
Site-Specific Cleavage of RNAs Derived from the PIM1 3′-UTR by a Metal-Free Artificial Ribonuclease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
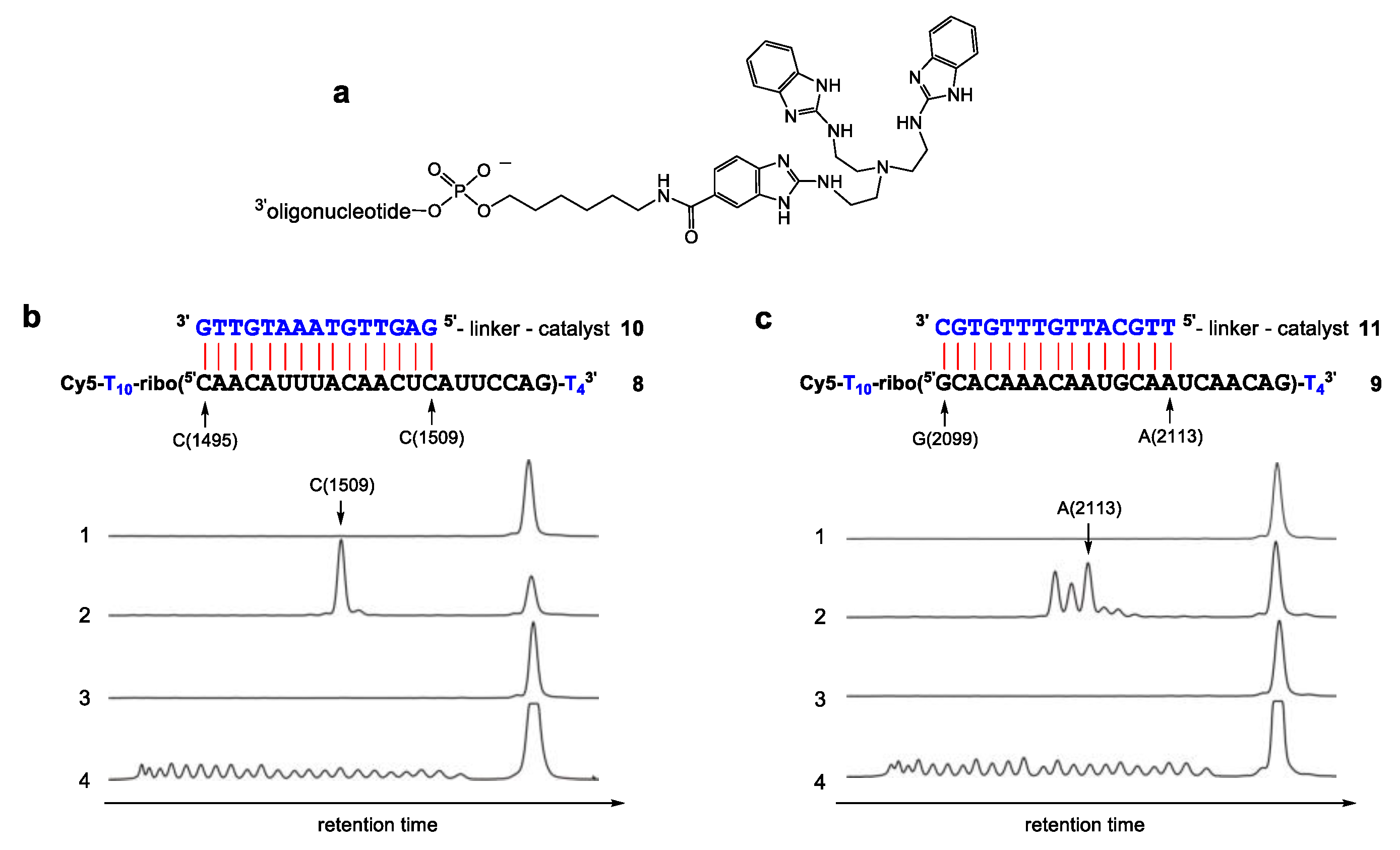
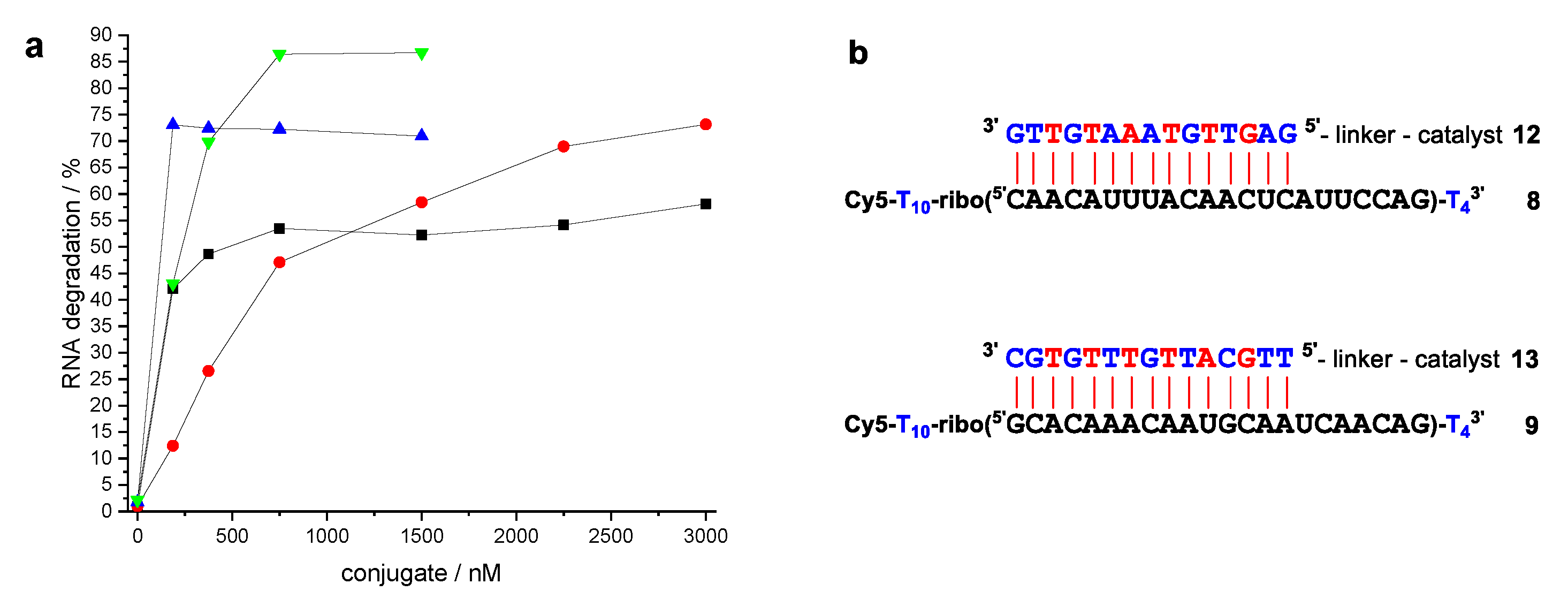
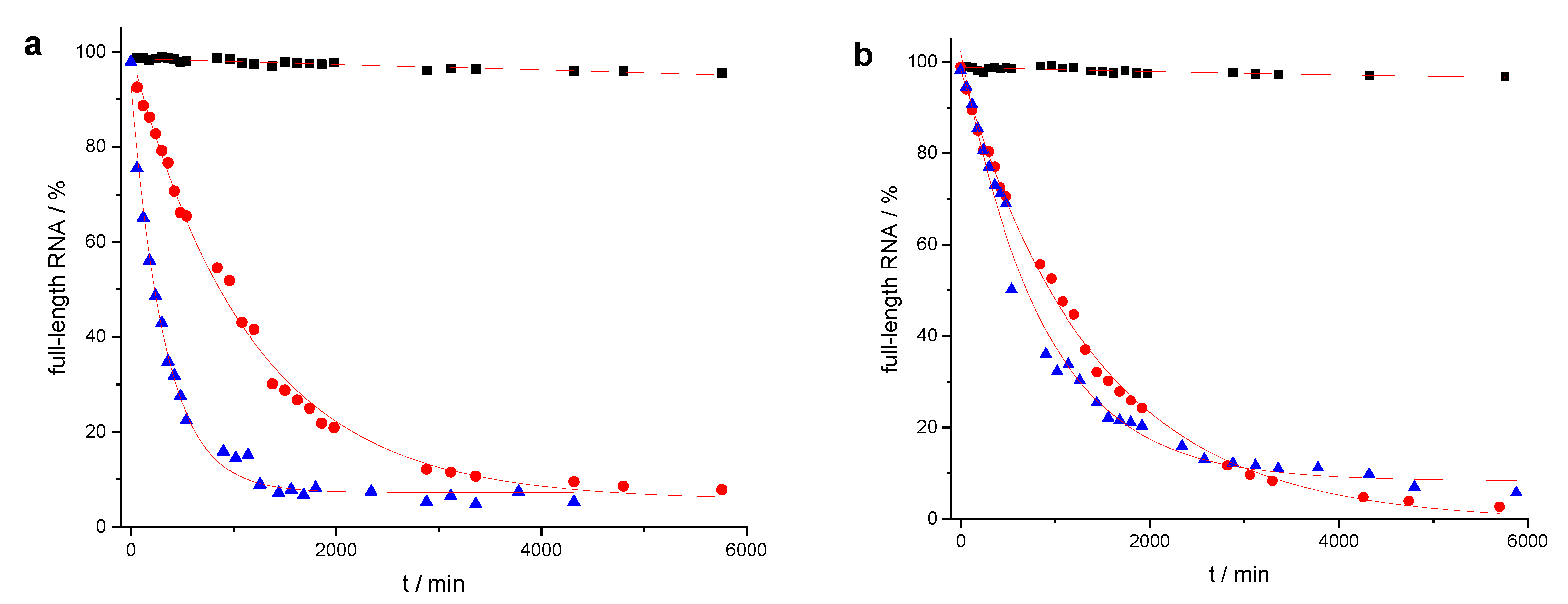
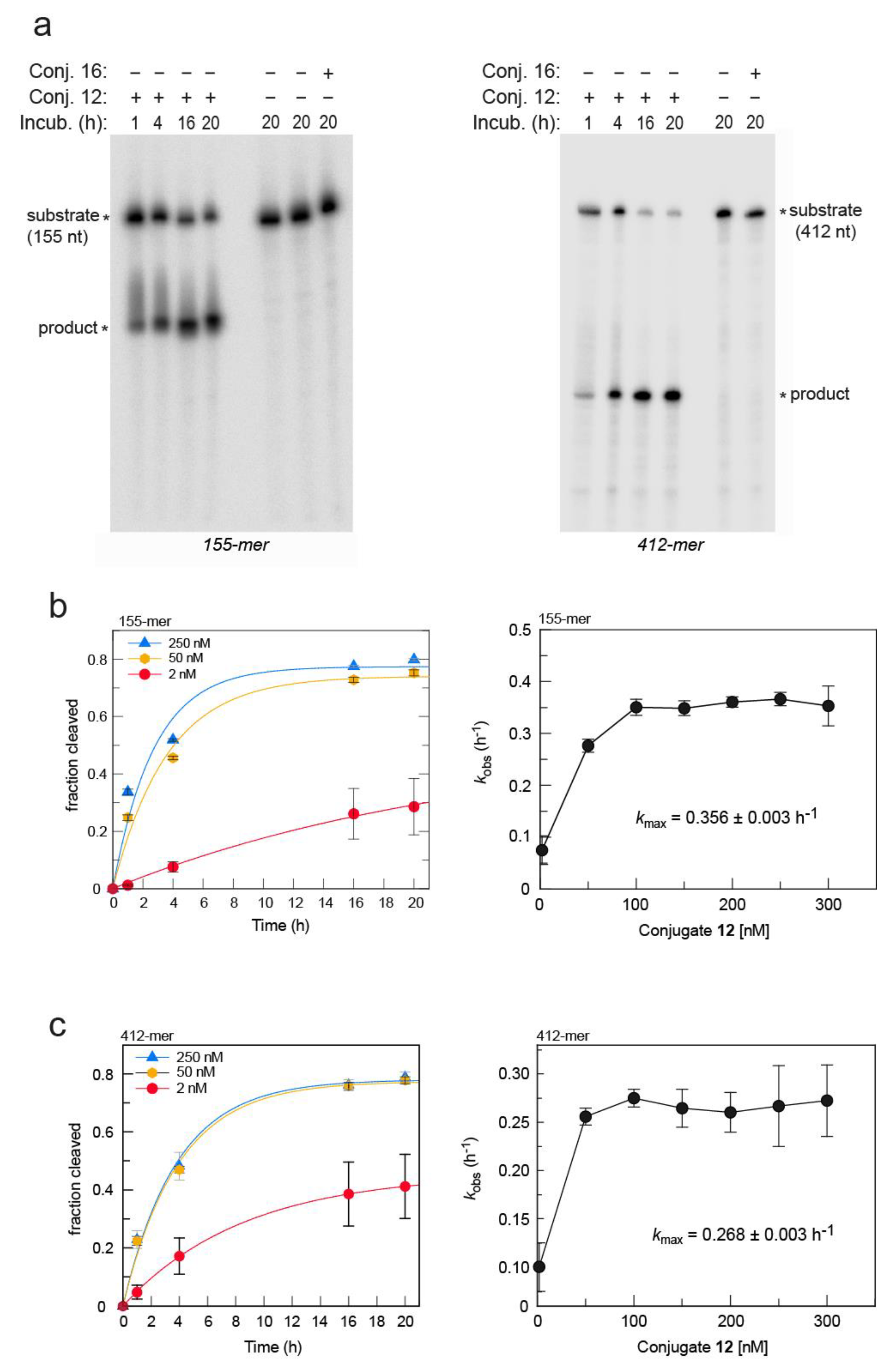
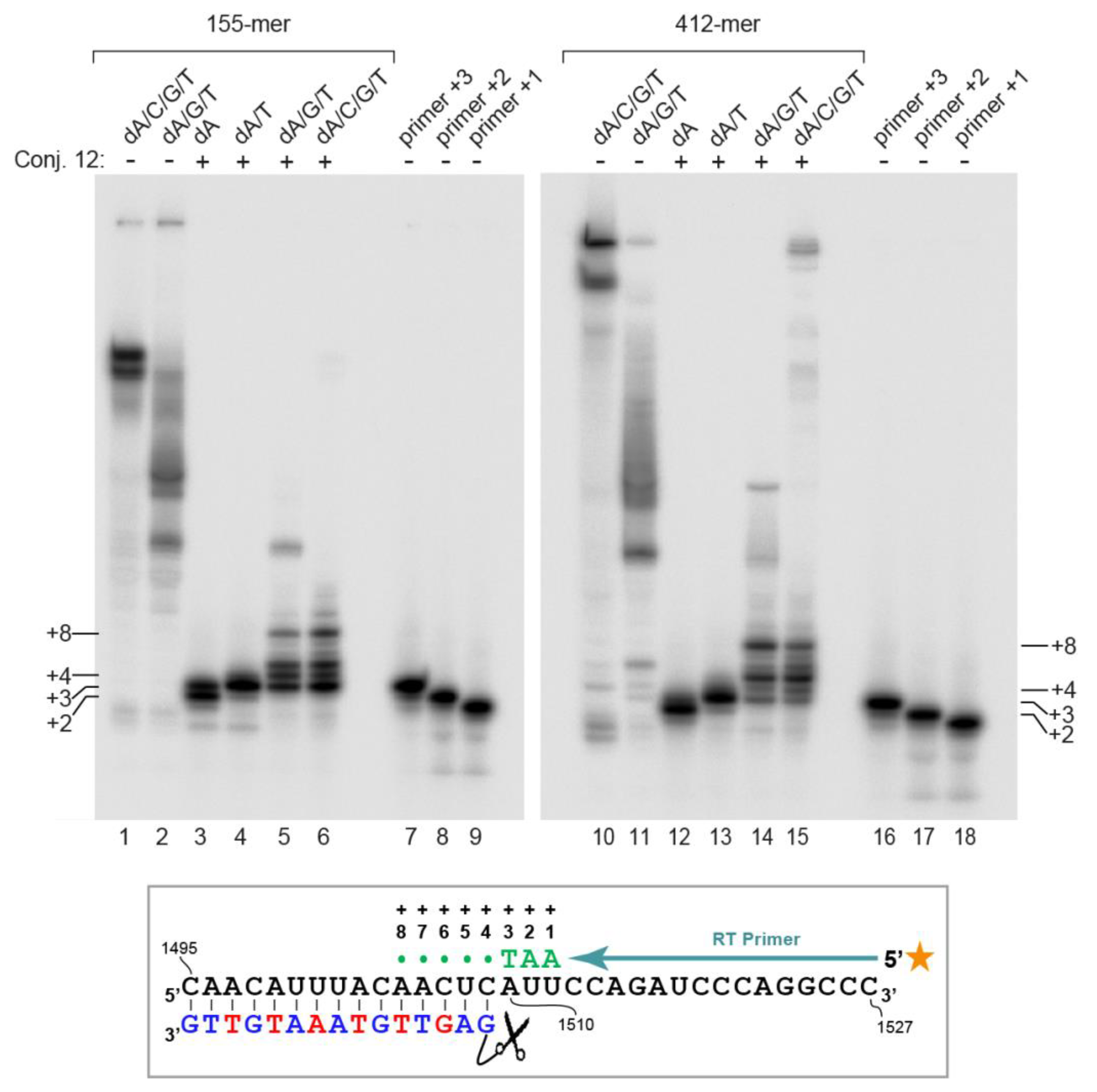
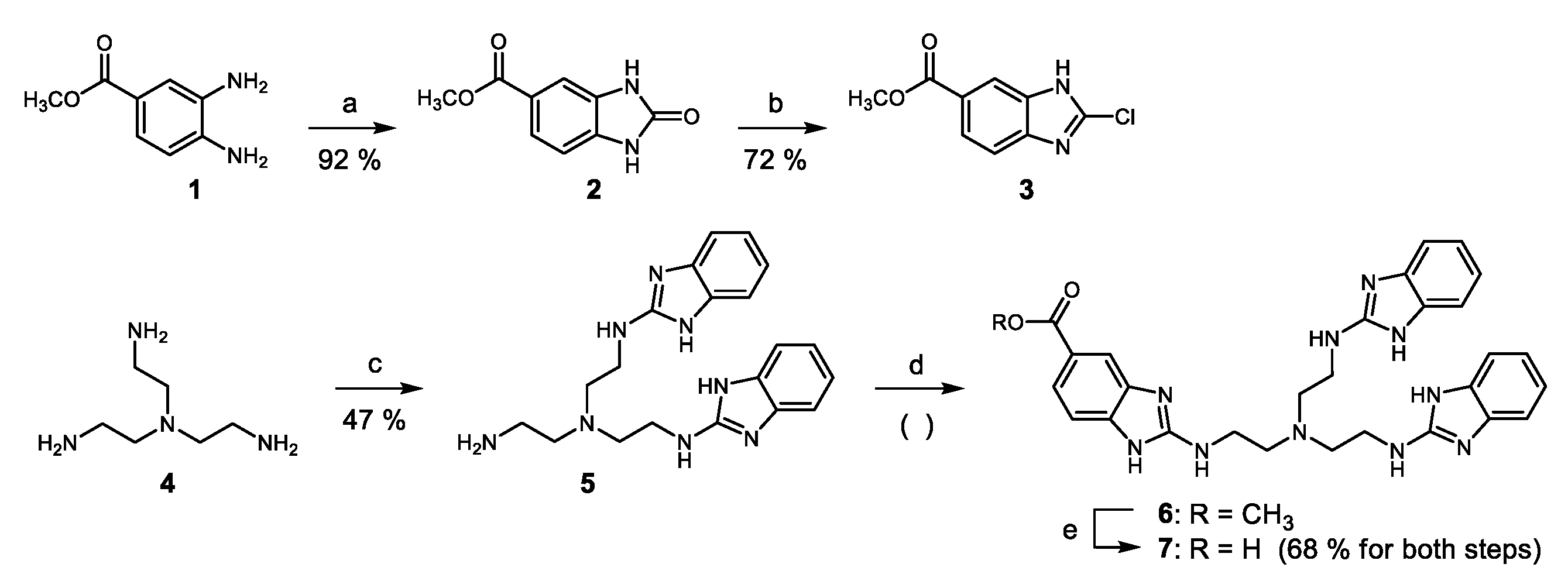
2. Results
3. Discussion
4. Materials and Methods
4.1. In Vitro Transcription of the 155-mer and 412-mer RNA Substrates 14 and 15
4.2. Dephosphorylation and 5′-[32P]-End-Labeling of RNA
4.3. In Vitro Cleavage Assay
4.4. Primer Extension
4.5. Splint Ligation Producing RNA 17
