
- Kadla, J.F.; Chang, H.M. The reactions of peroxides with lignin and lignin model compounds. In Oxidative Delignification Chemistry; ACS symposium series: Washington, DC, USA, 2001; Volume 785, pp. 108–129. [Google Scholar]
- Sales, F.G.; Maranhão, L.C.; Lima Filho, N.M.; Abreu, C.A. Kinetic evaluation and modeling of lignin catalytic wet oxidation to selective production of aromatic aldehydes. Ind. Eng. Chem. Res. 2006, 45, 6627–6631. [Google Scholar] [CrossRef]
- Robinette, H. Hydrogen Peroxide; Schumb, W.C., Satterfield, C.N., Wentworth, R.L., Eds.; Reinhold Publishing Corp.: New York, NY, USA, 1956; p. 195. [Google Scholar]
- Xiang, Q.; Lee, Y.Y. Oxidative cracking of precipitated hardwood lignin by hydrogen peroxide. Appl. Biochem. Biotechnol. 2000, 84, 153–162. [Google Scholar] [CrossRef]
- Lourenco, A.; Gominho, J.; Marques, A.V.; Pereira, H. Reactivity of syringyl and guaiacyl lignin units and delignification kinetics in the kraft pulping of Eucalyptus globulus wood using Py-GC–MS/FID. Bioresour. Technol. 2012, 123, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Doelle, K.; Bajrami, B. Sodium Hydroxide and Calcium Hydroxide Hybrid Oxygen Bleaching with System. In IOP Conference Series: Materials Science and Engineering; IOP publishing: Bristol, UK, 2018; p. 012136. [Google Scholar]
- Chinnappan, B.; Baskar, S.; Dhillon, R. Biomass Conversion: The Interface of Biotechnology, Chemistry and Materials Science; Springer Science & Business Media: New York, NY, USA, 2012; pp. 383–400. [Google Scholar]
- Sixta, H. Pulp properties and applications. In Handbook of Pulp; Wiley-VCH: Weinheim, Germany, 2006; p. 197. [Google Scholar]
- Gonzalez-Vila, F.J.; Almendros, G.; Del Río, J.C.; Martın, F.; Gutiérrez, A.; Romero, J. Ease of delignification assessment of wood from different Eucalyptus species by pyrolysis (TMAH)-GC/MS and CP/MAS 13C-NMR spectrometry. J. Anal. Appl. Pyrolysis 1999, 49, 295–305. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Rodríguez, I.M.; del Río, J.C. Chemical characterization of lignin and lipid fractions in industrial hemp bast fibers used for manufacturing high-quality paper pulps. J. Agric. Food Chem. 2006, 54, 2138–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C.C. Hydrolytic depolymerization of hydrolysis lignin: Effects of catalysts and solvents. Bioresour. Technol. 2015, 190, 416–419. [Google Scholar] [CrossRef]
- Adler, E. Lignin chemistry-past, present and future. Wood Sci. Technol. 1977, 11, 169–218. [Google Scholar] [CrossRef]
- Marzzacco, C.J. The enthalpy of decomposition of hydrogen peroxide: A general chemistry calorimetry experiment. J. Chem. Educ. 1999, 76, 1517. [Google Scholar] [CrossRef]
- Shimada, K.; Hosoya, S.; Ikeda, T. Condensation reactions of softwood and hardwood lignin model compounds under organic acid cooking conditions. J. Wood Chem. Technol. 1997, 1, 57–72. [Google Scholar] [CrossRef]
