
Discovery and Characterisation of Dual Inhibitors of Tryptophan 2,3-Dioxygenase (TDO2) and Indoleamine 2,3-Dioxygenase 1 (IDO1) Using Virtual Screening



Abstract
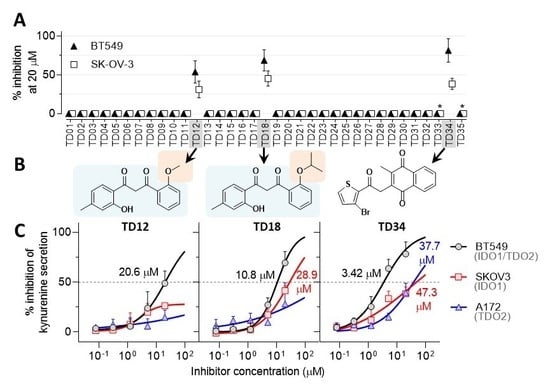
:
1. Introduction
2. Results and Discussion
2.1. Validation of Virtual Screening Methods
2.2. Molecular Docking
2.3. Pharmacophore Modelling
2.4. 3D Shape Similarity
2.5. Virtual Screening of the IBS Library
2.6. Cell-Based Assays for Testing the Inhibitory Activity of Test Compounds
2.7. Testing the in Silico Hits for Inhibition of IDO1 and TDO2
2.8. Suitability of the Identified Hits for Drug Development—Chemical Space
2.9. Molecular Modelling for Mechanistic Insights
3. Materials and Methods
3.1. Virtual Screening
3.2. Cell Based Kynurenine Assay and Measurement of Cellular Metabolic Activity
3.3. Western Blotting
3.4. Density Functional Theory
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan Metabolism as a Common Therapeutic Target in Cancer, Neurodegeneration and Beyond. Nature Rev. Drug Dis. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Adams, S.; Braidy, N.; Bessede, A.; Brew, B.J.; Grant, R.; Teo, C.; Guillemin, G.J. The Kynurenine Pathway in Brain Tumor Pathogenesis. Cancer Res. 2012, 72, 5649–5657. [Google Scholar] [CrossRef]
- Dobrovolsky, V.N.; Bowyer, J.F.; Pabarcus, M.K.; Heflich, R.H.; Williams, L.D.; Doerge, D.R.; Arvidsson, B.; Bergquist, J.; Casida, J.E. Effect of Arylformamidase (Kynurenine Formamidase) Gene Inactivation in Mice on Enzymatic Activity, Kynurenine Pathway Metabolites and Phenotype. Biochim. et Biophys. Acta 2005, 1724, 163. [Google Scholar] [CrossRef]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolušić, E.; Frédérick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of Tumoral Immune Resistance by Inhibition of Tryptophan 2,3-Dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef]
- Katz, J.B.; Muller, A.J.; Prendergast, G.C. Indoleamine 2,3-Dioxygenase in T-Cell Tolerance and Tumoral Immune Escape. Immunol. Rev. 2008, 222, 206–221. [Google Scholar] [CrossRef]
- Yu, C.; Fu, S.; Chen, X.; Ye, J.; Ye, Y.; Kong, L.; Zhu, Z. The Clinicopathological and Prognostic Significance of IDO1 Expression in Human Solid Tumors: Evidence from a Systematic Review and Meta-Analysis. Cell. Phys. Biochem. 2018, 49, 134–143. [Google Scholar] [CrossRef]
- Seegers, N.; van Doornmalen, A.M.; Uitdehaag, J.C.M.; de Man, J.; Buijsman, R.C.; Zaman, G.J.R. High-Throughput Fluorescence-Based Screening Assays for Tryptophan-Catabolizing Enzymes. J. Biomol. Screen. 2014, 19, 1266–1274. [Google Scholar] [CrossRef]
- Tomek, P.; Palmer, B.D.; Kendall, J.D.; Flanagan, J.U.; Ching, L. Formation of Fluorophores from the Kynurenine Pathway Metabolite N-Formylkynurenine and Cyclic Amines Involves Transamidation and Carbon-Carbon Bond Formation at the 2-Position of the Amine. Biochim. Biophys. Acta 2015, 1850, 1772. [Google Scholar] [CrossRef]
- Tomek, P.; Palmer, B.; Flanagan, J.; Fung, S.; Bridewell, D.; Jamie, J.; Ching, L. Formation of an N-Formylkynurenine-Derived Fluorophore and its use for Measuring Indoleamine 2,3-Dioxygenase 1 Activity. Anal. Bioanal. Chem. 2013, 405, 2515–2524. [Google Scholar] [CrossRef]
- Dolušić, E.; Larrieu, P.; Blanc, S.; Sapunaric, F.; Pouyez, J.; Moineaux, L.; Colette, D.; Stroobant, V.; Pilotte, L.; Colau, D.; et al. Discovery and Preliminary SARs of Keto-Indoles as Novel Indoleamine 2,3-Dioxygenase (IDO) Inhibitors. Eur. J. Med. Chem. 2011, 46, 3058–3065. [Google Scholar]
