
Synthesis of AMPSA Polymeric Derivatives Monitored by Electrical Conductivity and Evaluation of Thermosensitive Properties of Resulting Microspheres


Abstract
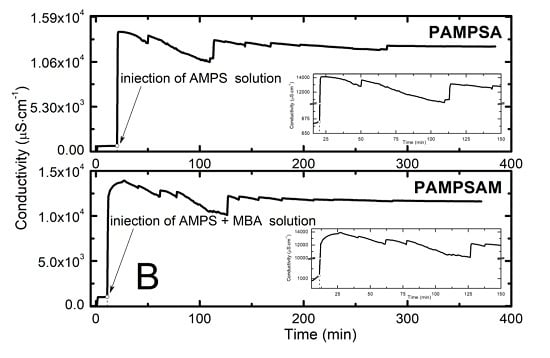
:
1. Introduction
2. Results
2.1. Synthesis
2.2. Conductivity Measurement Analysis
2.3. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy Analysis (ATR-FTIR)
2.4. Nuclear Magnetic Resonance Spectroscopy Analysis (1H-NMR)
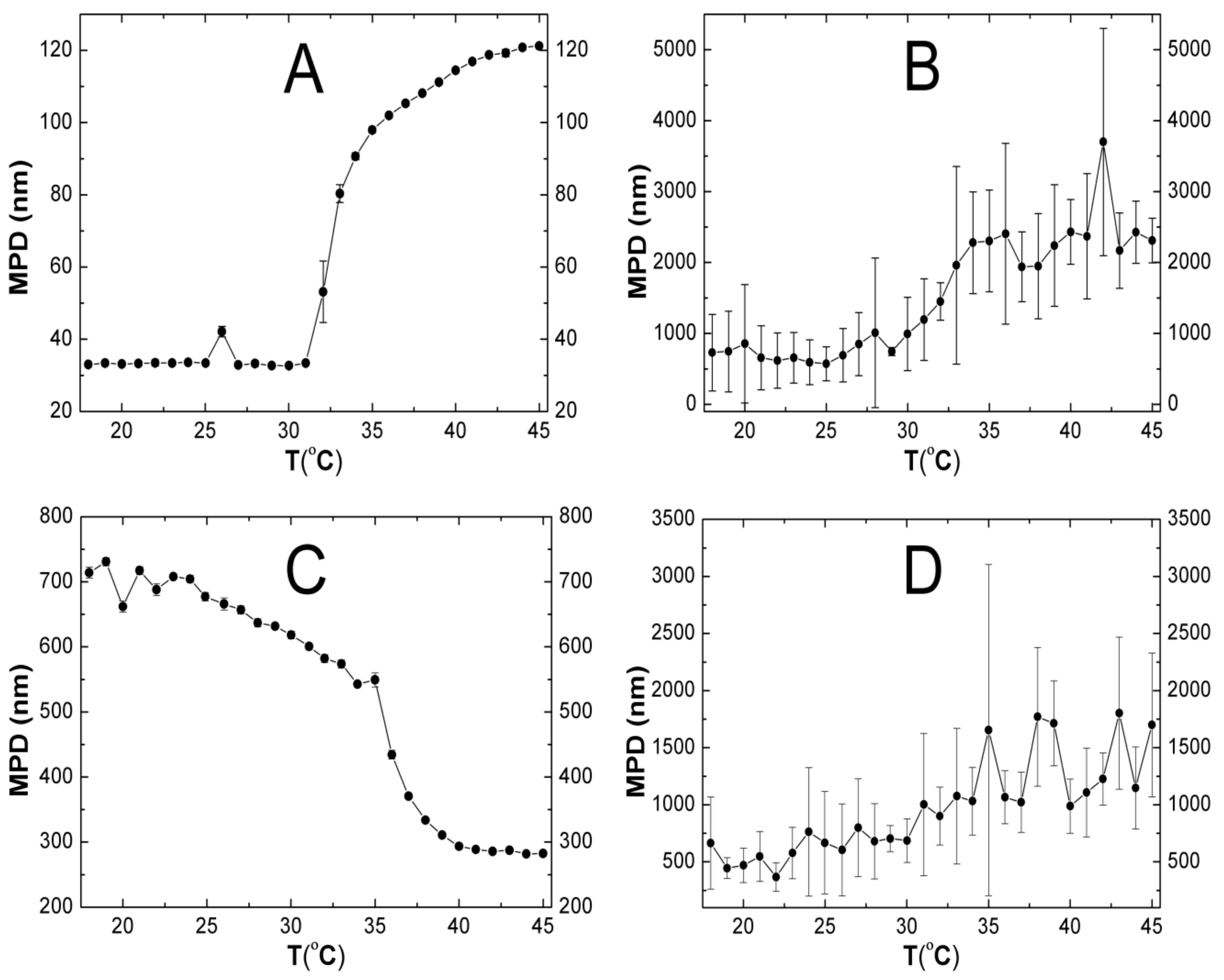
2.5. Hydrodynamic Diameter (HD)
2.6. Polydispersity Index (PDI)
2.7. Zeta Potential (ZP)
2.8. Scanning Electron Microscopy with Energy Dispersive Spectroscopy System (SEM-EDS) Analysis
3. Discussion
3.1. Synthesis
3.2. Conductivity
3.3. ATR-FTIR
3.4. H-NMR
3.5. HD
3.6. PDI
3.7. ZP
3.8. SEM-EDS
4. Materials and Methods
4.1. Materials
4.2. Synthesis
4.3. Conductivity Measurements
4.4. Attenuated Total Reflection Fourier Transformed Infrared Spectroscopy measurements (ATR-FTIR)
4.5. Nuclear Magnetic Resonance Spectroscopy measurements (1H-NMR)
4.6. Hydrodynamic diameter (HD)and polydispersity index(PDI) measurements
4.7. Zeta Potential (ZP) Measurements
4.8. Scanning Electron Microscopy With Energy Dispersive Spectroscopy System (SEM-EDS) Measurements
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nakajima, K.; Honda, S.; Nakamura, Y.; Redondo, F.L.H.; Kohsaka, S.; Yamato, M.; Kikuchi, A.; Okano, T. Intact microglia are cultured and non-invasively harvested without pathological activation using a novel cultured cell recovery method. Biomaterials 2001, 22, 1213–1223. [Google Scholar] [CrossRef]
- Shimizu, T.; Yamato, M.; Kikuchi, A.; Okano, T. Cell sheet engineering for myocardial tissue reconstruction. Biomaterials 2003, 24, 2309–2316. [Google Scholar] [CrossRef]
- Pelton, R.H.; Chibante, P. Preparation of aqueous latices with N-isopropylacrylamide. Colloid. Surf. A 1986, 20, 247–256. [Google Scholar] [CrossRef]
- Pelton, R.H.; Pelton, H.M.; Morphesis, A.; Rowells, R.L. Particle sizes and electrophoretic mobilities of poly(N-isopropylacrylamide) latex. Langmuir 1989, 5, 816–818. [Google Scholar] [CrossRef]
- Ougizawa, T.; Inoue, T.; Kammer, H.W. UCST and LCST behavior in polymer blends. Macromolecules 1985, 18, 2089–2092. [Google Scholar] [CrossRef]
- Heyda, J.; Soll, S.; Yuan, J.; Dzubiella, J. Thermodynamic description of the LCST of charged thermoresponsive copolymers. Macromolecules 2014, 47, 2096–2102. [Google Scholar] [CrossRef]
- Saikia, A.K.; Aggarwal, S.; Mandal, U.K. Preparation and controlled drug release characteristics of thermoresponsive PEG/Poly (NIPAM-co-AMPS) hydrogels. Int. J. Polym. Mat. 2012, 62, 39–44. [Google Scholar] [CrossRef]