- Kline, L.M.; Hayes, D.G.; Womac, A.R.; Labbe, N. Simplified determination of lignin content in hard and soft woods via UV-spectrophotometric analysis of biomass dissolved in ionic liquids. BioResources 2010, 5, 1366–1383. [Google Scholar]
- Fan, M.; Dai, D.; Huang, B. Fourier transform infrared spectroscopy for natural fibers. In Proceedings of the Fourier Transform-materials Analysis, InTech Janeza Trdine, Rijeka, Croatia, 23 May 2012; pp. 45–68. [Google Scholar]
- Kubo, S.; Kadla, J.F. Hydrogen bonding in lignin: A Fourier transform infrared model compound study. Biomacromolecules 2005, 6, 2815–2821. [Google Scholar] [CrossRef]
- Baumberger, S.; Abaecherli, A.; Fasching, M.; Gellerstedt, G.; Gosselink, R.; Hortling, B.; Li, J.; Saake, B.; de Jong, E. Molar mass determination of lignins by size-exclusion chromatography: Towards standardisation of the method. Holzforschung 2007, 61, 459–468. [Google Scholar] [CrossRef]
- Sarkanen, K.V.; Ludwig, C.H. Lignins. Occurrence, formation, structure, and reactions; Wiley-Inter-science: New York, NY, USA, 1971; pp. 433–479. [Google Scholar]
- Chen, C.; Jin, D.; Ouyang, X.; Zhao, L.; Qiu, X.; Wang, F. Effect of structural characteristics on the depolymerization of lignin into phenolic monomers. Fuel 2018, 223, 366–372. [Google Scholar] [CrossRef]
- Pu, Y.; Cao, S.; Ragauskas, A.J. Application of quantitative 31P NMR in biomass lignin and biofuel precursors characterization. Energy Environ. Sci. 2011, 4, 3154–3166. [Google Scholar] [CrossRef]
- Zakis, G.F. Functional Analysis of Lignins and Their Derivatives; Tappi Press: Atlanta, GA, USA, 1994; pp. 30–102. [Google Scholar]
- Lucia, L.A.; Goodell, M.M.; Chakar, F.S.; Ragauskas, A.J. Breaking the Oxygen Delignification Barrier: Lignin Reactivity and Inactivity; ACS Symposium Series: Atlanta, GA, USA, 2000; pp. 92–107. [Google Scholar]
- Wen, J.L.; Yuan, T.Q.; Sun, S.L.; Xu, F.; Sun, R.C. Understanding the chemical transformations of lignin during ionic liquid pretreatment. Green Chem. 2014, 16, 181–190. [Google Scholar] [CrossRef]
- Granata, A.; Argyropoulos, D.S. 2-Chloro-4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaphospholane, a reagent for the accurate determination of the uncondensed and condensed phenolic moieties in lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]
- Brebu, M.; Vasile, C. Thermal degradation of lignin-A review. Cellul. Chem. Technol. 2010, 44, 353–363. [Google Scholar]
- Wittkowski, R.; Ruther, J.; Drinda, H.; Rafiei-Taghanaki, F. Formation of Smoke Flavor Compounds by Thermal Lignin Degradation; ACS symposium series: Washington, DC, USA, 1992; pp. 232–490. [Google Scholar]
- Domínguez, J.C.; Oliet, M.; Alonso, M.V.; Gilarranz, M.A.; Rodríguez, F. Thermal stability and pyrolysis kinetics of organosolv lignins obtained from Eucalyptus globulus. Ind. Crop. Prod. 2008, 27, 150–156. [Google Scholar] [CrossRef]
