
Speciation of Chromium in Alkaline Soil Extracts by an Ion-Pair Reversed Phase HPLC-ICP MS Method


Abstract
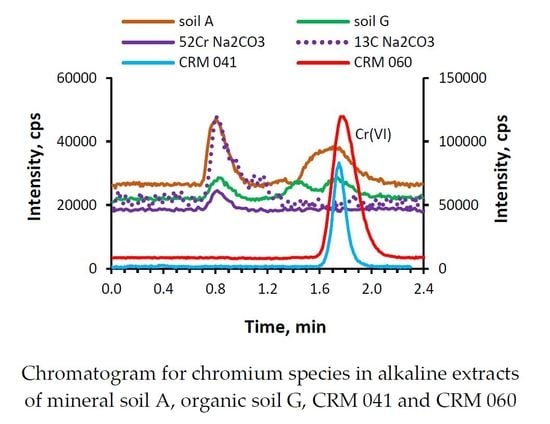
:
1. Introduction
2. Results and Discussion
2.1. Optimization of Ultrasound Assisted Extraction Procedure of Chromium Species from Soil
2.2. Optimization of the ICP MS Determination of Chromium
2.3. Optimization of the IP RP-HPLC-ICP MS Procedure
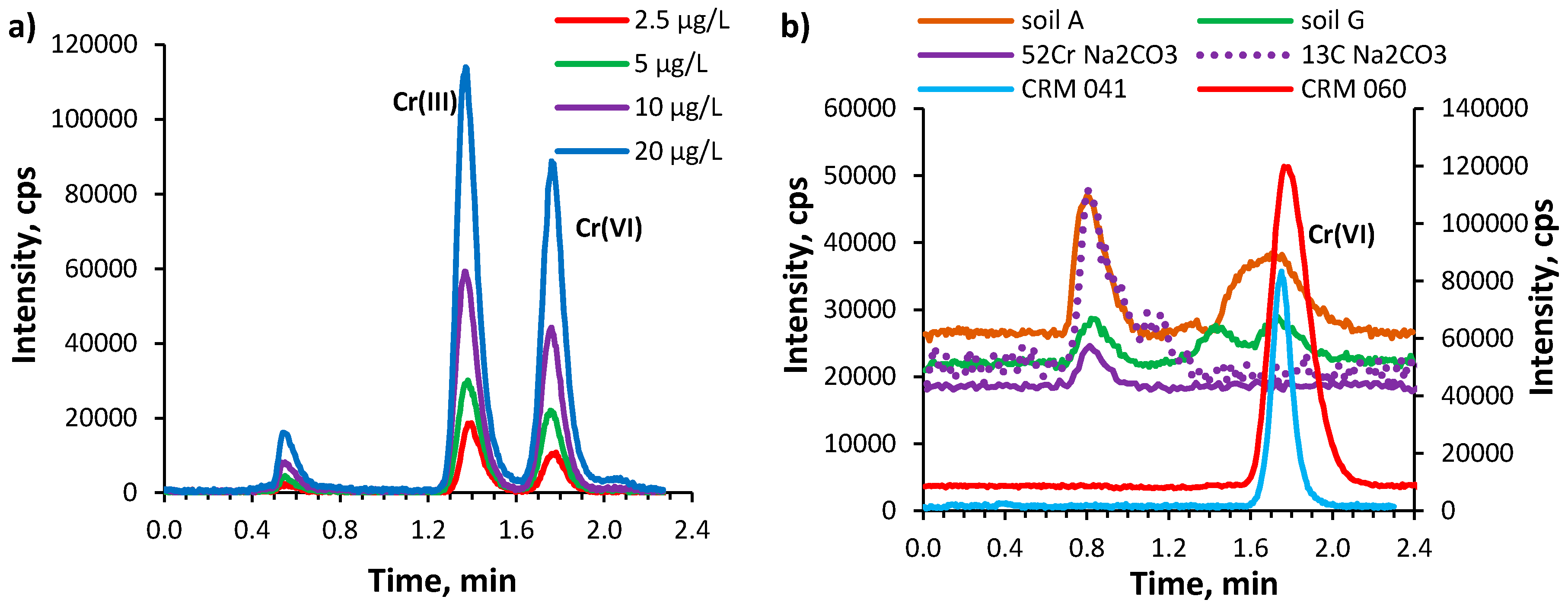
2.4. Analytical Characteristic of the IP RP HPLC-ICP MS Method
2.5. Chromium Speciation Analysis in Soil Extracts by IP RP HPLC-ICP MS
3. Materials and Methods
3.1. Instrumentation
3.2. Reagents
3.3. Materials and Samples
3.4. Procedures
3.4.1. Extraction Procedure
3.4.2. Direct Determination of Cr in Soil Extract by ICP MS
3.4.3. Determination of Chromium Species in Soil Extracts by IP RP-HPLC-ICP MS
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dhal, B.; Thatoi, H.N.; Das, N.N.; Pandey, B.D. Chemical and microchemical remediation of hexavalent chromium from contaminated soil and mining/metallurgical solid waste: A review. J. Hazard. Mat. 2013, 250–251, 272–291. [Google Scholar] [CrossRef]
- Kotaś, J.; Stasicka, Z. Chromium occurrence in the environment and methods of its speciation. Environ. Pollut. 2000, 107, 263–283. [Google Scholar] [CrossRef]
- Ščančar, J.; Milacic, R. A critical overview of Cr speciation analysis based on high performance liquid chromatography and spectrometric techniques. J. Anal. Atom. Spectrom. 2014, 29, 427–443. [Google Scholar] [CrossRef]
- Karaś, K.; Frankowski, M. Analysis of hazardous elements in children toys: Multi-elemental determination by chromatography and spectrometry methods. Molecules 2018, 23, 3017. [Google Scholar] [CrossRef] [PubMed]
- Hoet, P. Speciation of chromium in occupational exposure and clinical aspects. In Handbook of Elemental Speciation II: Species in the Environment, Food, Medicine & Occupational Health; Cornelis, R., Crews, H., Caruso, J., Heumann, K.G., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2005; pp. 135–157. [Google Scholar]
- IARC. Evaluation of Carcinogenic Risk to Humans: Chromium and Chromium Compounds: Biological Data Relevant to the Evaluation of Carcinogenic Risk to Humans; IARC Monographs; International Agency for Research on Cancer: Lyon, France, 1990; Volume 49, pp. 49–214. [Google Scholar]
- Bartlett, R.J. Chromium cycling in soils and water: Links, gaps, and methods. Environ. Health Perspect. 1991, 92, 17–24. [Google Scholar] [CrossRef]
