
The Effect of Biotinylated PAMAM G3 Dendrimers Conjugated with COX-2 Inhibitor (celecoxib) and PPARγ Agonist (Fmoc-L-Leucine) on Human Normal Fibroblasts, Immortalized Keratinocytes and Glioma Cells in Vitro

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
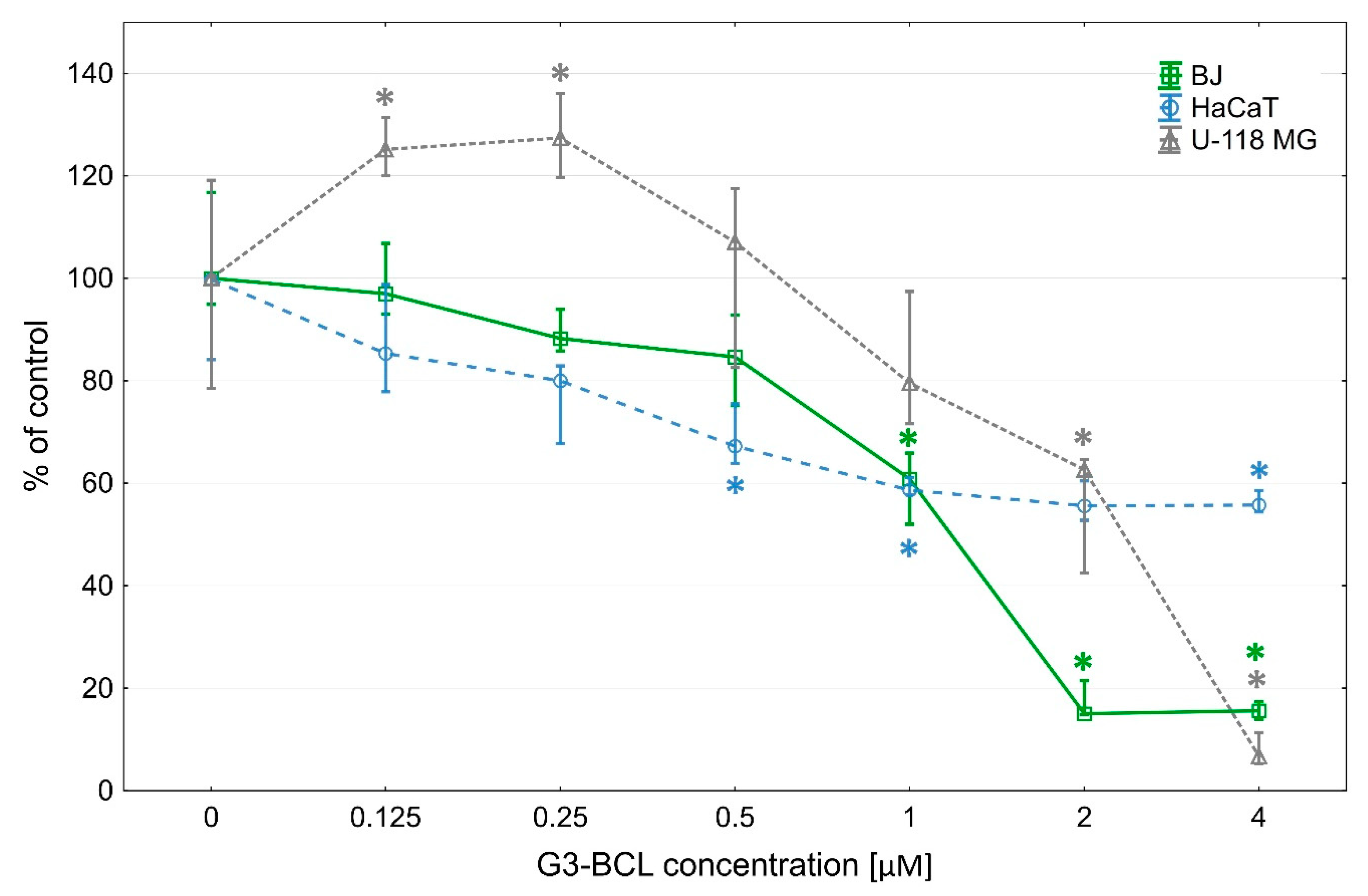
2.1. Cytotoxicity of G3-BCL Conjugate
2.2. Proliferation
2.3. Cellular Accumulation of G3-BCLF
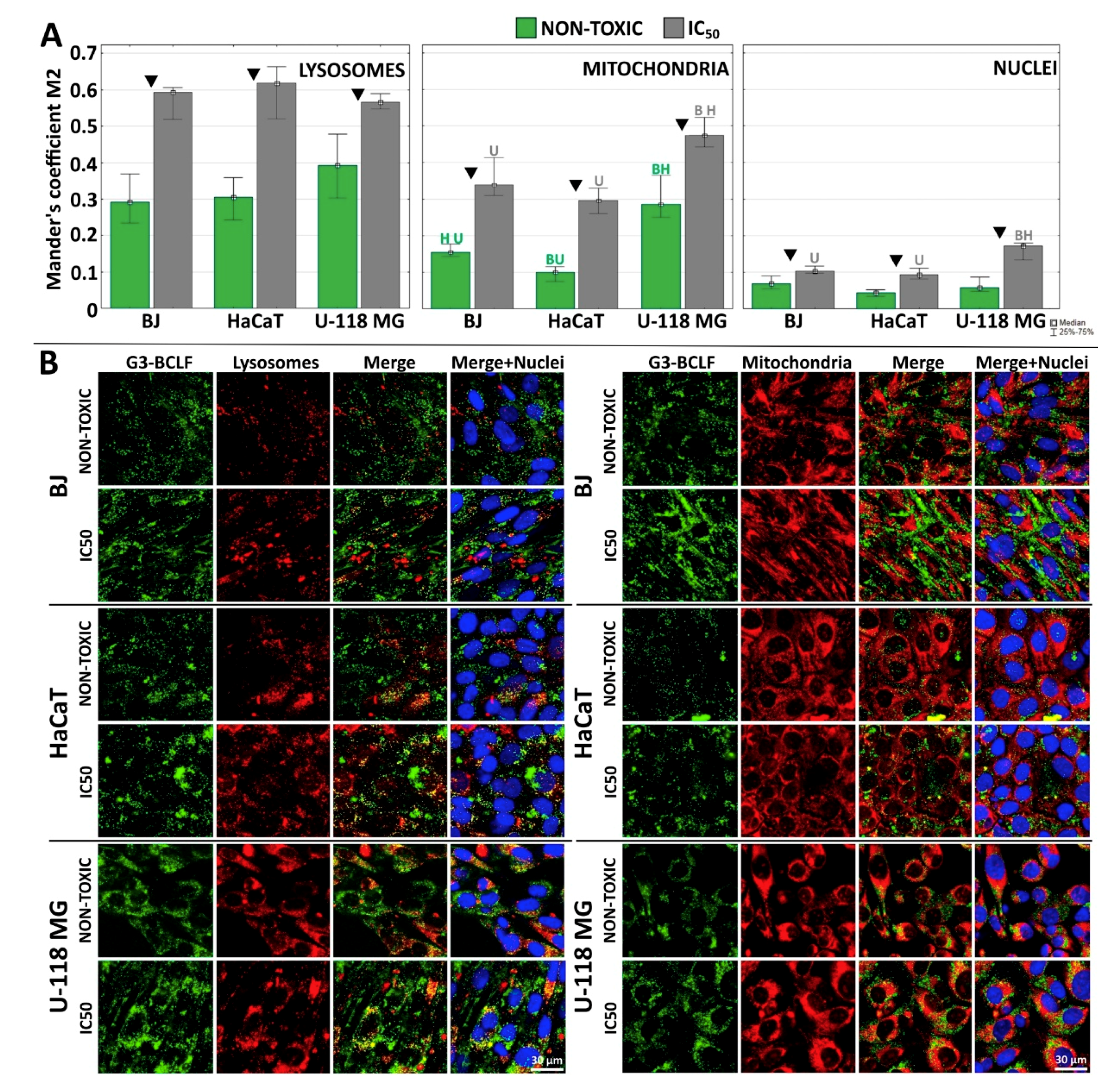
2.4. Subcellular Localization of G3-BCLF
2.5. Intracellular ATP Level
2.6. Apoptosis and Late Apoptosis/Necrosis
2.7. Effect of G3-BCL on Cell Migration
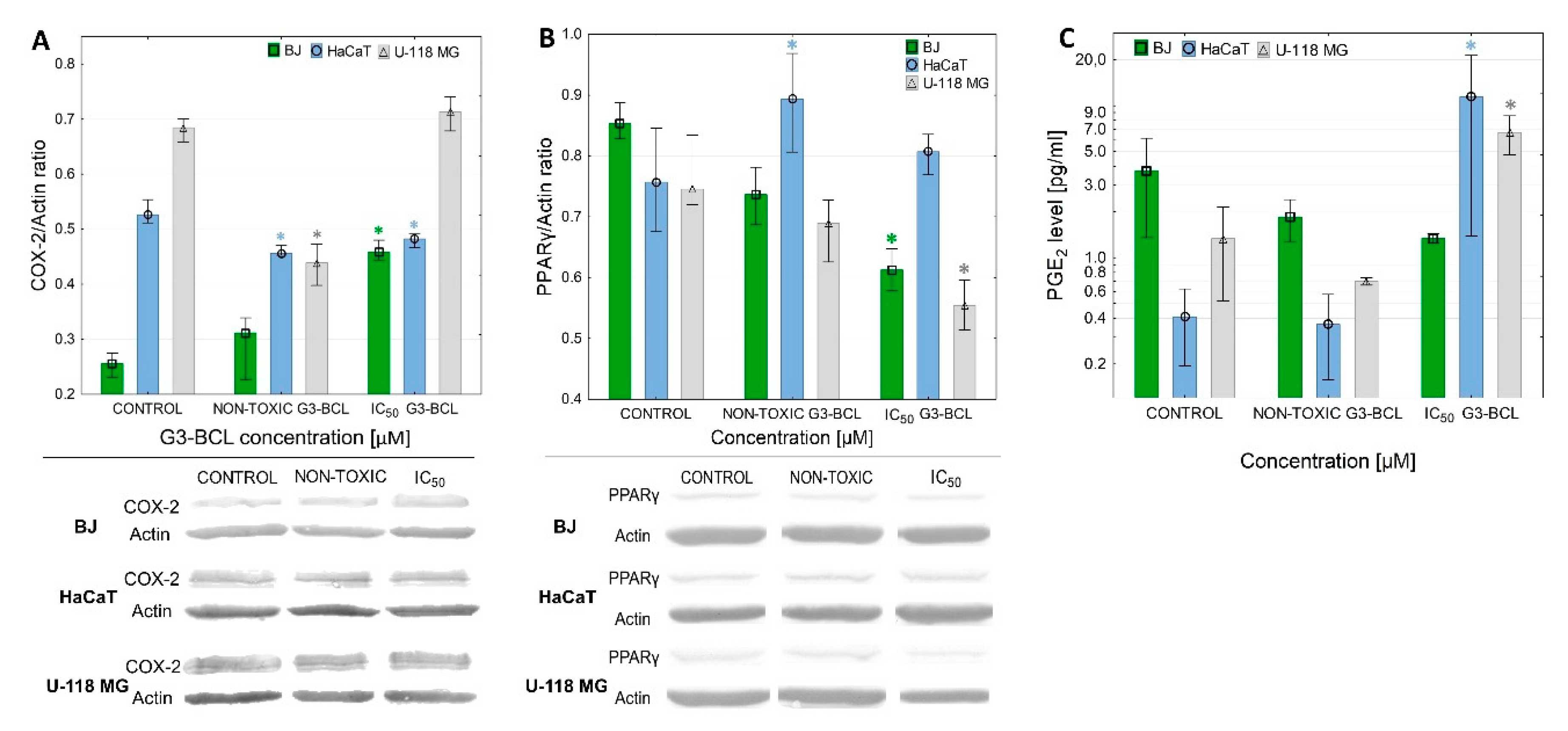
2.8. COX-2 Expression and PGE2 Production
3. Materials and Methods
3.1. Dendrimers
3.2. Materials
3.3. Cell cultures
3.3.1. Cytotoxicity
3.3.2. Proliferation
3.3.3. Cellular Uptake of G3-BCLF
3.3.4. Intracellular Location of G3-BCLF
3.3.5. Cellular ATP Level
3.3.6. Apoptosis and Late Apoptosis/Necrosis
3.3.7. Cell Mobility
3.3.8. COX-2 and PPARγ Expression and PGE2 Production
3.3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Demuth, T.; Berens, M.E. Molecular Mechanisms of Glioma Cell Migration and Invasion. J. Neurooncol. 2004, 70, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C. Temozolomide: Therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev. Neurother. 