
β-Amino- and Alkoxy-Substituted Disilanides


 , and
, and
Abstract
:
1. Introduction
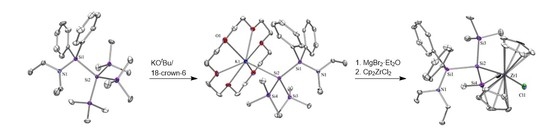
2. Results and Discussion
3. Materials and Methods
3.1. General Remarks
3.2. X-Ray Structure Determination
3.2.1. N,N-Diethylaminodiphenylsilyltris(trimethylsilyl)silane (1)
3.2.2. N,N-Diethylaminodiphenylsilylbis(trimethylsilyl)silyl Potassium 18-Crown-6 (1a)
3.2.3. N,N-Diethylaminodiphenylsilylbis(trimethylsilyl)silyl Magnesium Bromide (1b)
3.2.4. Methoxydimethylsilyltris(trimethylsilyl)silane (2)
3.2.5. 2-Methoxydimethylsilyltris(trimethylsilyl)silyl Potassium 18-Crown-6 (2a)
3.2.6. N,N-Diethylaminodiphenylsilylbis(trimethylsilyl)silyl Zirconocene Chloride (3a)
3.2.7. N,N-Diethylaminodiphenylsilylbis(trimethylsilyl)silyl Hafnocene Chloride (3b)
3.2.8. 1,2-Bis(N,N-diethylaminodiphenylsilyl)-1,1,2,2,-Tetrakis(trimethylsilyl)disilane (4)
3.2.9. N,N-Diethylaminodiphenylsilyl(fluorodiphenylsilyl)bis(trimethylsilyl)silane (5)
3.2.10. N,N-Diethylaminodiphenylsilyl(fluorodiphenylsilyl)(trimethylsilyl)silyl Potassium 18-Crown-6 (5a)
3.2.11. N,N-Diethylaminodiphenylsilyltris(trimethylsilyl)germane (6)
3.2.12. N,N-Diethylaminodiphenylsilylbis(trimethylsilyl)germyl Potassium 18-Crown-6 (6a)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lickiss, P.D.; Smith, C.M. Silicon derivatives of the metals of groups 1 and 2. Coord. Chem. Rev. 1995, 145, 75–124. [Google Scholar] [CrossRef]
- Tamao, K.; Kawachi, A. Silyl Anions. Adv. Organomet. Chem. 1995, 38, 1–58. [Google Scholar]
- Belzner, J.; Dehnert, U. Alkaline and Alkaline Earth Silyl Compounds—Preparation and Structure. In The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2003; pp. 779–825. ISBN 978-0-470-85725-0. [Google Scholar]
- Lerner, H.-W. Silicon derivatives of group 1, 2, 11 and 12 elements. Coord. Chem. Rev. 2005, 249, 781–798. [Google Scholar] [CrossRef]
- Marschner, C. Silicon Centered Anions. In Organosilicon Compounds: Theory and Experiment (Synthesis); Lee, V.Y., Ed.; Academic Press: London, UK, 2017; Volume 1, ISBN 978-0-12-801991-7. [Google Scholar]
- Marschner, C. Preparation and Reactions of Polysilanyl Anions and Dianions. Organometallics 2006, 25, 2110–2125. [Google Scholar] [CrossRef]
- Gilman, H.; Steudel, W. Diphenylsilyllithium, a new type of organosilyl-metallic compound. Chem. Ind. 1959, 1094. [Google Scholar]
- Gilman, H.; Holmes, J.M.; Smith, C.L. Branched-chain Methylated Polysilanes Containing a Silyl-lithium Group. Chem. Ind. 1965, 848–849. [Google Scholar]
- Lampland, N.L.; Pindwal, A.; Yan, K.; Ellern, A.; Sadow, A.D. Rare Earth and Main Group Metal Poly(hydrosilyl) Compounds. Organometallics 2017, 36, 4546–4557. [Google Scholar] [CrossRef]
- Tamao, K.; Kawachi, A.; Ito, Y. The first stable functional silyl anions: (aminosilyl)lithiums. J. Am. Chem. Soc. 1992, 114, 3989–3990. [Google Scholar] [CrossRef]
- Tamao, K.; Kawachi, A.; Tanaka, Y.; Ohtani, H.; Ito, Y. Synthetic applications of functionalized silyl anions: Aminosilyl anions as hydroxy anion equivalent. Tetrahedron 1996, 52, 5765–5772. [Google Scholar] [CrossRef]
- Tamao, K.; Kawachi, A. The Chemistry of Silylenoids: Preparation and Reactivity of (Alkoxysilyl)lithium Compounds. Angew. Chem. Int. Ed. Engl. 1995, 34, 818–820. [Google Scholar] [CrossRef]
- Likhar, P.R.; Zirngast, M.; Baumgartner, J.; Marschner, C. Preparation and structural characterisation of methoxybis(trimethylsilyl)silyl potassium and its condensation product. Chem. Commun. 2004, 1764–1765. [Google Scholar] [CrossRef] [PubMed]
