
Lignin-Only Polymeric Materials Based on Unmethylated Unfractionated Kraft and Ball-Milled Lignins Surpass Polyethylene and Polystyrene in Tensile Strength


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Macromolecular Lignin Configuration: Original Impressions
1.2. Macromolecular Lignin Configuration: Current Perspectives
1.3. Plastics with 100 wt% Lignin-Derivative Contents
2. Results and Discussion
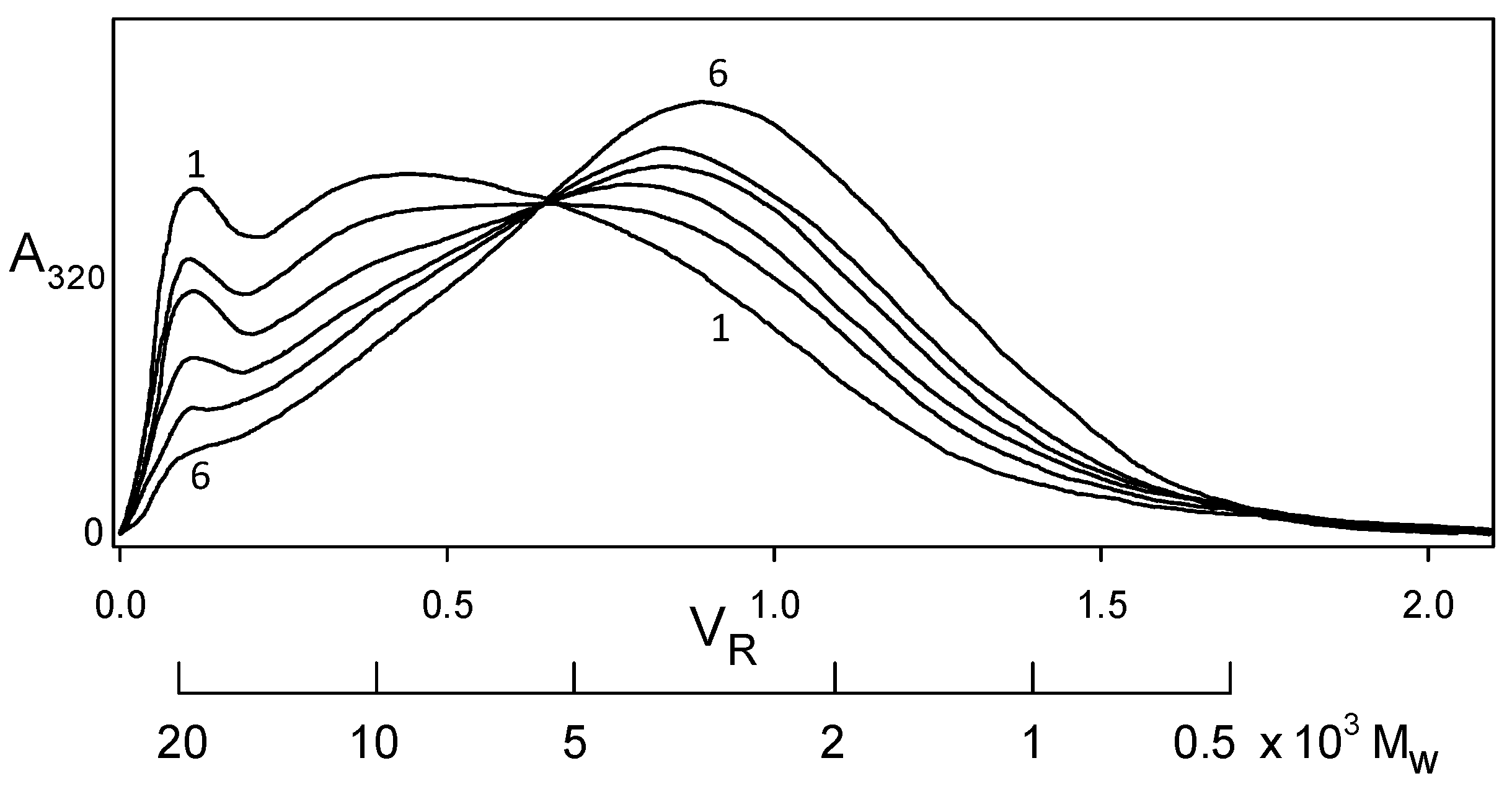
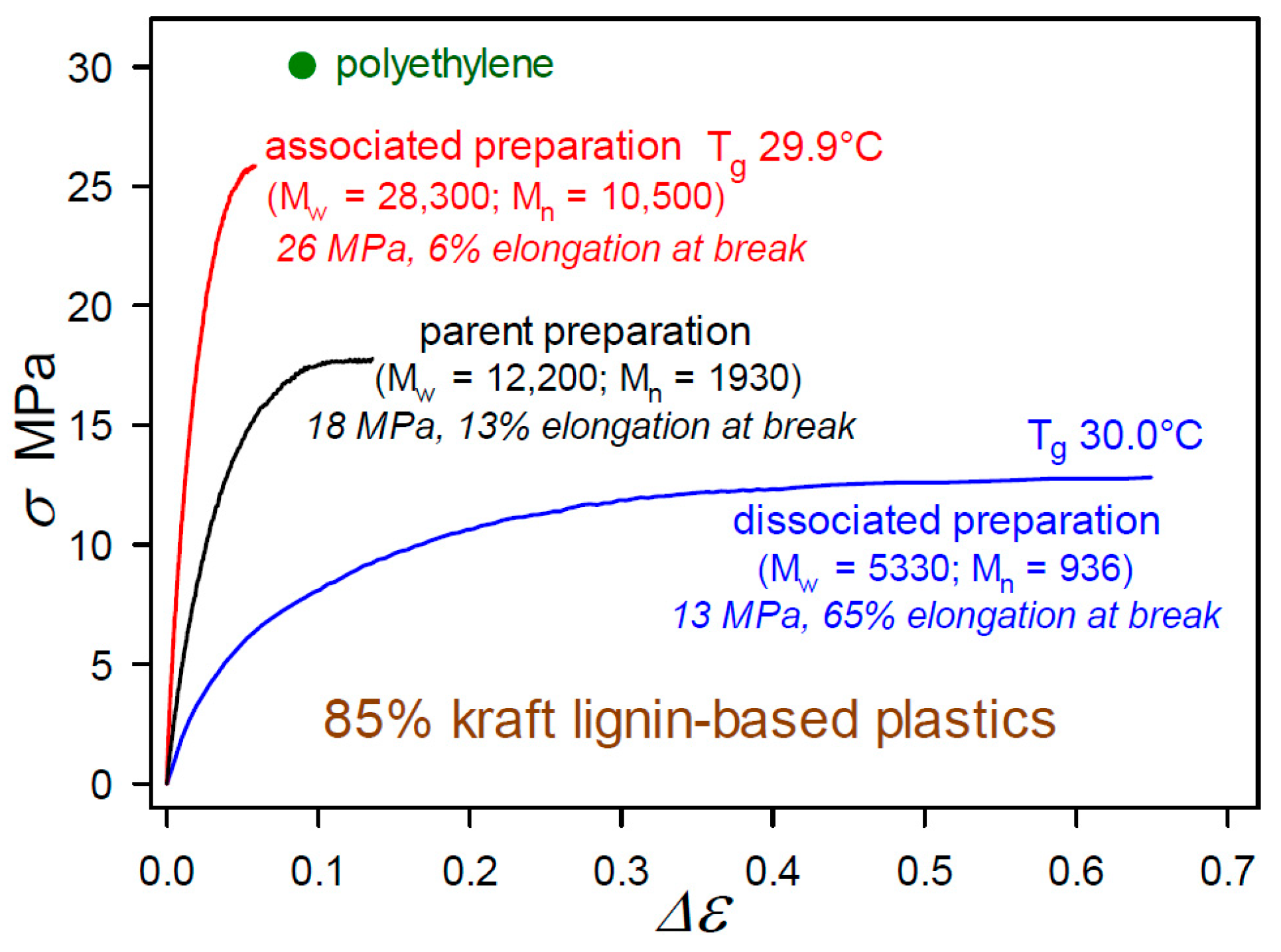
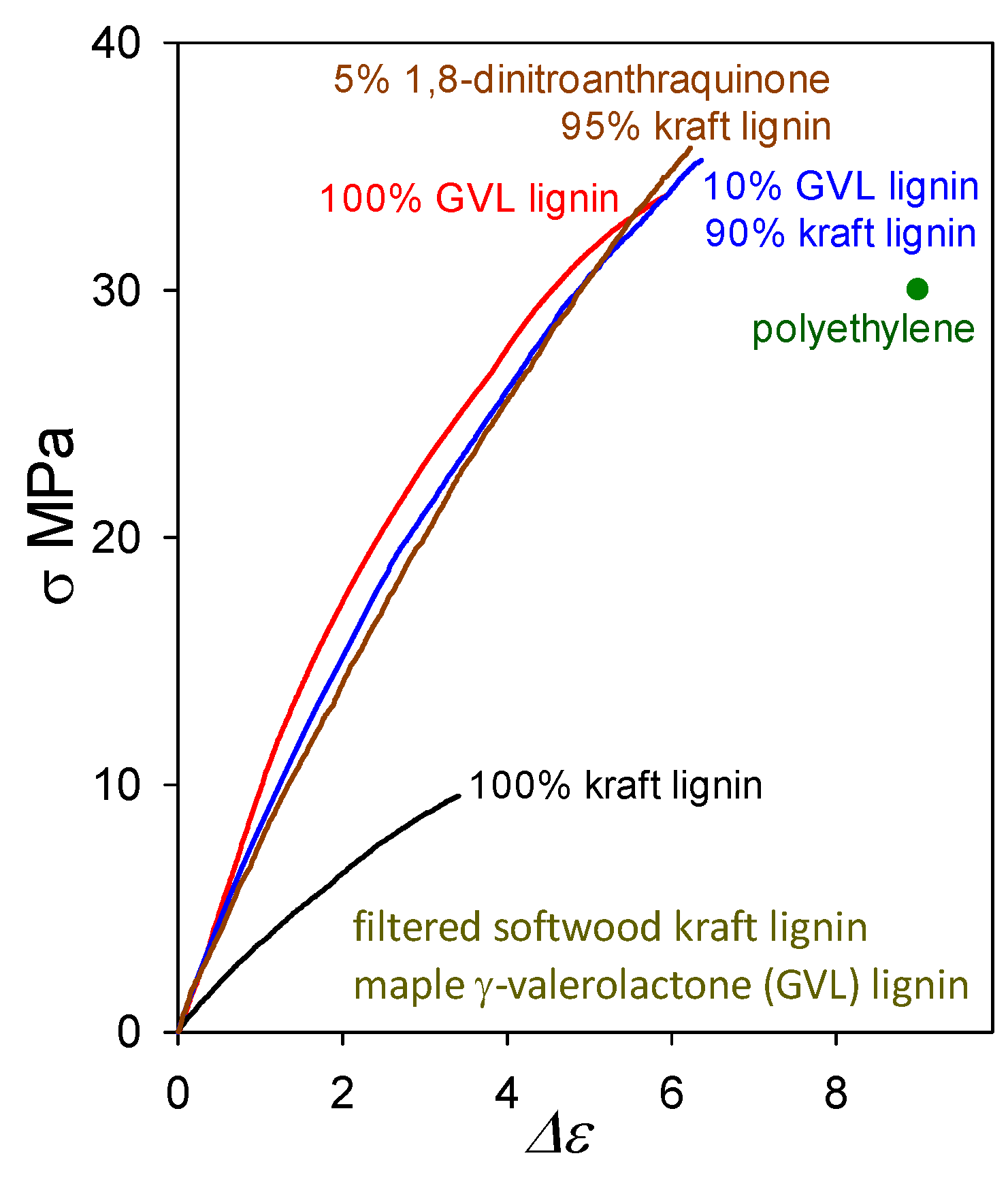
2.1. Plastics with Unmethylated Kraft Lignin Contents above 90 wt%
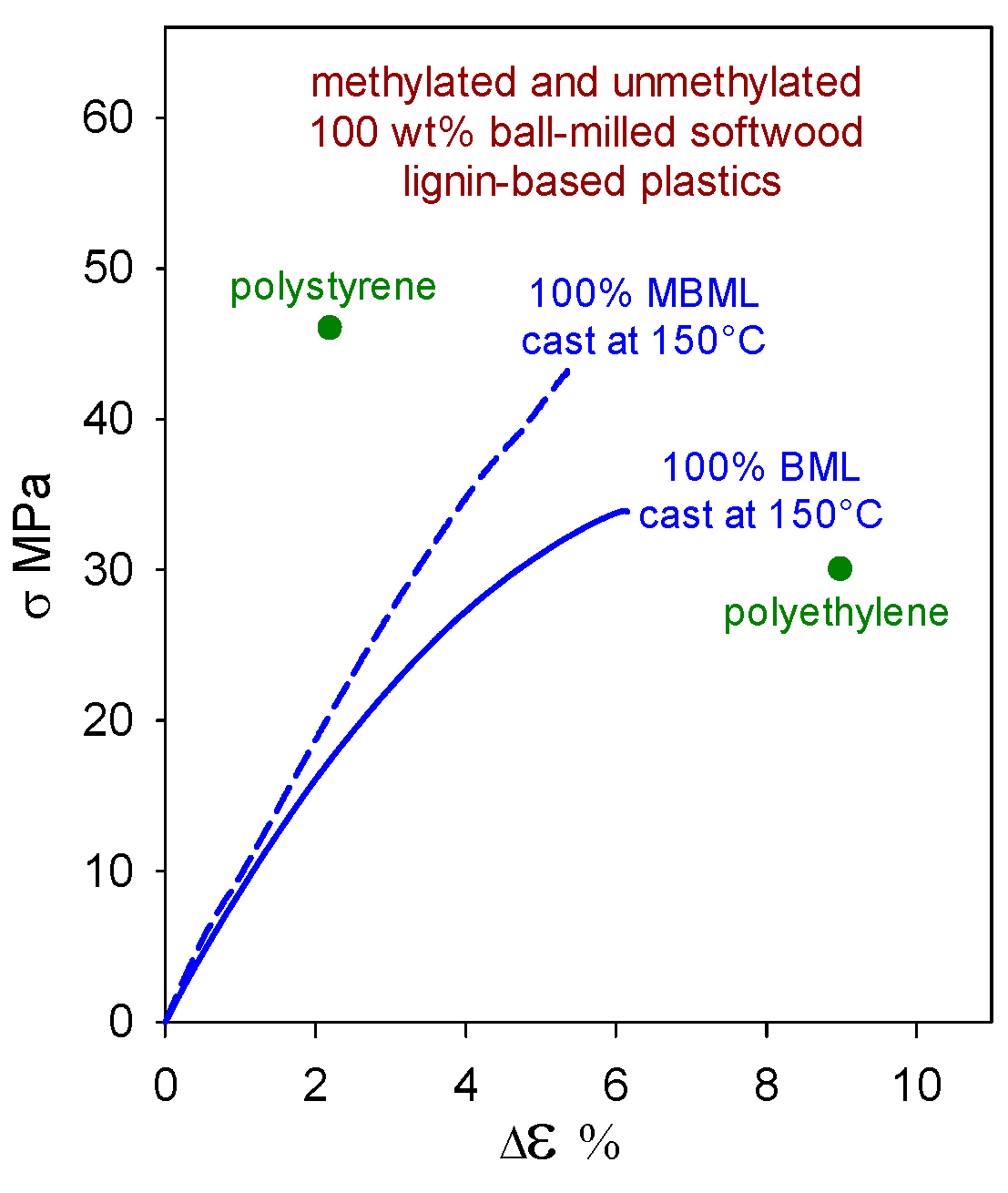
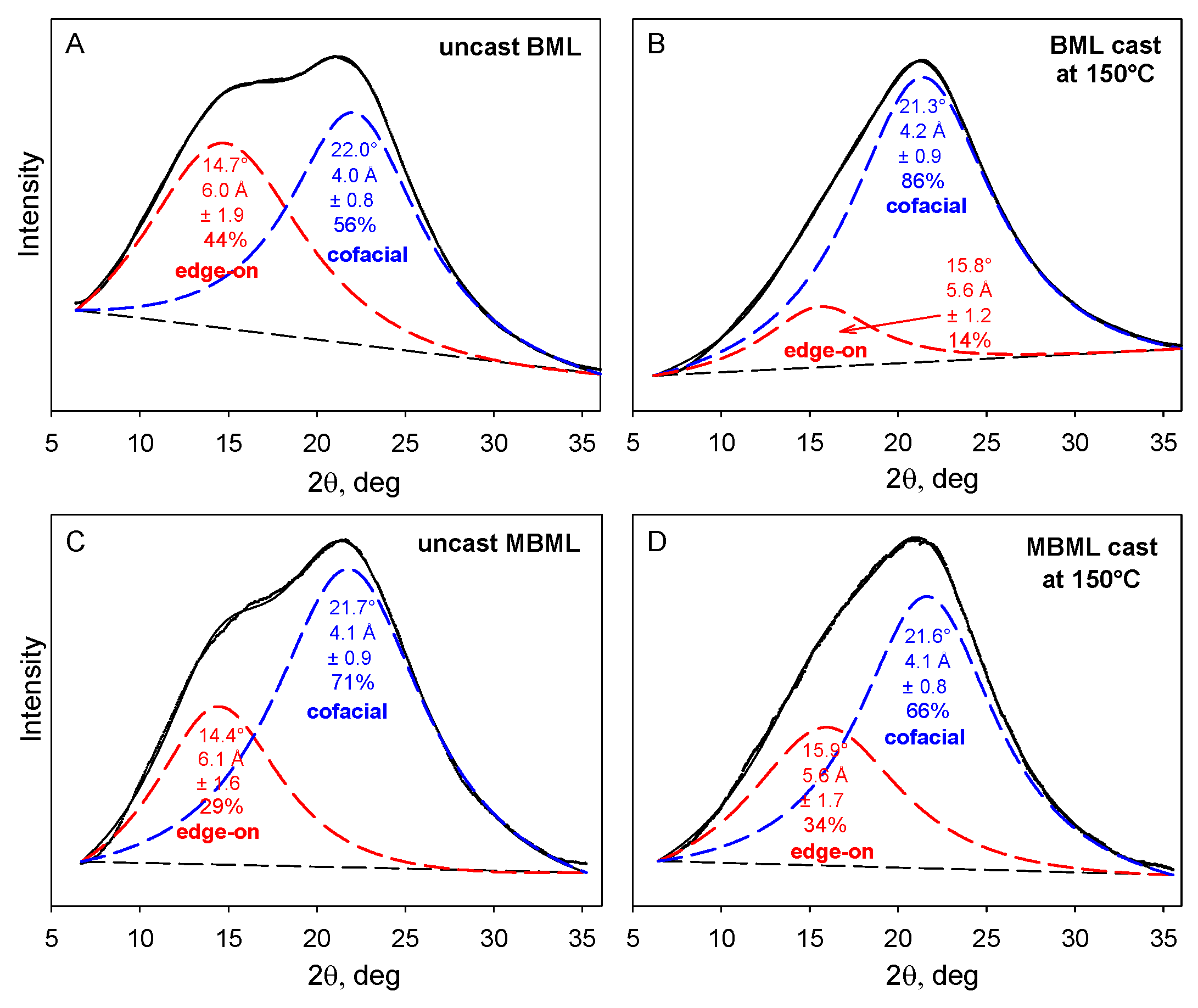
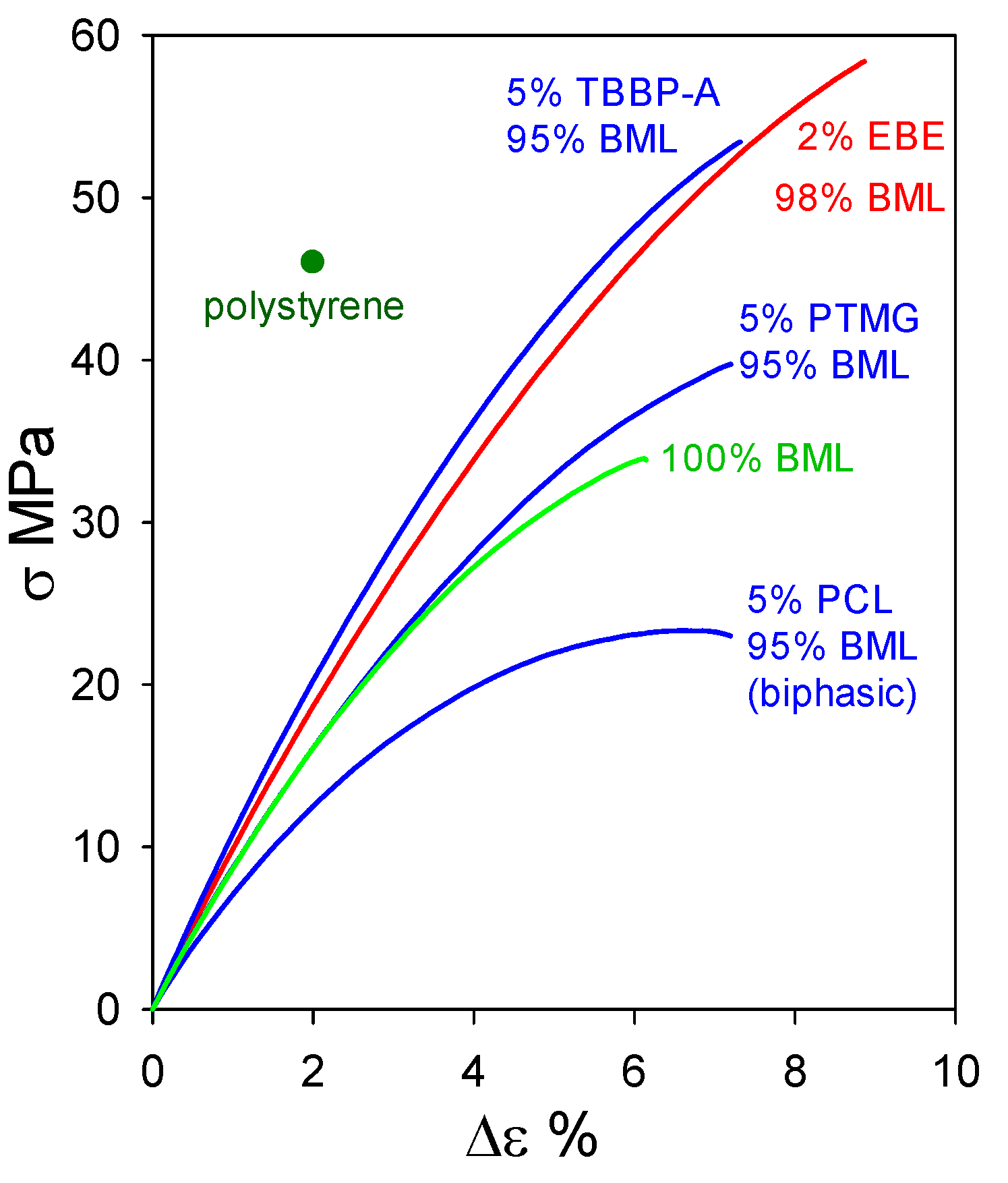
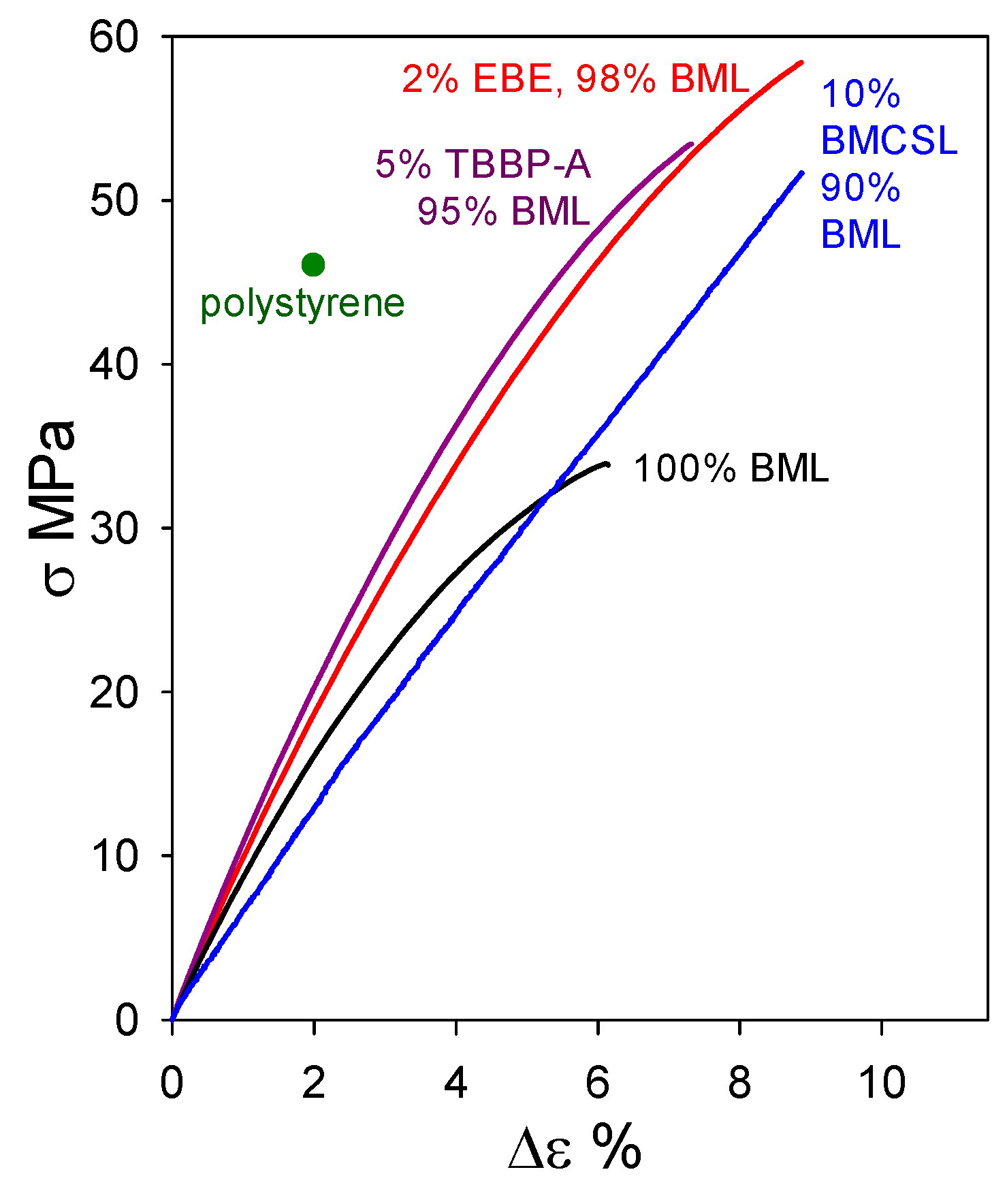
2.2. Plastics with Unmethylated Ball-Milled Lignin Contents above 95 wt%
3. Materials and Methods
3.1. Materials
3.2. Casting
3.3. Tensile Testing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beckham, G.T. Lignin Valorization: Emerging Approaches; Royal Society of Chemistry: London, UK, 2018; pp. 1–528. [Google Scholar]