- Smith, J.R.; Evans, K.J.; Wright, A.; Willows, R.D.; Jamie, J.F.; Griffith, R. Novel Indoleamine 2,3-Dioxygenase-1 Inhibitors from a Multistep in Silico Screen. Bioorg. Med. Chem. 2012, 20, 1354–1363. [Google Scholar] [CrossRef]
- Cheong, J.E.; Ekkati, A.; Sun, L. A Patent Review of IDO1 Inhibitors for Cancer. Exp. Opin. Ther. Pat. 2018, 28, 317–330. [Google Scholar] [CrossRef]
- Austin, C.J.D.; Rendina, L.M. Targeting Key Dioxygenases in Tryptophan–kynurenine Metabolism for Immunomodulation and Cancer Chemotherapy. Drug Dis. Today 2015, 20, 609–617. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, Y.; Yin, Z.; Feng, S.; Sun, L.; Li, Z. Research Progress of Indoleamine 2,3-Dioxygenase Inhibitors. Fut. Med. Chem. 2015, 7, 185–201. [Google Scholar] [CrossRef]
- Jacobs, K.R.; Castellano-Gonzalez, G.; Guillemin, G.J.; Lovejoy, D.B. Major Developments in the Design of Inhibitors along the Kynurenine Pathway. Cur. Med. Chem. 2017, 24, 2471–2495. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The Rationale of Indoleamine 2,3-Dioxygenase Inhibition for Cancer Therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients with Advanced Solid Tumors: Phase I Results from a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, JCO2018789602. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.; Kim, T.M.; et al. Epacadostat Plus Pembrolizumab Versus Placebo Plus Pembrolizumab in Patients with Unresectable Or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Li, H.; Ning, S.; Ghandi, M.; Kryukov, G.V.; Gopal, S.; Deik, A.; Souza, A.; Pierce, K.; Keskula, P.; Hernandez, D.; et al. The landscape of cancer cell line metabolism. Nat. Med. 2019, 25, 850–860. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2–KYN–AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharm. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Winters, M.; DuHadaway, J.B.; Pham, K.N.; Lewis-Ballester, A.; Badir, S.; Wai, J.; Sheikh, E.; Yeh, S.; Prendergast, G.C.; Muller, A.J.; et al. Diaryl Hydroxylamines as Pan Or Dual Inhibitors of Indoleamine 2,3-Dioxygenase-1, Indoleamine 2,3-Dioxygenase-2 and Tryptophan Dioxygenase. Eur. J. Med. Chem. 2019, 162, 455–464. [Google Scholar] [CrossRef]
- Yang, L.; Chen, J.; Chen, Y.; He, J.; Njoya, E.M.; Liu, S.; Xie, C.; Huang, W.; Wang, Z.; Wang, F.; et al. 4,6-Substituted-1H-Indazoles as Potent IDO1/TDO Dual Inhibitors. Bioorg. Med. Chem. 2019, 27, 1087–1098. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, S.; Fang, X.; Guo, L.; Hu, N.; Guo, Z.; Li, X.; Yang, S.; He, J.C.; Kuang, C.; et al. N-Benzyl/Aryl Substituted Tryptanthrin as Dual Inhibitors of Indoleamine 2,3-Dioxygenase and Tryptophan 2,3-Dioxygenase. J. Med. Chem. 2019, 62, 9161–9174. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications. Nat. Rev. Drug Dis. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Lavecchia, A.; Di Giovanni, C. Virtual Screening Strategies in Drug Discovery: A Critical Review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Richards, T.; Brin, E. Cell Based Functional Assays for IDO1 Inhibitor Screening and Characterization. Oncotarget 2018, 9, 30814–30820. [Google Scholar] [CrossRef]
- Tomek, P.; Palmer, B.D.; Flanagan, J.U.; Sun, C.; Raven, E.L.; Ching, L. Discovery and Evaluation of Inhibitors to the Immunosuppressive Enzyme Indoleamine 2,3-Dioxygenase 1 (IDO1): Probing the Active Site-Inhibitor Interactions. Eur. J. Med. Chem. 2017, 126, 983–996. [Google Scholar] [CrossRef]