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trawick, B.N.; Daniher, A.T.; Bashkin, J.K. Inorganic mimics of ribonucleases and ribozymes: From random cleavage to sequence-specific chemistry to catalytic antisense drugs. Chem. Rev. 1998, 98, 939–960. [Google Scholar] [CrossRef] [PubMed]
- Häner, R. Artificial ribonucleases. Chimia 2001, 55, 1035–1037. [Google Scholar]
- Komiyama, M.; Sumaoka, J.; Kuzuya, A.; Yamamoto, Y. Sequence-selective artificial ribonucleases. Methods Enzymol. 2001, 341, 455–468. [Google Scholar] [PubMed]
- Morrow, J.R.; Iranzo, O. Synthetic metallonucleases for RNA cleavage. Curr. Opin. Chem. Biol. 2004, 8, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Niittymäki, T.; Lönnberg, H. Artificial ribonucleases. Org. Biomol. Chem. 2006, 4, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ghidini, A.; Murtola, M.; Strömberg, R. Oligonucleotide based artificial ribonucleases (OBANs). In DNA in Supramolecular Chemistry and Nanotechnology; Stulz, E., Clever, G.H., Eds.; John Wiley: Chichester, UK, 2015; pp. 158–171. ISBN 9781118696880. [Google Scholar]
- Hall, J.; Hüsken, D.; Pieles, U.; Moser, H.E.; Häner, R. Efficient sequence-specific cleavage of RNA using novel europium complexes conjugated to oligonucleotides. Chem. Biol. 1994, 1, 185–190. [Google Scholar] [CrossRef]