- Metze, D.; Jakubowski, N.; Klockow, D. Speciation of chromium in environment and food. In Handbook of Elemental Speciation II: Species in the Environment, Food, Medicine & Occupational Health; Cornelis, R., Crews, H., Caruso, J., Heumann, K.G., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2005; pp. 120–134. [Google Scholar]
- Leśniewska, B.; Krymska, M.; Świerad, E.; Wiater, J.; Godlewska-Żyłkiewicz, B. An ultrasound-assisted procedure for fast screening of mobile fractions of Cd, Pb and Ni in soil. Insight into method optimization and validation. Environ. Sci. Pollut. Res. 2016, 23, 25093–25104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leśniewska, B.; Kisielewska, K.; Wiater, J.; Godlewska-Żyłkiewicz, B. Fast and simple procedure for fractionation of zinc in soil using an ultrasound probe and FAAS detection. Validation of the analytical method and evaluation of the uncertainty budget. Environ. Monit. Assess. 2016, 188, 29. [Google Scholar] [CrossRef] [PubMed]
- Orecchio, S.; Amorello, D.; Barreca, S.; Pettignano, A. Speciation of vanadium in urban, industrial and volcanic soils by a modified Tessier method. Environ. Sci. Process. Impacts 2016, 18, 323–329. [Google Scholar] [CrossRef]
- Lillengen, B.; Wibetoe, G. Graphite furnace atomic absorption spectrometry used for determination of total EDTA and acetic acid extractable chromium and cobalt in soils. Anal. Bioanal. Chem. 2002, 372, 187–195. [Google Scholar] [CrossRef]
- Rüdel, H.; Terytze, K. Determination of extractable chromium(VI) in soils using a photometric method. Chemosphere 1999, 39, 697–708. [Google Scholar] [CrossRef]
- Seby, F.; Vacchina, V. Critical assessment of hexavalent chromium species from different solid environmental, industrial and food matrices. Trends Anal. Chem. 2018, 104, 54–68. [Google Scholar] [CrossRef]
- Ščančar, J.; Zupančič, M.; Milačič, R. Development of analytical procedure for the determination of exchangeable Cr(VI) in soils by anion-exchange fast protein liquid chromatography with electrothermal atomic absorption spectrometry detection. Water Air Soil Pollut. 2007, 185, 121–129. [Google Scholar] [CrossRef]
- Zuliani, T.; Ščančar, J.; Milačič, R. The use of stable isotopes for Cr(VI) determination in silty-clay soil solution. Anal. Bioanal. Chem. 2013, 405, 7231–7240. [Google Scholar] [CrossRef]