2010, 10, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, S.; Tsugu, H.; Komatsu, F.; Ohmura, T.; Ohta, M.; Sakamoto, S.; Fukushima, T.; Inoue, T. Efficacy of Temozolomide Treatment in Patients with High-grade Glioma. Anticancer Res. 2009, 29, 911–917. [Google Scholar] [PubMed]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Pan, I.-W.; Ferguson, S.D.; Lam, S. Patient and treatment factors associated with survival among adult glioblastoma patients: A USA population-based study from 2000–2010. J. Clin. Neurosci. 2015, 22, 1575–1581. [Google Scholar] [CrossRef]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; del Carmen Patino, M.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [Green Version]
- Newcomb, E.W.; Madonia, W.J.; Pisharody, S.; Lang, F.F.; Koslow, M.; Miller, D.C. A Correlative Study of p53 Protein Alteration and p53 Gene Mutation in Glioblastoma Multiforme. Brain Pathol. 1993, 3, 229–235. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Alphandéry, E. Glioblastoma Treatments: An Account of Recent Industrial Developments. Front. Pharmacol. 2018, 9, 879. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, V.N.; Wu, J.; Hei, T.K. Regulation of human glioblastoma cell death by combined treatment of cannabidiol, γ-radiation and small molecule inhibitors of cell signaling pathways. Oncotarget 2017, 8, 74068–74095. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, F.G.; DuBois, R.N. Connecting COX-2 and Wnt in cancer. Cancer Cell 2006, 9, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Guan, J.; Li, C.; Gunter, S.; Nusrat, L.; Ng, S.; Dhand, K.; Morshead, C.; Kim, A.; Das, S. Aberrantly activated Cox-2 and Wnt signaling interact to maintain cancer stem cells in glioblastoma. Oncotarget 2017, 8, 82217–82230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecarpentier, Y.; Claes, V.; Vallée, A.; Hébert, J.-L. Thermodynamics in cancers: Opposing interactions between PPAR gamma and the canonical WNT/beta-catenin pathway. Clin. Trans. Med. 2017, 6, 14. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Opposite Interplay between the Canonical WNT/β-Catenin Pathway and PPAR Gamma: A Potential Therapeutic Target in Gliomas. Neurosci. Bull. 2018, 34, 573–588. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. Crosstalk between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress during Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef]
- Shono, T.; Tofilon, P.J.; Bruner, J.M.; Owolabi, O.; Lang, F.F. Cyclooxygenase-2 Expression in Human Gliomas: Prognostic Significance and Molecular Correlations. Cancer Res. 2001, 61, 4375–4381. [Google Scholar]
- New, P. Cyclooxygenase in the Treatment of Glioma: Its Complex Role in Signal Transduction. Cancer Control 2004, 11, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.T. Cyclooxygenase-2 in oncogenesis. Clin. Chim. Acta 2011, 412, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Shi, Z.; Jiang, J. Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov. Today 2017, 22, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Tegeder, I.; Pfeilschifter, J.; Geisslinger, G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001, 15, 2057–2072. [Google Scholar] [CrossRef]
- Schönthal, A.H. Direct non-cyclooxygenase-2 targets of celecoxib and their potential relevance for cancer therapy. Br. J. Cancer 2007, 97, 1465–1468. [Google Scholar] [CrossRef]
- Grösch, S.; Maier, T.J.; Schiffmann, S.; Geisslinger, G. Cyclooxygenase-2 (COX-2)–Independent Anticarcinogenic Effects of Selective COX-2 Inhibitors. J. Natl. Cancer Inst. 2006, 98, 736–747. [Google Scholar] [CrossRef]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef]
- Finch, E.R.; Tukaramrao, D.B.; Goodfield, L.L.; Quickel, M.D.; Paulson, R.F.; Prabhu, K.S. Activation of PPARγ by endogenous prostaglandin J2 mediates the antileukemic effect of selenium in murine leukemia. Blood 2017, 129, 1802–1810. [Google Scholar] [CrossRef]