- Takegami, Y.; Yokoyama, Y.; Norisugi, O.; Nagatsuma, M.; Takata, K.; Rehman, M.U.; Matsunaga, K.; Yokoi’, H.; Fujiki, S.; Makino, T.; et al. Synthesis and characterization of high-quality skin-cooling sheets containing thermosensitive poly(N-isopropylacrylamide). J. Biom. Mat. Res. B: Appl. Biomater. 2011, 98B, 110–113. [Google Scholar] [CrossRef]
- Indulekha, S.; Arunkumar, P.; Bahadur, D.; Srivastava, R. Thermoresponsive polymeric gel as an on demand transdermal drug delivery system for pain management. Mater. Sci. Eng. C 2016, 62, 113–122. [Google Scholar] [CrossRef]
- Liu, L.; Wang, W.; Ju, X.J.; Xie, R.; Chu, L.Y. Smart thermo-triggered squirting capsules for nanoparticle delivery. Soft Matt. 2010, 6, 3759–3763. [Google Scholar] [CrossRef]
- Gong, C.; Qi, T.; Wei, X.; Qu, Y.; Wu, Q.; Luo, F.; Qian, Z. Thermosensitive polymeric hydrogels as drug delivery systems. Curr. Med. Chem. 2012, 20, 79–94. [Google Scholar] [CrossRef]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef]
- Gupta, P.; Vermani, K.; Garg, S. Hydrogels: From controlled release to pH-responsive drug delivery. Drug Discov. Today 2002, 7, 569–579. [Google Scholar] [CrossRef]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Advc. Drug Deliv. Rev. 2001, 53, 321–339. [Google Scholar] [CrossRef]
- Shin, H.S.; Kim, S.Y.; Lee, Y.M. Indomethacin release behaviors from pH and thermoresponsive poly-(vinyl alcohol) and poly-(acrylic acid) IPN hydrogels for site-specific drug delivery. J. Appl. Polym. Sci. 1997, 65, 685–693. [Google Scholar] [CrossRef]
- Wei, H.; Cheng, C.; Chang, C.; Chen, W.Q.; Cheng, S.X.; Zhang, X.Z.; Zhuo, R.-X. Synthesis and applications of shell cross-linked thermoresponsive hybrid micelles based on poly-(N-isopropylacrylamide-co-3-(trimethoxysilyl)-propyl-methacrylate)-b-poly-(methyl methacrylate). Langmuir 2008, 24, 4564–4570. [Google Scholar] [CrossRef]
- Loh, X.J.; Wu, Y.L.; Seow, W.T.J.; Norimzan, M.N.I.; Zhang, Z.X.; Xu, F.J.; Kang, E.T.; Neoh, K.G.; Li, J. Micellization and phase transition behavior of thermosensitive poly-(N-isopropylacrylamide)-poly(ε-caprolactone)-poly-(N-isopropylacrylamide) triblock copolymers. Polymer 2008, 49, 5084–5094. [Google Scholar] [CrossRef]
- Gil, E.S.; Hudson, S.M. Stimuli-responsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Taghizadeh, B.; Taranejoo, S.; Monemian, S.A.; Moghaddam, Z.S.; Daliri, K.; Derakhshankhah, H.; Derakhshani, Z. Classification of stimuli–responsive polymers as anticancer drug delivery systems. Drug Deliv. 2015, 22, 145–155. [Google Scholar] [CrossRef]
- Oerlemans, C.; Bult, W.; Bos, M.; Storm, G.; Frank, J.; Nijsen, W.; Hennink, W.E. Polymeric micelles in anticancer terapy: Targeting, imaging and triggered release. Pharm. Res. 2010, 27, 2569–2589. [Google Scholar] [CrossRef]