- Panichev, N.; Mandiwana, K.; Foukaridis, G. Electrothermal atomic absorption spectrometric determination of Cr(VI) in soil after leaching of Cr(VI) species with carbon dioxide. Anal. Chim. Acta 2003, 491, 81–89. [Google Scholar] [CrossRef]
- James, B.R.; Petura, J.C.; Vitale, R.J.; Mussoline, G.R. Hexavalent chromium extraction from soils: A comparison of five methods. Environ. Sci. Technol. 1995, 29, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Huo, D.; Lu, Y.; Kingston, H.M. Determination and correction of analytical biases and study on chemical mechanisms in the analysis of Cr(VI) in soil samples using EPA protocols. Environ. Sci. Technol. 1998, 32, 3418–3423. [Google Scholar] [CrossRef]
- Malherbe, J.; Isaure, M.P.; Seby, F.; Watson, R.P.; Rodrigez-Gonzalez, P.; Stutzman, P.E.; Davis, C.W.; Maurizio, C.; Unceta, N.; Sieber, J.R.; et al. Evaluation of hexavalent chromium extraction method EPA method 3060A for soils using XANES spectroscopy. Environ. Sci. Technol. 2011, 45, 10492–10500. [Google Scholar] [CrossRef]
- Tirez, K.; Scharf, H.; Calzolari, D.; Cleven, R.; Kisser, M.; Luck, D. Validation of a European standard for the determination of hexavalent chromium in solid material. J. Environ. Monit. 2007, 9, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Wolle, M.M.; Rahman, G.M.M.; Kingston, H.M.S.; Pamuku, M. Optimization and validation of strategies for quantifying chromium species in soil based on speciated isotope dilution mass spectrometry with mass balance. J. Anal. Atom. Spectrom. 2014, 29, 1640–1647. [Google Scholar] [CrossRef]
- Korolczuk, M.; Grabarczyk, M. Evaluation of ammonia buffer containing EDTA as an extractant for Cr(VI) from solid samples. Talanta 2005, 66, 1320–1325. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Korolczuk, M.; Tyszczuk, K. Extraction and determination of hexavalent chromium in soil samples. Anal. Bioanal. Chem. 2006, 386, 357–362. [Google Scholar] [CrossRef]
- Leśniewska, B.; Gontarska, M.; Godlewska-Żyłkiewicz, B. Selective separation of chromium species from soils by single-step extraction methods: A critical appraisal. Water Air Soil Pollut. 2017, 228, 274. [Google Scholar] [CrossRef] [PubMed]
- Huo, D.; Kingston, H.M. Correction of species transformations in the analysis of Cr(VI) in solid environmental samples using speciated isotope dilution mass spectrometry. Anal. Chem. 2000, 72, 5047–5054. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-Y.; Jiang, S.-J.; Sahayam, A.C. Speciation of chromium and vanadium in environmental samples using HPLC-DRC-ICP-MS. J. Anal. Atom. Spectrom. 2007, 22, 636–641. [Google Scholar] [CrossRef]