- Knopfová, L.; Šmarda, J. The use of Cox-2 and PPARγ signaling in anti-cancer therapies (Review). Exp. Ther. Med. 2010, 1, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.B.; Zhu, C.; Yong, S.K.; Gao, Q.; Wong, M.C. Enhanced sensitivity of celecoxib in human glioblastoma cells: Induction of DNA damage leading to p53-dependent G1 cell cycle arrest and autophagy. Mol. Cancer 2009, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Mondal, P.; Ghosh, S.; Mehta, V.S.; Sen, E. PPAR γ regulated CIDEA affects pro-apoptotic responses in glioblastoma. Cell Death Discov. 2015, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Strakova, N.; Ehrmann, J.; Dzubak, P.; Bouchal, J.; Kolar, Z. The Synthetic Ligand of Peroxisome Proliferator-Activated Receptor-γ Ciglitazone Affects Human Glioblastoma Cell Lines. J. Pharmacol. Exp. Ther. 2004, 309, 1239–1247. [Google Scholar] [CrossRef]
- Im, C.-N. Combination Treatment with PPARγ Ligand and Its Specific Inhibitor GW9662 Downregulates BIS and 14-3-3 Gamma, Inhibiting Stem-Like Properties in Glioblastoma Cells. Available online: https://www.hindawi.com/journals/bmri/2017/5832824/ (accessed on 27 September 2019).