- Xin, Y.; Yin, M.; Zhao, L.; Meng, F.; Luo, L. Recent progress on nanoparticle-based drug delivery systems for cancer therapy. Cancer Biol. Med. 2017, 14, 228–241. [Google Scholar] [CrossRef] [Green Version]
- Kohori, F.; Sakai, K.; Aoyagi, T.; Yokoyama, M.; Yamato, M.; Sakurai, T. Control of adriamycin cytotoxic activity using thermally responsive polymeric micelles composed of poly(N-isopropylacrylamide- co-N,N-dimethylacrylamide)-b-poly(d,l-lactide). Colloids Surf. B Biointerfaces 1999, 16, 195–205. [Google Scholar] [CrossRef]
- Liu, S.Q.; Tong, Y.W.; Yang, Y.Y. Thermally sensitive micelles self-assembled from poly(N-isopropylacrylamide-co-N,N-dimethylacrylamide)-b-poly(d,l-lactide-co-glycolide) for controlled delivery of paclitaxel. Mol. BioSyst. 2005, 1, 158–165. [Google Scholar] [CrossRef]
- Liu, B.; Yang, M.; Li, R.; Ding, Y.; Qian, X.; Yu, L.; Jiang, X. The antitumor effect of novel docetaxel-loaded thermosensitive micelles. Eur. J. Pharm. Biopharm. 2008, 69, 527–534. [Google Scholar] [CrossRef]
- Qu, T.; Wang, A.; Yuan, J.; Shi, J.; Gao, Q. Preparation and characterization of thermo-responsive amphiphilic triblock copolymer and its self-assembled micelle for controlled drug release. Colloids Surf. B: Biointerfaces 2009, 72, 94–100. [Google Scholar] [CrossRef]
- Nakayama, M.; Okano, T.; Miyazaki, T.; Kohori, F.; Sakai, K.; Yokoyama, M. Molecular design of biodegradable polymeric micelles for temperature-responsive drug release. J. Control. Release 2006, 115, 46–56. [Google Scholar] [CrossRef]
- Wei, H.; Cheng, S.X.; Zhang, X.Z.; Zhuo, R.X. Thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers. Prog. Polym. Sci. 2009, 34, 893–910. [Google Scholar] [CrossRef]
- Molina, M.; Giulbudagian, M.; Calderón, M. Positively charged thermoresponsive nanogels for anticancer drug delivery. Macromol. Chem. Phys. 2014, 215, 2414–2419. [Google Scholar] [CrossRef]
- Watkins, K.A.; Chen, R. pH-responsive, lysine-based hydrogels for the oral delivery of a wide size range of molecules. Int. J. Pharm. 2015, 478, 496–503. [Google Scholar] [CrossRef]
- Reyes-Ortega, F. 3-pH-responsive polymers: Properties, synthesis and applications. In Smart Polymers and Their Applications, 1st ed.; Aguilar De Armas, M.R., Román, J.S., Eds.; Woodhead Publishing in Materials: Cambridge, UK, 2014; pp. 45–92. ISBN 9780857096951. [Google Scholar]
- Roy, S.G.; De, P. pH responsive polymers with amino acids in the side chains and their potential applications. J. Appl. Polym. Sci. 2014, 131, 41084–41096. [Google Scholar] [CrossRef]
- Schilli, Ch.M.; Zhang, M.; Rizzardo, E.; Thang, S.H.; Chong, Y.; Edwards, K.; Karlsson, G.; Müller, A.H.E. A new double- responsive block copolymer synthesized via RAFT. polymerization: Poly(N-isopropylacrylamide)-block-poly(acrylic acid). Macromolecules 2004, 37, 7861–7866. [Google Scholar] [CrossRef]