- Gou, M.; Ran, X.; Martin, D.W.; Liu, C.-J. The scaffold proteins of lignin biosynthetic cytochrome P450 enzymes. Nat. Plants 2018, 4, 299–310. [Google Scholar] [CrossRef]
- Sarkanen, S.; Chen, Y.-r.; Wang, Y.-Y. Journey to polymeric materials composed exclusively of simple lignin derivatives. ACS Sustain. Chem. Eng. 2016, 4, 5223–5229. [Google Scholar] [CrossRef]
- Moacanin, J.; Felicetta, V.F.; Haller, W.; McCarthy, J.L. Molecular weights of lignin sulfonates by light scattering. J. Am. Chem. Soc. 1955, 77, 3470–3475. [Google Scholar] [CrossRef]
- Yean, W.Q.; Rezanowich, A.; Goring, D.A.I. The molecular weight and configuration of sodium lignin sulphonate from spruce wood. In Chim. Biochim. Lignine Cellulose Hémicelluloses; Actes Symp. Internat.: Grenoble, France, 1964; pp. 327–343. [Google Scholar]
- Goring, D.A.I. The lignin macromolecule. Pulp Paper Mag. Canada 1957, 58, 165–171. [Google Scholar]
- Gupta, P.R.; Goring, D.A.I. Physicochemical studies of alkali lignins. III. Size and shape of the macromolecule. Can. J. Chem. 1960, 38, 270–279. [Google Scholar] [CrossRef]
- Rezanowich, A.; Goring, D.A.I. Polyelectrolyte expansion of a lignin sulfonate microgel. J. Colloid Sci. 1960, 15, 452–471. [Google Scholar] [CrossRef]
- Chen, Y.-r.; Sarkanen, S. From the macromolecular behavior of lignin components to the mechanical properties of lignin-based plastics. Cellulose Chem. Technol. 2006, 40, 149–163. [Google Scholar]
- Saraf, V.P.; Glasser, W.G. Engineering plastics from lignin. III. Structure property relationships in solution cast polyurethane films. J. Appl. Polym. Sci. 1984, 29, 1831–1841. [Google Scholar] [CrossRef]
- Yoshida, H.; Mörck, R.; Kringstad, K.P.; Hatakeyama, H. Kraft lignin in polyurethanes. II. Effects of the molecular weight of kraft lignin on the properties of polyurethanes from a kraft lignin-polyether triol-polymeric MDI system. J. Appl. Polym. Sci. 1990, 40, 1819–1832. [Google Scholar] [CrossRef]