- Harloff, J.; Popowski, E.; Reinke, H. Substituted lithium trimethylsiloxysilanides LiSiRR’(OSiMe3) - Investigations of their synthesis, stability and reactivity. J. Organomet. Chem. 2007, 692, 1421–1441. [Google Scholar] [CrossRef]
- Fischer, R.; Baumgartner, J.; Kickelbick, G.; Marschner, C. The First Stable β-Fluorosilylanion. J. Am. Chem. Soc. 2003, 125, 3414–3415. [Google Scholar] [CrossRef] [PubMed]
- Zirngast, M.; Flock, M.; Baumgartner, J.; Marschner, C. Formation of Formal Disilene Fluoride Adducts. J. Am. Chem. Soc. 2008, 130, 17460–17470. [Google Scholar] [CrossRef]
- Molev, G.; Bravo-Zhivotovskii, D.; Karni, M.; Tumanskii, B.; Botoshansky, M.; Apeloig, Y. Synthesis, Molecular Structure, and Reactivity of the Isolable Silylenoid with a Tricoordinate Silicon. J. Am. Chem. Soc. 2006, 128, 2784–2785. [Google Scholar] [CrossRef]
- Auer, D.; Kaupp, M.; Strohmann, C. “Unexpected” 29Si NMR Chemical Shifts in Heteroatom-Substituted Silyllithium Compounds: A Quantum-Chemical Analysis. Organometallics 2004, 23, 3647–3655. [Google Scholar] [CrossRef]
- Balatoni, I.; Hlina, J.; Zitz, R.; Pöcheim, A.; Baumgartner, J.; Marschner, C. Disilene Fluoride Adducts versus β-Halooligosilanides. Inorg. Chem. 2019. [Google Scholar] [CrossRef]
- Zirngast, M.; Baumgartner, J.; Marschner, C. Preparation, Structure and Reactivity of Et2N(Me3Si)2SiK. Eur. J. Inorg. Chem. 2008, 1078–1087. [Google Scholar] [CrossRef]
- Marschner, C. A New and Easy Route to Polysilanylpotassium Compounds. Eur. J. Inorg. Chem. 1998, 221–226. [Google Scholar] [CrossRef]
- Kayser, C.; Fischer, R.; Baumgartner, J.; Marschner, C. Tailor-made Oligosilyl Potassium Compounds. Organometallics 2002, 21, 1023–1030. [Google Scholar] [CrossRef]
- Barrett, A.G.M.; Head, J.; Smith, M.L.; Stock, N.S.; White, A.J.P.; Williams, D.J. Fleming−Tamao Oxidation and Masked Hydroxyl Functionality: Total Synthesis of (+)-Pramanicin and Structural Elucidation of the Antifungal Natural Product (−)-Pramanicin. J. Org. Chem. 1999, 64, 6005–6018. [Google Scholar] [CrossRef]
- Suji, H.; Fukazawa, A.; Yamaguchi, S.; Toshimitsu, A.; Tamao, K. All-Anti-Pentasilane: Conformation Control of Oligosilanes Based on the Bis(tetramethylene)-Tethered Trisilane Unit. Organometallics 2004, 23, 3375–3377. [Google Scholar] [CrossRef]
- Uhlig, W. Tailor-made synthesis of functionally substituted oligosilanes from silyl triflates and (aminosilyl)lithium compounds. Z. Naturforsch. B Chem. Sci. 2003, 58, 183–190. [Google Scholar] [CrossRef]
- Ishida, S.; Otsuka, K.; Toma, Y.; Kyushin, S. An Organosilicon Cluster with an Octasilacuneane Core: A Missing Silicon Cage Motif. Angew. Chem. Int. Ed. 2013, 52, 2507–2510. [Google Scholar] [CrossRef]
- Klink, R.; Schrenk, C.; Schnepf, A. Si(SiMe3)2SiPh3 – a ligand for novel sub-valent tin cluster compounds. Dalton Trans. 2014, 43, 16097–16104. [Google Scholar] [CrossRef]
- Farwell, J.D.; Lappert, M.F.; Marschner, C.; Strissel, C.; Tilley, T.D. The first structurally characterised oligosilylmagnesium compound. J. Organomet. Chem. 2000, 603, 185–188. [Google Scholar] [CrossRef]
- Gaderbauer, W.; Zirngast, M.; Baumgartner, J.; Marschner, C.; Tilley, T.D. Synthesis of Polysilanylmagnesium Compounds. Organometallics 2006, 25, 2599–2606. [Google Scholar] [CrossRef]