- Zhang, Y.; Kang, S.A.; Mukherjee, T.; Bale, S.; Crane, B.R.; Begley, T.P.; Ealick, S.E. Crystal Structure and Mechanism of Tryptophan 2,3-Dioxygenase, a Heme Enzyme Involved in Tryptophan Catabolism and in Quinolinate Biosynthesis. Biochemistry 2007, 46, 145–155. [Google Scholar] [CrossRef]
- Peng, Y.; Ueng, S.; Tseng, C.; Hung, M.; Song, J.; Wu, J.; Liao, F.; Fan, Y.; Wu, M.; Hsiao, W.; et al. Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J. Med. Chem. 2016, 59, 282–293. [Google Scholar] [CrossRef]
- Luo, S.; Xu, K.; Xiang, S.; Chen, J.; Chen, C.; Guo, C.; Tong, Y.; Tong, L. High-resolution Structures of Inhibitor Complexes of Human Indoleamine 2,3-dioxygenase 1 in a New Crystal Form. Acta Crystal. Sect. F 2018, 74, 717–724. [Google Scholar] [CrossRef]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. The ChEMBL Database in 2017. Nucl. Acids Res. 2015, 43, W61–W620. [Google Scholar]
- Pantouris, G.; Loudon-Griffiths, J.; Mowat, C.G. Insights into the Mechanism of Inhibition of Tryptophan 2,3-Dioxygenase by Isatin Derivatives. J. Enz. Inhib. Med. Chem. 2016, 31, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Magid, A.F. Targeting the Inhibition of Tryptophan 2,3-Dioxygenase (TDO-2) for Cancer Treatment. ACS Med. Chem. Let. 2017, 8, 11–13. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Lin, S.; Liao, F.; Hsiao, W.; Lee, L.; Peng, Y.; Hsieh, C.; Wu, M.; Song, J.; Yueh, A.; et al. Identification of Substituted Naphthotriazolediones as Novel Tryptophan 2,3-Dioxygenase (TDO) Inhibitors through Structure-Based Virtual Screening. J. Med. Chem. 2015, 58, 7807–7819. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Majjigapu, S.M.; Grosdidier, A.; Bron, S.; Stroobant, V.; Pilotte, L.; Colau, D.; Vogel, P.; van den Eynde, B.J.; Zoete, V.; et al. Rational Design of 4-Aryl-1,2,3-Triazoles for Indoleamine 2,3-Dioxygenase 1 Inhibition. J. Med. Chem. 2012, 55, 5270–5290. [Google Scholar]
- InterBioScree. Available online: https://www.ibscreen.com/ (accessed on 20 October 2018).
- Lipinski, C.A. Lead- and Drug-Like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Takao, M.; Okamoto, A.; Nikaido, T.; Urashima, M.; Takakura, S.; Saito, M.; Saito, M.; Okamoto, S.; Takikawa, O.; Sasaki, H.; et al. Increased Synthesis of Indoleamine-2,3-Dioxygenase Protein is Positively Associated with Impaired Survival in Patients with Serous-Type, but Not with Other Types of, Ovarian Cancer. Oncol. Rep. 2007, 17, 1333. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An Endogenous Tumour-Promoting Ligand of the Human Aryl Hydrocarbon Receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A Web-Based Suite of Genomic and Pharmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef]
- Yue, E.W.; Douty, B.; Wayland, B.; Bower, M.; Liu, X.; Leffet, L.; Wang, Q.; Bowman, K.J.; Hansbury, M.J.; Liu, C.; et al. Discovery of Potent Competitive Inhibitors of Indoleamine 2,3-Dioxygenase with in Vivo Pharmacodynamic Activity and Efficacy in a Mouse Melanoma Model. J. Med. Chem. 2009, 52, 7364–7367. [Google Scholar] [CrossRef]
- Salter, M.; Hazelwood, R.; Pogson, C.I.; Iyer, R.; Madge, D.J. The Effects of a Novel and Selective Inhibitor of Tryptophan 2,3-Dioxygenase on Tryptophan and Serotonin Metabolism in the Rat. Biochem. Pharm. 1995, 49, 1435–1442. [Google Scholar] [CrossRef]
- eMolecules. Available online: https://www.emolecules.com/ (accessed on 14 June 2019).