- Grommes, C.; Landreth, G.E.; Sastre, M.; Beck, M.; Feinstein, D.L.; Jacobs, A.H.; Schlegel, U.; Heneka, M.T. Inhibition of in Vivo Glioma Growth and Invasion by Peroxisome Proliferator-Activated Receptor γ Agonist Treatment. Mol. Pharmacol. 2006, 70, 1524–1533. [Google Scholar] [CrossRef]
- Grommes, C.; Conway, D.S.; Alshekhlee, A.; Barnholtz-Sloan, J.S. Inverse association of PPARγ agonists use and high grade glioma development. J. Neurooncol. 2010, 100, 233–239. [Google Scholar] [CrossRef]
- Rocchi, S.; Picard, F.; Vamecq, J.; Gelman, L.; Potier, N.; Zeyer, D.; Dubuquoy, L.; Bac, P.; Champy, M.F.; Plunket, K.D.; et al. A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Mol. Cell 2001, 8, 737–747. [Google Scholar] [CrossRef]
- Uram, Ł.; Filipowicz, A.; Misiorek, M.; Pieńkowska, N.; Markowicz, J.; Wałajtys-Rode, E.; Wołowiec, S. Biotinylated PAMAM G3 dendrimer conjugated with celecoxib and/or Fmoc-l-Leucine and its cytotoxicity for normal and cancer human cell lines. Eur. J. Pharm. Sci. 2018, 124, 1–9. [Google Scholar] [CrossRef]
- Michael, M.S.; Badr, M.Z.; Badawi, A.F. Inhibition of cyclooxygenase-2 and activation of peroxisome proliferator-activated receptor-γ synergistically induces apoptosis and inhibits growth of human breast cancer cells. Int. J. Mol. Med. 2003, 11, 733–736. [Google Scholar] [CrossRef]
- Mustafa, A.; Kruger, W.D. Suppression of Tumor Formation by a Cyclooxygenase-2 Inhibitor and a Peroxisome Proliferator-Activated Receptor γ Agonist in an In vivo Mouse Model of Spontaneous Breast Cancer. Clin. Cancer Res. 2008, 14, 4935–4942. [Google Scholar] [CrossRef]
- Sun, W.-H.; Chen, G.-S.; Ou, X.-L.; Yang, Y.; Luo, C.; Zhang, Y.; Shao, Y.; Xu, H.-C.; Xiao, B.; Xue, Y.-P.; et al. Inhibition of COX-2 and activation of peroxisome proliferator-activated receptor gamma synergistically inhibits proliferation and induces apoptosis of human pancreatic carcinoma cells. Cancer Lett. 2009, 275, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.P.; Kurian, K.M. Biological Rationale for the Use of PPARγ Agonists in Glioblastoma. Front. Oncol. 2014, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Martorana, G.E.; Ferrari, F.; De, P.S.; Rossi, C.; Giardina, B. Mitochondrial respiratory chain dysfunction, a non-receptor-mediated effect of synthetic PPAR-ligands: Biochemical and pharmacological implications. Biochem. Biophys. Res. Commun. 2004, 319, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Giardina, B. Mitochondria, PPARs, and Cancer: Is Receptor-Independent Action of PPAR Agonists a Key? PPAR Res. 2008, 2008, 256251. [Google Scholar] [CrossRef]
- Walker, C.; Biasucci, L.M. Cardiovascular safety of non-steroidal anti-inflammatory drugs revisited. Postgrad. Med. 2018, 130, 55–71. [Google Scholar] [CrossRef]
- Whilding, L.M.; Maher, J. CAR T-cell immunotherapy: The path from the by-road to the freeway? Mol. Oncol. 2015, 9, 1994–2018. [Google Scholar] [CrossRef]
- Jahan, S.T.; Sadat, S.M.A.; Walliser, M.; Haddadi, A. Targeted Therapeutic Nanoparticles: An Immense Promise to Fight against Cancer. J. Drug Deliv. 2017, 2017, 9090325. [Google Scholar] [CrossRef]
- Jain, K.; Kesharwani, P.; Gupta, U.; Jain, N.K. Dendrimer toxicity: Let’s meet the challenge. Int. J. Pharm. 2010, 394, 122–142. [Google Scholar] [CrossRef]