- Kang, S.I.; Bae, Y.H. pH-Induced solubility transition of sulfonamide-based polymers. J. Controlled Release 2002, 80, 145–155. [Google Scholar] [CrossRef]
- Zhu, L.; Smith, P.P.; Boyes, S.G. pH-responsive polymers for imaging acidic biological environments in tumors. J. Polym. Sci. Part. B: Polym. Phys. 2013, 51, 1062–1067. [Google Scholar] [CrossRef] [Green Version]
- Musiał, W.; Michálek, J. The influence of low process temperature on the hydrodynamic radius of polyNIPAM-co-PEG thermosensitive nanoparticles presumed as drug carriers for bioactive proteins. Acta Pol. Pharm. 2015, 72, 161–169. [Google Scholar]
- Gola, A.; Knysak, T.; Musiał, W. The Influence of Anionic Initiator on the Selected Properties of Poly-N-Isopropyl Acrylamide Evaluated for Controlled Drug Delivery. Molecules 2017, 22, 23. [Google Scholar] [CrossRef]
- Kabiri, K.; Zohuriaan-Mehr, M.J.; Mirzadeh, H.; Kheirabadi, M. Solvent- ion- and pH-specific swelling of poly(2-acrylamido-2- methylpropane sulfonic acid) superabsorbing gels. J. Polym. Res. 2010, 17, 203–212. [Google Scholar] [CrossRef]
- Gospodinova, N.; Tomšík, E.; Omelchenko, O. How strong are strong poly(sulfonic acids)? An example of the poly(2-acrylamido-2-methyl-1-propanesulfonic acid). Europ. Polym. J. 2016, 74, 130–135. [Google Scholar] [CrossRef]
- Yoshida, H.; Furukawa, Y.; Tasumi, M. Structural studies and vibrational analyses of stable and less stable conformers of 1,3,5-hexatriene based on ab initio MO calculations. J. Mol. Struct. 1989, 194, 279–299. [Google Scholar] [CrossRef]
- Scherer, J.R.; Potts, W.J. Normal Coordinates for the Out-of-Plane Deformations of Vinyl Bromide. J. Chem. Phys. 1959, 30, 1527–1529. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of infrared spectra, a practical approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2000; pp. 10815–10837. ISBN 0471976709. [Google Scholar]
- Mishra, P.K.; Ekielski, A. The self-assembly of lignin and its application in nanoparticle synthesis: A Short Review. Nanomaterials 2019, 9, 243. [Google Scholar] [CrossRef]
- Mishra, P.K.; Wimmer, R. Aerosol assisted self-assembly as a route to synthesize solid and hollow spherical lignin colloids and its utilization in layer by layer deposition. UltrasonicsSonochemistry 2017, 35, 45–50. [Google Scholar] [CrossRef]
- Matthews, B.A.; Rhodes, C.T. Use of the Derjaguin, Landau, Verwey and Overbeek theory to interpret pharmaceutical suspension stability. J. Pharm. Sci. 1970, 59, 521–525. [Google Scholar] [CrossRef]
- Pelton, R. Temperature-sensitive aqueous microgels. Adv. Colloid Interface Sci. 2000, 85, 1–33. [Google Scholar] [CrossRef]
- McPhee, W.; Tam, K.C.; Pelton, R. Poly(N-isopropylacrylamide) latices prepared with sodium dodecyl sulfate. J. Colloid Interface Sci. 1993, 156, 24–30. [Google Scholar] [CrossRef]