- El-Saied, H.; Nada, A.A.M. The thermal behaviour of lignins from wasted black pulping liquors. Polym. Degrad. Stab. 1993, 40, 417–421. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Li, M.; Wyman, C.E.; Cai, C.M.; Ragauskas, A.J. Fast fractionation of technical lignins by organic cosolvents. ACS Sustain. Chem. Eng. 2018, 6, 6064–6072. [Google Scholar] [CrossRef]
- Zhao, J.; Xiuwen, W.; Hu, J.; Liu, Q.; Shen, D.; Xiao, R. Thermal degradation of softwood lignin and hardwood lignin by TG-FTIR and Py-GC/MS. Polym. Degrad. Stab. 2014, 108, 133–138. [Google Scholar] [CrossRef]
- Ház, A.; Jablonský, M.; Orságová, A.; Šurina, I. Characterization of lignins by py-GC/MS. In Proceedings of the 4th International Scientific Conference, Renewable Energy Sources, Tatranské Matliare, Slovakia, 21–23 May 2013; pp. 55–59. [Google Scholar]
- Siddiqui, H.; Mahmood, N.; Yuan, Z.; Crapulli, F.; Dessbesell, L.; Rizkalla, A.; Ray, A.; Xu, C.C. Sustainable bio-based phenol-formaldehyde resoles using hydrolytically depolymerized kraft lignin. Molecules 2017, 22, 1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gierer, J. Chemistry of delignification. Wood Sci. Technol. 1986, 20, 1–33. [Google Scholar] [CrossRef]
- Sun, R.; Tomkinson, J.; Zhu, W.; Wang, S.Q. Delignification of maize stems by peroxy mono-sulfuric acid, peroxy formic acid, peracetic acid, and hydrogen peroxide. 1. Physicochemical and structural characterization of the solubilized lignins. J. Agric. Food Chem. 2000, 48, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C. Lignin reactions in delignification with peroxyacetic acid. In Chemistry of Delignification with Oxygen, Ozone and Peroxides; Gratzl, J., Nakano, J., Singh, R.P., Eds.; Uni Publishers Co., Ltd.: Tokyo, Japan, 1980; pp. 217–228. [Google Scholar]
- Oki, T.; Ishikawa, H.; Okubo, K. Oxidative degradation of dihydrodehydrodiisoeugenol and its methyl derivative [of lignin] by peroxide and oxygen alkali methods. J. Jpn. Wood Res. Soc. 1980, 26, 463–470. [Google Scholar]
- Evstigneev, E.I. Oxidation of hydrolysis lignin with hydrogen peroxide in acid solutions. Russ. J. Appl. Chem. 2013, 86, 258–265. [Google Scholar] [CrossRef]
- Hosseinaei, O.; Harper, D.; Bozell, J.; Rials, T. Improving processing and performance of pure lignin carbon fibers through hardwood and herbaceous lignin blends. Int. J. Mol. Sci. 2017, 18, 1410. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Softwood | Hardwood | |
|---|---|---|
| Chemical Composition (wt%) | ||
| Cellulose | 40–44 | 40–44 |
| Hemicellulose | 30–32 | 15–35 |
| Lignin | 25–32 | 18–25 |
| Extractives | 5 | 2 |
| Lignin Linkage Types (number/100 phenylpropane units) | ||
| β-O-4 | 43–50 | 50–65 |
| α-O-4 | 6–8 | 4–8 |
| β-5 + α-O-4 | 9–12 | 4–6 |
| β–β | 2–4 | 3–7 |
| 5–5′ | 10–25 | 4–10 |
| 4-O-5′ | 4 | 6–7 |
| β-1 | 3–7 | 5–7 |
| C-6, C-2 | 3 | 2–3 |
| Elemental Composition, wt% (d.b.) | Original Lignin | De-Polymerized Lignins | Original Lignin | De-Polymerized Lignins | ||||
|---|---|---|---|---|---|---|---|---|
| SKL | 1:1 | 1:0.75 | 1:0.5 | HKL | 1:1 | 1:0.75 | 1:0.5 | |
| C | 65.2 | 50.5 | 53.9 | 57.4 | 63.2 | 47.7 | 49.9 | 52.5 |