- Ghosh, I.; Jain, R.K.; Glasser, W.G. Multiphase materials with lignin. XV. Blends of cellulose acetate butyrate with lignin esters. J. Appl. Polym. Sci. 1999, 74, 448–457. [Google Scholar] [CrossRef]
- Zhuang, W. Lignins and derivatives: GPC/SEC analysis. In Encyclopedia of Chromatography, 3rd ed.; Vol. II. Cazes, J., Ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 1359–1368. [Google Scholar]
- Sarkanen, S.; Teller, D.C.; Abramowski, E.; McCarthy, J.L. Kraft lignin component conformation and associated complex configuration in aqueous alkaline solution. Macromolecules 1982, 15, 1098–1104. [Google Scholar] [CrossRef]
- Mlynár, J.; Sarkanen, S. Renaissance in ultracentrifugal sedimentation equilibrium calibrations of size exclusion chromatographic elution profiles. In Strategies in Size Exclusion Chromatography; Potschka, M., Dubin, P.L., Eds.; ACS Symposium Series 635; American Chemical Society: Washington, DC, USA, 1996; pp. 379–400. [Google Scholar]
- Davis, J.R. Tensile Testing of Plastics. In Tensile Testing, 2nd ed.; ASM International: Materials Park, OH, USA, 2004; pp. 137–154. [Google Scholar]
- Wang, Y.-Y.; Chen, Y.-r.; Sarkanen, S. Blend configuration in functional polymeric materials with a high lignin content. Faraday Discuss. 2017, 202, 43–59. [Google Scholar] [CrossRef]
- Li, Y.; Sarkanen, S. Miscible blends of kraft lignin derivatives with low-Tg polymers. Macromolecules 2005, 38, 2296–2306. [Google Scholar] [CrossRef]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the structure of softwood kraft lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
- Gellerstedt, G. Softwood kraft lignin: Raw material for the future. Ind. Crops Prod. 2015, 77, 845–854. [Google Scholar] [CrossRef]
- Dutta, S.; Garver, T.M., Jr.; Sarkanen, S. Modes of association between kraft lignin components. ACS Symp. Ser. 1989, 397, 155–176. [Google Scholar]
- Sarkanen, S.; Teller, D.C.; Hall, J.; McCarthy, J.L. Associative effects among Organosolv lignin components. Macromolecules 1981, 14, 426–434. [Google Scholar] [CrossRef]