- Cui, H.; Zhang, H.; Liu, Y.; Gu, Q.; Xu, J.; Huang, X.; She, Z. Ethylnaphthoquinone Derivatives as Inhibitors of Indoleamine-2, 3-Dioxygenase from the Mangrove Endophytic Fungus Neofusicoccum Austral SYSU-SKS024. Fitoterapia 2018, 125, 281–285. [Google Scholar] [CrossRef]
- Matuszek, A.M.; Reynisson, J. Defining Known Drug Space using DFT. Mol. Inf. 2016, 35, 46–53. [Google Scholar] [CrossRef]
- Beall, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for their Exclusion in Bioassays. J. Med. Chem. 2010, 2010, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Logan, G.; Reynisson, J. Wine Compounds as a Source for HTS Screening Collections. A Feasibility Study. Mol. Inf. 2012, 31, 847–855. [Google Scholar] [CrossRef]
- Eurtivong, C.; Reynisson, J. The Development of a Weighted Index to Optimise Compound Libraries for High Throughput Screening. Mol. Inf. 2019, 38, e1800068. [Google Scholar] [CrossRef]
- Lewis-Ballester, A.; Forouhar, F.; Kim, S.; Lew, S.; Wang, Y.; Karkashon, S.; Seetharaman, J.; Batabyal, D.; Chiang, B.; Hussain, M.; et al. Molecular Basis for Catalysis and Substrate-Mediated Cellular Stabilization of Human Tryptophan 2,3-Dioxygenase. Sci. Rep. 2016, 6, 35169. [Google Scholar] [CrossRef] [Green Version]
- MicahNelp, M.T.; Kates, P.A.; Hunt, J.T.; Newitt, J.A.; Balog, A.; Maley, D.; Zhu, X.; Abell, L.; Allentoff, A.; Borzilleri, R.; et al. Immune-Modulating Enzyme Indoleamine 2,3-Dioxygenase is Effectively Inhibited by Targeting its Apo-Form. Proc. Natl. Acad. Sci. USA 2018, 115, 3249–3254. [Google Scholar]
- Meng, B.; Wu, D.; Gu, J.; Ouyang, S.; Ding, W.; Liu, Z. Structural and Functional Analyses of Human Tryptophan 2,3-dioxygenase. Proteins: Struct. Funct. Bioinf. 2014, 82, 3210–3216. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comp. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [Green Version]
- QikProp Version 3.2; Schrödinger: New York, NY, USA, 2009.
- Ioakimidis, L.; Thoukydidis, L.; Naeem, S.; Mirza, A.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comp. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Dixon, S.; Smondyrev, A.; Knoll, E.; Rao, S.; Shaw, D.; Friesner, R. PHASE: A New Engine for Pharmacophore Perception, 3D QSAR Model Development, and 3D Database Screening: 1. Methodology and Preliminary Results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Cho, A.E.; Guallar, V.; Berne, B.J.; Friesner, R. Importance of Accurate Charges in Molecular Docking: Quantum Mechanical/Molecular Mechanical (QM/MM) Approach. J. Comput. Chem. 2005, 26, 915–931. [Google Scholar] [CrossRef] [Green Version]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A High-performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quant. Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Murphy, R.B.; Philipp, D.M.; Friesner, R.A. A Mixed Quantum Mechanics/Molecular Mechanics (QM/MM) Method for Large-scale Modeling of Chemistry in Protein Environments. J. Comput. Chem. 2000, 21, 1442–1457. [Google Scholar] [CrossRef]
- Fung, S.S.; Wang, H.; Tomek, P.; Squire, C.J.; Flanagan, J.U.; Palmer, B.D.; Bridewell, D.J.A.; Tijono, S.M.; Jamie, J.F.; Ching, L. Discovery and Characterisation of Hydrazines as Inhibitors of the Immune Suppressive Enzyme, Indoleamine 2,3-Dioxygenase 1 (IDO1). Bioorg. Med. Chem. 2013, 21, 7595–7603. [Google Scholar] [CrossRef]