- Derouiche, Y.; Lickiss, P.D. Preparation and reactions of tris(trimethylsilyl)silyl silicon derivatives and related tetrasilylsilanes. J. Organomet. Chem. 1991, 407, 41–49. [Google Scholar] [CrossRef]
- Kayser, C.; Marschner, C. Oligosilylanions and their Reactions with Zirconocene and Hafnocene Dichlorides. Monatsh. Chem. 1999, 130, 203–206. [Google Scholar]
- Kayser, C.; Frank, D.; Baumgartner, J.; Marschner, C. Reactions of oligosilyl potassium compounds with Group 4 metallocene dichlorides. J. Organomet. Chem. 2003, 667, 149–153. [Google Scholar] [CrossRef]
- Kayser, C.; Kickelbick, G.; Marschner, C. Einfache Synthese von Oligosilyl-α,ω-dikaliumverbindungen. Angew. Chem. 2002, 114, 1031–1034. [Google Scholar] [CrossRef]
- Zirngast, M.; Flörke, U.; Baumgartner, J.; Marschner, C. Oligosilylated group 4 titanocenes in the oxidation state +3. Chem. Commun. 2009, 37, 5538–5540. [Google Scholar] [CrossRef] [PubMed]
- Arp, H.; Zirngast, M.; Marschner, C.; Baumgartner, J.; Rasmussen, K.; Zark, P.; Müller, T. Synthesis of Oligosilanyl Compounds of Group 4 Metallocenes with the Oxidation State +3. Organometallics 2012, 31, 4309–4319. [Google Scholar] [CrossRef] [PubMed]
- Zirngast, M.; Flock, M.; Baumgartner, J.; Marschner, C. Group 4 Metallocene Complexes of Disilenes, Digermenes, and a Silagermene. J. Am. Chem. Soc. 2009, 131, 15952–15962. [Google Scholar] [CrossRef]
- Bock, H.; Meuret, J.; Klaus, R. Structures of charge-perturbed or sterically overcrowded molecules. 22. Structures and properties of supersilyl compounds (R3Si)3SiSi(SiR3)3 and (R3Si)3SiC6H4Si(SiR3)3. Angew. Chem. Int. Ed. Engl. 1993, 32, 414–416. [Google Scholar] [CrossRef]
- Fronczek, F.R.; Lickiss, P.D. Structure of the highly crowded alkyne di[tris(trimethylsilyl)methyl]acetylene and the octasilane hexakis(trimethylsilyl)disilane. Acta Cryst. C 1993, 49, 331–333. [Google Scholar] [CrossRef]
- Kyushin, S.; Sakurai, H.; Betsuyaku, T.; Matsumoto, H. Highly Stable Silyl Radicals (EtnMe3-nSi)3Si• (n = 1−3). Organometallics 1997, 16, 5386–5388. [Google Scholar] [CrossRef]
- Fischer, R.; Konopa, T.; Baumgartner, J.; Marschner, C. Small Cyclosilanes: Syntheses and Reactions toward Mono- and Dianions. Organometallics 2004, 23, 1899–1907. [Google Scholar] [CrossRef]
- Kravchenko, V.; Bravo-Zhivotovskii, D.; Tumanskii, B.; Botoshansky, M.; Segal, N.; Molev, G.; Kosa, M.; Apeloig, Y. Kinetic stabilization of polysilyl radicals. In Organosilicon Chemistry VI; Auner, N., Weis, J., Eds.; Wiley-VCH: Weinheim, Germany, 2005; Volume 1, pp. 48–58. [Google Scholar]
- Krempner, C. Polysilane Dendrimers. Polymers 2012, 4, 408–447. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.; Baumgartner, J.; Marschner, C. Silylgermylpotassium Compounds. Organometallics 2005, 24, 1263–1268. [Google Scholar] [CrossRef]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of Nuclear Magnetic Resonance Signals by Polarization Transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Helmer, B.J.; West, R. Enhancement of 29Si NMR Signals by Proton Polarization Transfer. Organometallics 1982, 1, 877–879. [Google Scholar] [CrossRef]
- SAINTPLUS: Software Reference Manual; Version 6.45; Bruker-AXS: Madison, WI, USA, 1997–2003.
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. A 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS; Version 2.10; Bruker AXS Inc.: Madison, WI, USA, 2003; Available online: https://xray.chem.tamu.edu/pdf/manuals/bruker_guide.pdf (accessed on 9 July 2008).