- Kratz, K.; Eimer, W. Swelling properties of colloidal poly(N-Isopropylacrylamide) microgels in solution. Ber. Bunsenges. Phys. Chem. 1998, 102, 848–854. [Google Scholar] [CrossRef]
- Oh, K.S.; Bae, Y.C. Swelling behavior of submicron gel particles. J. Appl. Polym. Sci. 1998, 69, 109–114. [Google Scholar] [CrossRef]
- Ito, K. Competition between thermal decomposition and hydrolysis of 2,2′-azobis (2-amidinopropane) in aqueous solution. J. Polym. Sci. Polym. Chem. 1973, 11, 1673–1681. [Google Scholar] [CrossRef]
- Musiał, W.; Kokol, V.; Fecko, T.; Voncina, B. Morphological patterns of poly(N-isopropylacrylamide) derivatives synthesized with EGDMA, DEGDMA, and TEGDMA crosslinkers for application as thermosensitive drug carriers. Chem. Papers 2010, 64, 791–798. [Google Scholar] [CrossRef]
- Available online: https://www.iso.org/standard/65410.html (accessed on 23 March 2019).
Sample Availability: Samples of the compounds PNA, PAMPSA, PNAM, PAMPSAM are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Polymer Microparticle System | Monomer [mol] | Cationic Initiator [mol] | Cross-Linker [mol] | |
|---|---|---|---|---|
| NIPA | AMPS | AMP | MBA | |
| PNA | 4.433 × 10−2 | - | 2.238 × 10−3 | - |
| PAMPSA | - | 2.453 × 10−2 | 1.232 × 10−3 | - |
| PNAM | 4.429 × 10−2 | - | 2.224 × 10−3 | 3.256 × 10−3 |
| PAMPSAM | - | 2.416 × 10−2 | 1.239 × 10−3 | 3.282 × 10−3 |
| Type of Polymer Microparticles System | Zeta Potential [mV] | pH | |||
|---|---|---|---|---|---|
| Measurement Temperatures [°C] | |||||
| 18 | 25 | 32 | 45 | 25 | |
| PNA | −1.36 ± 0.87 | −0.34 ± 0.83 | 1.65 ± 0.75 | 47.32 ± 1.88 | 7.39 |
| PAMPSA | −0.01 ± 0.37 | 0.15 ± 0.22 | −0.23 ± 0.05 | 0.06 ± 0.34 | 4.42 |
| PNAM | 8.90 ± 0.11 | 10.00 ± 0.33 | 11.66 ± 0.82 | 30.16 ± 1.54 | 7.50 |
| PAMPSAM | −0.09 ± 0.11 | −0.13 ± 0.01 | −0.13 ± 0.32 | −0.04 ± 0.11 | 3.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gola, A.; Sacharczuk, M.; Musiał, W. Synthesis of AMPSA Polymeric Derivatives Monitored by Electrical Conductivity and Evaluation of Thermosensitive Properties of Resulting Microspheres. Molecules 2019, 24, 1164. https://doi.org/10.3390/molecules24061164
Gola A, Sacharczuk M, Musiał W. Synthesis of AMPSA Polymeric Derivatives Monitored by Electrical Conductivity and Evaluation of Thermosensitive Properties of Resulting Microspheres. Molecules. 2019; 24(6):1164. https://doi.org/10.3390/molecules24061164
Chicago/Turabian StyleGola, Agnieszka, Magdalena Sacharczuk, and Witold Musiał. 2019. "Synthesis of AMPSA Polymeric Derivatives Monitored by Electrical Conductivity and Evaluation of Thermosensitive Properties of Resulting Microspheres" Molecules 24, no. 6: 1164. https://doi.org/10.3390/molecules24061164