- Peskin, A.V.; Winterbourn, C.C. A Microtiter Plate Assay for Superoxide Dismutase using a Water-Soluble Tetrazolium Salt (WST-1). Clin. Chim. Acta 2000, 293, 157–166. [Google Scholar] [CrossRef]
- Leung, E.Y.; Kim, J.E.; Askarian-Amiri, M.; Joseph, W.R.; McKeage, M.J.; Baguley, B.C. Hormone Resistance in Two MCF-7 Breast Cancer Cell Lines is Associated with Reduced mTOR Signaling, Decreased Glycolysis, and Increased Sensitivity to Cytotoxic Drugs. Front. Oncol. 2014, 4, 221. [Google Scholar] [CrossRef] [Green Version]
- Kirtikara, K.; Vichai, V. Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening. Nature Prot. 2006, 1, 1112–1116. [Google Scholar]
- Leung, E.Y.; Askarian-Amiri, M.; Finlay, G.J.; Rewcastle, G.W.; Baguley, B.C. Potentiation of Growth Inhibitory Responses of the mTOR Inhibitor Everolimus by Dual mTORC1/2 Inhibitors in Cultured Breast Cancer Cell Lines. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Leung, E.Y.; Kim, J.E.; Askarian-Amiri, M.; Rewcastle, G.W.; Finlay, G.J.; Baguley, B.C. Relationships between Signaling Pathway Usage and Sensitivity to a Pathway Inhibitor: Examination of Trametinib Responses in Cultured Breast Cancer Lines. PLoS ONE 2014, 9, e105792. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01, Gaussian, Inc.: Wallingford, CT, USA, 2019.
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Corect Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional thermochemistryIII. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of Polarization Functions on Molecular-Orbital Hydrogenation Energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Wong, M.W. Vibrational Frequency Prediction using Density Functional Theory. Chem. Phys. Lett. 1996, 256, 391–399. [Google Scholar]
- Foresman, J.B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods. Gaussian Inc. 1996, 142–143. [Google Scholar]
Sample Availability: Samples of the compounds TD12, TD18 and TD34 are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Query | Typed Pharmacophore | Typed Atoms | Untyped Atoms |
|---|---|---|---|
| 1a | 0.76 | 0.95 | 0.95 |
| 1b | 0.65 | 0.90 | 0.87 |
| 1c | 0.78 | 0.96 | 0.97 |
| MW | PSA | #Rotor | DonorHB | AccptHB | Log P | KDI2a/b | |
|---|---|---|---|---|---|---|---|
| TD12 | 284.3 | 73.3 | 6 | 1 | 4.5 | 2.7 | 5.70/0.75 |
| TD18 | 312.4 | 72.2 | 7 | 1 | 4.5 | 3.5 | 5.67/0.71 |
| TD34 | 375.2 | 76.0 | 3 | 0 | 6.0 | 2.6 | 5.69/0.70 |
| Ligand | 6A4I | 5EK3 | 6E43 |
|---|---|---|---|
| TD12 | −7.9 | −6.4 | −7.2 |
| TD18 | −7.4 | −6.8 | −7.4 |
| TD34 | −3.9 | −6.3 | −8.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sari, S.; Tomek, P.; Leung, E.; Reynisson, J. Discovery and Characterisation of Dual Inhibitors of Tryptophan 2,3-Dioxygenase (TDO2) and Indoleamine 2,3-Dioxygenase 1 (IDO1) Using Virtual Screening. Molecules 2019, 24, 4346. https://doi.org/10.3390/molecules24234346
Sari S, Tomek P, Leung E, Reynisson J. Discovery and Characterisation of Dual Inhibitors of Tryptophan 2,3-Dioxygenase (TDO2) and Indoleamine 2,3-Dioxygenase 1 (IDO1) Using Virtual Screening. Molecules. 2019; 24(23):4346. https://doi.org/10.3390/molecules24234346
Chicago/Turabian StyleSari, Suat, Petr Tomek, Euphemia Leung, and Jóhannes Reynisson. 2019. "Discovery and Characterisation of Dual Inhibitors of Tryptophan 2,3-Dioxygenase (TDO2) and Indoleamine 2,3-Dioxygenase 1 (IDO1) Using Virtual Screening" Molecules 24, no. 23: 4346. https://doi.org/10.3390/molecules24234346