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POVRAY 3.6.; Persistence of Vision Pty Ltd.: Williamstown, VIC, Australia, 2004; Available online: http://www.povray.org/download/ (accessed on 9 July 2008).
- Gilman, H.; Smith, C.L. Tetrakis(trimethylsilyl)silane. J. Organomet. Chem. 1967, 8, 245–253. [Google Scholar] [CrossRef]
- Nützel, K. Methoden zur Herstellung und Umwandlung magnesiumorganischer Verbindungen. In Houben-Weyl Methoden der Organischen Chemie; Müller, E., Ed.; Thieme: Stuttgart, Germany, 1973; Volume 13/2a, p. 76. ISBN 978-0-86577-944-0. [Google Scholar]
- Heyn, B.; Hipfler, B.; Kreisel, G.; Schreer, H.; Walther, D. Anorganische Synthesechemie; Springer-Lehrbuch: Berlin, Germany, 1990; ISBN 978-3-540-16588-0. [Google Scholar]
- Brook, A.G.; Abdesaken, F.; Söllradl, H. Synthesis of some tris(trimethylsilyl)germyl compounds. J. Organomet. Chem. 1986, 299, 9–13. [Google Scholar] [CrossRef]
Sample Availability: No samples available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Si1-X | Si1-Si2 | Si2-SiMe3 | Si2-M |
|---|---|---|---|---|
| 1 | 1.717(3) | 2.3744(14) | 2.3540(15), 2.3657(15), 2.3706(14) | n.a. |
| 1a | 1.743(2) | 2.3293(10) | 2.3345(10), 2.3510(9) | 3.4990(10) (K) |
| 1b | 1.730(7) | 2.338(3) | 2.601(4) (Mg) | |
| 2a | 1.678(5) (X = O) | 2.329(2) | 2.331(2) 2.335(2) | 3.5064(19) (K) |
| FPh2Si(Me3Si)3SiK [a] | 1.639(2) (X = F) | 2.2970(15) | 2.341(1)/ 2.347(1) | 3.5609(14) (K) |
| ClPh2Si(Me3Si)3SiK [a] | 2.153(2) (X = Cl) | 2.315(2) | 2.356(2)/ 2.360(2) | n.a. |
| 3a | 1.741(6) | 2.387(3) | 2.863(2) (Zr) | |
| 3b | 1.7315(19) | 2.3937(9) | 2.8339(8) (Hf) | |
| 4 | 1.727(3) | 2.464(4) | 2.394(3), 2.410(3) | n.a. |
| Compound | δ 29Si1-X (NEt2) | δ 29Si2 | δ 29SiMe3 | δ 29Si Other |
|---|---|---|---|---|
| 1 | +2.0 | −133.4 | −10.1 | |
| 1a | +16.3 | −186.6 | −6.3 | |
| 1b | +8.7 | −165.5 | −8.3 | |
| 2 | +24.5 (OMe) | −136.5 | −10.4 | |
| 2a | +40.0 (OMe) | −201.8 | −4.5 | |
| FPh2Si(Me3Si)3SiK[a] | +41.4 (F) | −201.6 | −4.4 | |
| ClPh2Si(Me3Si)3SiK[a] | +39.7 (Cl) | −195.7 | −5.7 | |
| 3a | +7.6 | −89.5 | −6.7 | |
| 3b | +9.5 | −83.1 | −5.8 | |
| 4 | +4.8 | −115.4 | −7.9 | |
| 5 | +0.3 (d, JSiF = 6 Hz) | −131.2 (d, JSiF = 19 Hz) | −9.2 | +20.1 (SiPh2F) (d, JSiF = 318 Hz) |
| 5a | +14.9 | −198.9 (SiK) | −6.6 (d, 3JSi–F = 5Hz) | +36.0 (SiPh2F) (d, JSi–F = 359 Hz) |
| 6 | +4.4 | n.a. | −5.4 | |
| 6a | +17.6 | n.a | −5.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balatoni, I.; Hlina, J.; Zitz, R.; Pöcheim, A.; Baumgartner, J.; Marschner, C. β-Amino- and Alkoxy-Substituted Disilanides. Molecules 2019, 24, 3823. https://doi.org/10.3390/molecules24213823
Balatoni I, Hlina J, Zitz R, Pöcheim A, Baumgartner J, Marschner C. β-Amino- and Alkoxy-Substituted Disilanides. Molecules. 2019; 24(21):3823. https://doi.org/10.3390/molecules24213823
Chicago/Turabian StyleBalatoni, Istvan, Johann Hlina, Rainer Zitz, Alexander Pöcheim, Judith Baumgartner, and Christoph Marschner. 2019. "β-Amino- and Alkoxy-Substituted Disilanides" Molecules 24, no. 21: 3823. https://doi.org/10.3390/molecules24213823