- Bashkin, J.K.; Frolova, E.I.; Sampath, U. Sequence-specific cleavage of HIV mRNA by a ribozyme mimic. J. Am. Chem. Soc. 1994, 116, 5981–5982. [Google Scholar] [CrossRef]
- Matsumura, K.; Endo, M.; Komiyama, M. Lanthanide complex–oligo-DNA hybrid for sequence-selective hydrolysis of RNA. J. Chem. Soc. Chem. Commun. 1994, 2019–2020. [Google Scholar] [CrossRef]
- Magda, D.; Miller, R.A.; Sessler, J.L.; Iverson, B.L. Site-specific hydrolysis of RNA by europium(III) texaphyrin conjugated to a synthetic oligodeoxyribonucleotide. J. Am. Chem. Soc. 1994, 116, 7439–7440. [Google Scholar] [CrossRef]
- Murtola, M.; Wenska, M.; Strömberg, R. PNAzymes that are artificial RNA restriction enzymes. J. Am. Chem. Soc. 2010, 132, 8984–8990. [Google Scholar] [CrossRef] [PubMed]
- Gnaccarini, C.; Peter, S.; Scheffer, U.; Vonhoff, S.; Klussmann, S.; Göbel, M.W. Site-specific cleavage of RNA by a metal-free artificial nuclease attached to antisense oligonucleotides. J. Am. Chem. Soc. 2006, 128, 8063–8067. [Google Scholar] [CrossRef] [PubMed]
- Danneberg, F.; Ghidini, A.; Dogandzhiyski, P.; Kalden, E.; Strömberg, R.; Göbel, M.W. Sequence-specific RNA cleavage by PNA conjugates of the metal-free artificial ribonuclease tris(2-aminobenzimidazole). Beilstein J. Org. Chem. 2015, 11, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogandzhiyski, P.; Ghidini, A.; Danneberg, F.; Strömberg, R.; Göbel, M.W. Studies on tris(2-aminobenzimidazole)-PNA based artificial nucleases: A comparison of two analytical techniques. Bioconjugate Chem. 2015, 26, 2514–2519. [Google Scholar] [CrossRef] [PubMed]
- Canaple, L.; Hüsken, D.; Hall, J.; Häner, R. Artificial ribonucleases: Efficient and specific in vitro cleavage of human c-raf-1 RNA. Bioconjugate Chem. 2002, 13, 945–951. [Google Scholar] [CrossRef]
- Tursynbay, Y.; Zhang, J.; Li, Z.; Tokay, T.; Zhumadilov, Z.; Wu, D.; Xie, Y. Pim-1 kinase as cancer drug target: An update. Biomed. Rep. 2016, 4, 140–146. [Google Scholar] [CrossRef] [PubMed]
- NCBI Database. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NM_001243186.1 (accessed on 22 November 2018).