| H | 5.5 | 4.9 | 4.7 | 5.0 | 5.41 | 3.74 | 3.87 | 4.3 |
| N | 0.2 | 0 | 0.06 | 0.06 | 0.02 | 0.08 | 0.06 | 0.08 |
| S | 1.51 | 1.79 | 1.27 | 1.29 | 2.25 | 1.9 | 1.9 | 1.9 |
| Ash 1 | 0.83 | 0.83 | 0.83 | 0.83 | 0.44 | 0.44 | 0.44 | 0.44 |
| O 2 | 26.8 | 42.0 | 39.2 | 35.4 | 28.5 | 46.1 | 43.8 | 40.8 |
| Pk# | RT | Area% | Library/ID | Mw | Qual |
|---|---|---|---|---|---|
| 1 | 0.36 | 2.35 | Acetic acid, methyl ester | 74 | 72 |
| 2 | 0.84 | 7.5 | Acetic acid | 60 | 86 |
| 3 | 3.71 | 1.6 | Furfural | 96 | 91 |
| 4 | 5.44 | 1.7 | Propanedioic acid, dimethyl ester | 132 | 83 |
| 5 | 8.37 | 10.5 | Phenol, 2-methoxy-(guaiacol) | 124 | 97 |
| 6 | 10.63 | 3.01 | 2-Methoxy-5-methylphenol | 138 | 90 |
| 7 | 10.79 | 4.68 | 1,2-Benzenediol-(catechol) | 110 | 95 |
| 8 | 15.93 | 3.67 | 1,4-Benzenediol, 2-methoxy- | 140 | 95 |
| 9 | 17.6 | 2.53 | Benzoic acid, 4-hydroxy-3-methoxy-methyl ester | 182 | 96 |
| 10 | 17.78 | 2.07 | Homovanillyl alcohol | 168 | 80 |
| 11 | 18.32 | 10.1 | 3-Hydroxy-4-methoxybenzoic acid | 168 | 95 |
| 12 | 21.75 | 4 | 9,10-Anthracenedione | 208 | 95 |
| 13 | 35 | 2.21 | Urs-12-ene | 410 | 83 |
| Pk# | RT | Area% | Library/ID | Mw | Qual |
|---|---|---|---|---|---|
| 1 | 0.23 | 11.55 | Acetic acid, methyl ester | 74 | 64 |
| 2 | 0.61 | 14.13 | Acetic acid | 60 | 90 |
| 3 | 3.7 | 6.91 | Furfural | 96 | 94 |
| 4 | 5.45 | 8.93 | Propanedioic acid, dimethyl ester | 132 | 83 |
| 5 | 9.45 | 3.64 | 1-Butene, 3,3-dimethyl- | 84 | 50 |
| 6 | 15.92 | 2.69 | 1,4-Benzenediol, 2-methoxy- | 140 | 80 |
| 7 | 16.44 | 4.27 | 2-Naphthyl methyl ketone | 170 | 22 |
| 8 | 17.6 | 5.48 | Benzoic acid, 4-hydroxy-3-methoxy-methyl ester | 182 | 94 |
| 9 | 17.77 | 2.07 | 2-Propanone, 1-(4-hydroxy-3-methoxyphenyl)- | 180 | 80 |
| 10 | 18.18 | 4.5 | Benzoic acid, 4-hydroxy-3-methoxy- | 213 | 93 |
| SKL | HKL | |
|---|---|---|
| Elemental analysis (wt% d.b.) 1 | ||
| C | 65.2 | 63.2 |
| H | 5.52 | 5.41 |
| N | 0.20 | 0.02 |
| O 2 | 26.7 | 28.7 |
| Ash 3 | 0.83 | 0.44 |
| Total S | 1.51 | 2.25 |
| Mw, UV detector (Da) | 6041 | 2718 |
| PDI 4, UV detector | 3.3 | 3.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, Z.; Al Dajani, W.W.; Paleologou, M.; Xu, C. Sustainable Process for the Depolymerization/Oxidation of Softwood and Hardwood Kraft Lignins Using Hydrogen Peroxide under Ambient Conditions. Molecules 2020, 25, 2329. https://doi.org/10.3390/molecules25102329
Ahmad Z, Al Dajani WW, Paleologou M, Xu C. Sustainable Process for the Depolymerization/Oxidation of Softwood and Hardwood Kraft Lignins Using Hydrogen Peroxide under Ambient Conditions. Molecules. 2020; 25(10):2329. https://doi.org/10.3390/molecules25102329
Chicago/Turabian StyleAhmad, Zaid, Waleed Wafa Al Dajani, Michael Paleologou, and Chunbao (Charles) Xu. 2020. "Sustainable Process for the Depolymerization/Oxidation of Softwood and Hardwood Kraft Lignins Using Hydrogen Peroxide under Ambient Conditions" Molecules 25, no. 10: 2329. https://doi.org/10.3390/molecules25102329