- Sarkanen, S.; Teller, D.C.; Stevens, C.R.; McCarthy, J.L. Associative interactions between kraft lignin components. Macromolecules 1984, 17, 2588–2597. [Google Scholar] [CrossRef]
- Li, Y.; Mlynár, J.; Sarkanen, S. The first 85% kraft lignin-based thermoplastics. J. Polym. Sci. B: Polym. Phys. 1997, 35, 1899–1910. [Google Scholar] [CrossRef]
- Lindgren, K.; Samuelsson, Å.; Kulander, I. Techno economic evaluation of lignin extraction in a dissolving pulp biorefinery. Nordic Wood Biorefinery Conf. 2017, 118–123. [Google Scholar]
- Tomani, P. The Lignoboost process. Cellulose Chem. Technol. 2010, 44, 53–58. [Google Scholar]
- Shaanxi Dideu Medichem Co. Available online: https://www.alibaba.com (accessed on 21 November 2019).
- Rosen, M.; Kiani, A. The role of plastics compounding for injection molding. Plastics Eng. 2016, 72, 24–28. [Google Scholar] [CrossRef]
- Polystyrene (PS): Production, Market, Price and its Properties. Available online: https://www.plasticsinsight.com/resin-intelligence/resin-prices/polystyrene/ (accessed on 21 November 2019).
- Luterbacher, J.S.; Rand, J.M.; Alonso, D.M.; Han, J.; Youngquist, J.T.; Maravelias, C.T.; Pfleger, B.F.; Dumesic, J.A. Nonenzymatic sugar production from biomass using biomass-derived γ-valerolactone. Science 2014, 343, 277–280. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A. Comprehensive structural analysis of biorefinery lignins with a quantitative 13C NMR approach. RSC Adv. 2015, 5, 87187–87199. [Google Scholar] [CrossRef]
- Hu, Z.; Du, X.; Liu, J.; Chang, H.-m.; Jameel, H. Structural characterization of pine kraft lignin: Biochoice lignin vs Indulin AT. J. Wood Chem. Technol. 2016, 36, 432–446. [Google Scholar] [CrossRef]
- McClelland, D.J.; Galebach, P.H.; Motagamwala, A.H.; Wittrig, A.M.; Karlen, S.D.; Buchanan, J.S.; Dumesic, J.A.; Huber, G.W. Supercritical methanol depolymerization and hydrodeoxygenation of lignin and biomass over reduced copper porous metal oxides. Green Chem. 2019, 21, 2988–3005. [Google Scholar] [CrossRef]
Sample Availability: Not available. |













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-r.; Sarkanen, S.; Wang, Y.-Y. Lignin-Only Polymeric Materials Based on Unmethylated Unfractionated Kraft and Ball-Milled Lignins Surpass Polyethylene and Polystyrene in Tensile Strength. Molecules 2019, 24, 4611. https://doi.org/10.3390/molecules24244611
Chen Y-r, Sarkanen S, Wang Y-Y. Lignin-Only Polymeric Materials Based on Unmethylated Unfractionated Kraft and Ball-Milled Lignins Surpass Polyethylene and Polystyrene in Tensile Strength. Molecules. 2019; 24(24):4611. https://doi.org/10.3390/molecules24244611
Chicago/Turabian StyleChen, Yi-ru, Simo Sarkanen, and Yun-Yan Wang. 2019. "Lignin-Only Polymeric Materials Based on Unmethylated Unfractionated Kraft and Ball-Milled Lignins Surpass Polyethylene and Polystyrene in Tensile Strength" Molecules 24, no. 24: 4611. https://doi.org/10.3390/molecules24244611