- Thomas, M.; Lange-Grünweller, K.; Weirauch, U.; Gutsch, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 2012, 31, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, U.; Strick, A.; Ludwig, V.; Peter, S.; Kalden, E.; Göbel, M.W. Metal-free catalysts for the hydrolysis of RNA derived from guanidines, 2-aminopyridines, and 2-aminobenzimidazoles. J. Am. Chem. Soc. 2005, 127, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Zaramella, S.; Yeheskiely, E.; Strömberg, R. A method for solid-phase synthesis of oligo- nucleotide 50-peptide-conjugates using acid-labile alpha-amino protections. J. Am. Chem. Soc. 2004, 126, 14029–14035. [Google Scholar] [CrossRef] [PubMed]
- Wenska, M.; Alvira, M.; Steunenberg, P.; Stenberg, Å.; Murtola, M.; Strömberg, R. An activated triple bond linker enables ‘click’ attachment of peptides to oligonucleotides on solid support. Nucleic Acids Res. 2011, 39, 9047–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ViennaRNA Web Services. Available online: http://rna.tbi.univie.ac.at/ (accessed on 22 November 2018).
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Grünweller, A.; Wyszko, E.; Bieber, B.; Jahnel, R.; Erdmann, V.A.; Kurreck, J. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res. 2003, 31, 3185–3193. [Google Scholar] [CrossRef] [PubMed]
- Tubiana, M. Tumor cell proliferation kinetics and tumor growth rate. Acta Oncol. 1989, 28, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Matsushima, Y.; Sugimura, T.; Terada, M. A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res. 1991, 19, 2785. [Google Scholar] [CrossRef] [PubMed]
- Gößringer, M.; Helmecke, D.; Köhler, K.; Schön, A.; Kirsebom, L.A.; Bindereif, A.; Hartmann, R.K. Enzymatic RNA Synthesis using Bacteriophage T7 RNA Polymerase. In Handbook of RNA Biochemistry, 2nd ed.; Hartmann, R.K., Bindereif, A., Schön, A., Westhof, E., Eds.; WILEY-VCH: Weinheim, Germany, 2015; pp. 3–27. ISBN 978-3-527-32776-8. [Google Scholar]
- Smith, B.D.; Soellner, M.B.; Raines, R.T. Potent Inhibition of Ribonuclease A by Oligo(vinylsulfonic Acid). J. Biol. Chem. 2003, 23, 20934–20938. [Google Scholar] [CrossRef] [PubMed]
- Weinrich, T.; Jaumann, E.A.; Scheffer, U.; Prisner, T.F.; Göbel, M.W. A Cytidine Phosphoramidite with Protected Nitroxide Spin Label: Synthesis of a Full-Length TAR RNA and Investigation by in-Line Probing and EPR Spectroscopy. Chemistry 2018, 24, 6202–6207. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound 7 are available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zellmann, F.; Thomas, L.; Scheffer, U.; Hartmann, R.K.; Göbel, M.W. Site-Specific Cleavage of RNAs Derived from the PIM1 3′-UTR by a Metal-Free Artificial Ribonuclease. Molecules 2019, 24, 807. https://doi.org/10.3390/molecules24040807
Zellmann F, Thomas L, Scheffer U, Hartmann RK, Göbel MW. Site-Specific Cleavage of RNAs Derived from the PIM1 3′-UTR by a Metal-Free Artificial Ribonuclease. Molecules. 2019; 24(4):807. https://doi.org/10.3390/molecules24040807
Chicago/Turabian StyleZellmann, Felix, Laura Thomas, Ute Scheffer, Roland K. Hartmann, and Michael W. Göbel. 2019. "Site-Specific Cleavage of RNAs Derived from the PIM1 3′-UTR by a Metal-Free Artificial Ribonuclease" Molecules 24, no. 4: 807. https://doi.org/10.3390/molecules24040807