- Wolf, R.E.; Morrison, J.M.; Goldhaber, M.B. Simultaneous determination of Cr(III) and Cr(VI) using reversed-phased ion-pairing liquid chromatography with dynamic reaction cell inductively coupled plasma mass spectrometry. J. Anal. Atom. Spectrom. 2007, 22, 1051–1060. [Google Scholar] [CrossRef]
- Wolf, R.E.; Morman, S.A.; Hageman, P.L.; Hoefen, T.M.; Plumlee, G.S. Simultaneous speciation of arsenic, selenium, and chromium: Species stability, sample preservation, and analysis of ash and soil leachates. Anal. Bioanal. Chem. 2011, 401, 2733–2745. [Google Scholar] [CrossRef]
- Bendicho, C.; De La Calle, I.; Pena, F.; Costas, M.; Cabaleiro, N.; Lavilla, I. Ultrasound-assisted pre-treatment of solid samples in the context of green analytical chemistry. Trends Anal. Chem. 2012, 31, 50–60. [Google Scholar] [CrossRef]
- Krasnodębska-Ostręga, B.; Kaczorowska, M.; Golimowski, J. Ultrasound-assisted extraction for the evaluation of element mobility in bottom sediment collected at mining and smelting Pb-Zn ores area in Poland. Microchim. Acta 2006, 154, 39–43. [Google Scholar] [CrossRef]
- Kazi, T.G.; Jamali, M.K.; Siddiqui, A.; Kazi, G.H.; Arain, M.B.; Afridi, H.I. An ultrasonic assisted extraction method to release heavy metals from untreated sewage sludge samples. Chemosphere 2006, 63, 411–420. [Google Scholar] [CrossRef] [PubMed]
- D’Ilio, S.; Violante, N.; Majorani, C.; Petrucci, F. Dynamic reaction cell ICP-MS for determination of total As, Cr, Se and V in complex matrices: Still a challenge? A review. Anal. Chim. Acta 2011, 698, 6–13. [Google Scholar] [CrossRef]
- Vitale, R.J.; Mussoline, G.R.; Petura, J.C.; James, B.R. Hexavalent chromium extraction from soils: Evaluation of an alkaline digestion method. J. Environ. Qual. 1994, 23, 1249–1256. [Google Scholar] [CrossRef]
- James, B.R. The challenge of remediating chromium-contaminated soil. The complex chemistry of chromium compounds presents unique measurement and regulatory challenges. Environ. Sci. Technol. 1996, 30, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Pettine, M.; Millero, F.J.; La Noce, T. Chromium(III) interaction in seawater through its oxidation kinetics. Mar. Chem. 1991, 34, 29–46. [Google Scholar] [CrossRef]
- Guidotti, L.; Queipo Abad, S.; Rodríguez-González, P.; García Alonso, J.I.; Beone, G.M. Quantification of Cr(VI) in soil samples from a contaminated area in northern Italy by isotope dilution mass spectrometry. Environ. Sci. Pollut. Res. 2015, 22, 17569–17576. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPLC-ICP MS | |
|---|---|
| HPLC | |
| Column | C8 cartridge column (3 µm diameter particles, 4.6 mm × 33 mm) |
| Mobile phase | 1 mmol L−1 TBAH and 0.6 mmol L−1 EDTA, pH 7.2 ± 0.1 |
| Flow rate | 0.8 mL min−1 |
| Injection volume | 50 µL |
| Elution program | isocratic |
| Total analysis time | 2.4 min |
| ICP MS | |
| RF power | 1550 W |
| Plasma gas flow rate | 15 L min−1 |
| Auxiliary gas flow rate | 1 L min−1 |
| Nebulizer gas flow rate | 0.9 L min−1 |
| Sampling depth | 10 mm |
| Spray chamber temperature | 2 °C |
| Collision gas | He a,b, O2 a |
| Collision gas flow rate | He a,b: 4 mL min−1; O2 a: 0.4 mL min−1 |
| Octopole bias | −5V |
| Octopole RF | 190 V |
| Discrimination energy | He: 5 V a,b; O2:−7Va |
| Scan type | MS/MS a,b |
| Integration time | 0.1 s a, 0.5 s b |
| Monitored ion (m/z) | 52Cr+ (He mode) a,b, 52Cr16O+ (O2 mode) a, 13C+ (He mode) b, |
| Internal standard (m/z) | 115In+ a |
| Calibration Graph Equation | LOD, µg L−1 | LOQ, µg L−1 | Precision (RSD, %) | ||
|---|---|---|---|---|---|
| ICP MS | 1-25 µg L−1 standards (n = 3) | ||||
| He as collision gas | |||||
| Cr * | y = 0.0604x + 0.0074 | 0.027 | 0.081 | 1.1–3.2 | |
| Cr ** | y = 0.0618x + 0.0148 | 0.023 | 0.069 | 1.8–4.6 | |
| O2 as reaction gas | |||||
| Cr * | y = 0.0088x + 0.0007 | 0.085 | 0.255 | 1.5–4.5 | |
| Cr** | y = 0.0100x + 0.0023 | 0.083 | 0.250 | 1.8–4.4 | |
| HPLC-ICP MS | 10 µg L−1 Cr(III) and Cr(VI) (n = 6) | ||||
| He as collision gas | tret. | area of peak | |||
| Cr(III) ** | y = 159 809x + 14 411 | 0.08 | 0.23 | 0.9 | 3.3 |
| Cr(VI) ** | y = 153 210x + 7 103 | 0.09 | 0.26 | 0.2 | 2.5 |
| Sample | Total Content Cr ± SD, µg g−1 | Cr Mass Fraction ± SD, µg g−1, ICP MS | Cr in Total Content ± SD, % | Mass Fraction of Chromium, µg g−1 ± SD, HPLC-ICP MS | ||
|---|---|---|---|---|---|---|
| Cr(VI) | Cr(III) | |||||
| CRM 041 | ** 86.3 ± 2.96 | 86.3 ± 2.98 | 100 ± 3.4 | 86.3 ± 2.7 | - | 100.0 ± 3.2 |
| CRM 060 | ** 195 ± 9.02 | 178.6 ± 9.20 | 91.6 ± 4.7 | 191.3 ± 7.6 | 107.1 ± 4.2 | |
| Soil A | 69 ± 5 | 1.1 ± 0.1 | 1.5 ± 0.1 | 0.9 ± 0.2 | - | 87.3 ± 7.6 |
| Soil B | 42 ± 3 | 1.7 ± 0.2 | 4.0 ± 0.5 | 0.7 ± 0.1 | 41.8 ± 2.1 | |
| Soil E | 2336 ± 30 | 44.6 ± 5.9 | 1.9 ± 0.3 | 2.2 ± 0.4 | 0.2 ± 0.02 | 5.3 ± 0.9 |
| Soil F | 142 ± 6 | 2.1 ± 0.1 | 1.5 ± 0.07 | 0.42 ± 0.02 | 0.35 ± 0.05 | 36.3 ± 5.8 |
| Soil G | 284 ± 8 | 15.6 ± 1.5 | 5.4 ± 0.5 | 1.3 ± 0.7 | 0.9 ± 0.2 | 14.5 ± 3.5 |
| Sample | Added Cr(VI), µg L−1 | Recovery of Cr(VI) ± SD, %, n = 3 |
|---|---|---|
| Soil C (M) | 1.4 2.6 | 111.1 ± 2.1 115.4± 3.5 |
| Soil E (O) | 2.7 5.4 | 115.2 ± 3.1 113.6 ± 2.5 |
| Soil F (O) | 1.1 2.7 | 101.2 ± 2.4 104.4 ± 2.8 |
| Soil G (O) | 5 µg·L−1 Cr(VI) + * 5 µg·L−1 Cr(III) | 104.8 ± 4.9 * 89.9 ± 12.8 |
| Soil Spiked with BaCrO4 | Added Cr, µg g−1 | Mass Fraction of Cr, µg g−1 | Recovery, % | |||
|---|---|---|---|---|---|---|
| ICP-MS | HPLC-ICP MS | ICP-MS | HPLC-ICP MS | |||
| Mineral: air-dry | 43.1 | 40.2 ± 1.9 | 42.8 ± 1.5 | 106.5 ± 3.7 | 93.4 ± 4.5 | 99.3 ± 3.6 |
| moist | 33.7 ± 1.3 | 31.8 ± 0.4 | 94.4 ± 3.9 | 78.2 ± 3.0 | 78.2 ± 3.0 | |
| Organic: air-dry | 45.1 | 32.8 ± 1.3 | 31.6 ± 0.2 | 96.3 ± 0.6 | 72.8 ± 2.9 | 70.0 ± 0.5 |
| moist | 24.9 ± 1.7 | 23.6 ± 2.4 | 94.8 ± 3.0 | 55.2 ± 3.7 | 52.3 ± 5.2 | |
| Humic Acid + Cr, µg·L−1 | Determined Concentration, µg·L−1 | Recovery of Cr, % | |
|---|---|---|---|
| Cr(III) | Cr(VI) | ||
| Cr(III) | |||
| 100 | 0.27 | ||
| 200 | 0.72 | 0.35 | |
| 400 | 1.44 | 0.36 | |
| Cr(VI) | |||
| 100 | 42.2 | 36.9 | 79.1 |
| 200 | 27.4 | 75.6 | 51.5 |
| 400 | 32.4 | 79.2 | 27.9 |
| Sample | Organic Matter, % | pHKCl | Total Content of Mn ± SD, µg g−1, n = 3 | Total Content of Fe ± SD, mg g−1, n = 3 |
|---|---|---|---|---|
| Soil A (VL) | 1.9 | 7.8 | 132 ± 9 | 13.9 ± 0.7 |
| Soil B (M) | 2.9 | 7.3 | 570 ± 31 | 10.6 ± 0.5 |
| Soil C (M) | 3.1 | 7.4 | 445 ± 11 | 8.6 ± 0.3 |
| Soil D (M) | 4.3 | 7.4 | 453 ± 17 | 8.6 ± 0.3 |
| Soil E (VL) | 5.4 | 7.7 | 218 ± 13 | 6.1 ± 0.3 |
| Soil F (L) | 7.3 | 7.3 | 268 ± 8 | 4.1 ± 0.3 |
| Soil G (M) | 10.1 | 7.2 | 348 ± 6 | 14.2 ± 1.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leśniewska, B.; Godlewska-Żyłkiewicz, B. Speciation of Chromium in Alkaline Soil Extracts by an Ion-Pair Reversed Phase HPLC-ICP MS Method. Molecules 2019, 24, 1172. https://doi.org/10.3390/molecules24061172
Leśniewska B, Godlewska-Żyłkiewicz B. Speciation of Chromium in Alkaline Soil Extracts by an Ion-Pair Reversed Phase HPLC-ICP MS Method. Molecules. 2019; 24(6):1172. https://doi.org/10.3390/molecules24061172
Chicago/Turabian StyleLeśniewska, Barbara, and Beata Godlewska-Żyłkiewicz. 2019. "Speciation of Chromium in Alkaline Soil Extracts by an Ion-Pair Reversed Phase HPLC-ICP MS Method" Molecules 24, no. 6: 1172. https://doi.org/10.3390/molecules24061172