- Dwivedi, N.; Shah, J.; Mishra, V.; Amin, M.C.I.M.; Iyer, A.K.; Tekade, R.K.; Kesharwani, P. Dendrimer-mediated approaches for the treatment of brain tumor. J. Biomater. Sci. Polym. Ed. 2016, 27, 557–580. [Google Scholar] [CrossRef]
- Florendo, M.; Figacz, A.; Srinageshwar, B.; Sharma, A.; Swanson, D.; Dunbar, G.L.; Rossignol, J. Use of Polyamidoamine Dendrimers in Brain Diseases. Molecules 2018, 23, 2238. [Google Scholar] [CrossRef]
- Karim, R.; Palazzo, C.; Evrard, B.; Piel, G. Nanocarriers for the treatment of glioblastoma multiforme: Current state-of-the-art. J. Control Release 2016, 227, 23–37. [Google Scholar] [CrossRef]
- Sarin, H.; Kanevsky, A.S.; Wu, H.; Brimacombe, K.R.; Fung, S.H.; Sousa, A.A.; Auh, S.; Wilson, C.M.; Sharma, K.; Aronova, M.A.; et al. Effective transvascular delivery of nanoparticles across the blood-brain tumor barrier into malignant glioma cells. J. Transl. Med. 2008, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Gonawala, S.; Ali, M.M. Application of Dendrimer-based Nanoparticles in Glioma Imaging. J. Nanomed. Nanotechnol. 2017, 8. [Google Scholar] [CrossRef]
- Castro, R.I.; Forero-Doria, O.; Guzmán, L.; Castro, R.I.; Forero-Doria, O.; Guzmán, L. Perspectives of Dendrimer-based Nanoparticles in Cancer Therapy. Anais Academia Brasileira Ciências 2018, 90, 2331–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripodo, G.; Mandracchia, D.; Collina, S.; Rui, M.; Rossi, D. New Perspectives in Cancer Therapy: The Biotin-Antitumor Molecule Conjugates. Med. Chem. 2014, 8, 1–4. [Google Scholar]
- Holban, A.M.; Grumezescu, A.M. Nanoarchitectonics for Smart Delivery and Drug Targeting, 1st ed.; Elsevier: Amsterdam, Netherlands, 2016; ISBN 978-0-323-47347-7. [Google Scholar]
- Spector, R.; Mock, D.M. Biotin transport and metabolism in the central nervous system. Neurochem. Res. 1988, 13, 213–219. [Google Scholar] [CrossRef]
- Hemmer, R.; Hall, A.; Spaulding, R.; Rossow, B.; Hester, M.; Caroway, M.; Haskamp, A.; Wall, S.; Bullen, H.A.; Morris, C.; et al. Analysis of Biotinylated Generation 4 Poly(amidoamine) (PAMAM) Dendrimer Distribution in the Rat Brain and Toxicity in a Cellular Model of the Blood-Brain Barrier. Molecules 2013, 18, 11537–11552. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhao, X.; Chen, J.; Chen, J.; Kuznetsova, L.; Wong, S.S.; Ojima, I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconj. Chem. 2010, 21, 979–987. [Google Scholar] [CrossRef] [Green Version]
- Veszelka, S.; Meszaros, M.; Kiss, L.; Kota, Z.; Pali, T.; Hoyk, Z.; Bozso, Z.; Fulop, L.; Toth, A.; Rakhely, G.; et al. Biotin and Glutathione Targeting of Solid Nanoparticles to Cross Human Brain Endothelial Cells. Curr. Pharm. Des. 2017, 23, 4198–4205. [Google Scholar] [CrossRef] [Green Version]
- Uram, Ł.; Szuster, M.; Filipowicz, A.; Zaręba, M.; Wałajtys-Rode, E.; Wołowiec, S. Cellular uptake of glucoheptoamidated poly(amidoamine) PAMAM G3 dendrimer with amide-conjugated biotin, a potential carrier of anticancer drugs. Bioorg. Med. Chem. 2017, 25, 706–713. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Geeviman, K.; Ramulu, C.; Babu, P.P. The nonsteroidal anti-inflammatory drug celecoxib suppresses the growth and induces apoptosis of human glioblastoma cells via the NF-κB pathway. J. Neurooncol. 2012, 106, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-Y.; Xiao, J.; Liu, Q.; Xia, K. Parecoxib inhibits glioblastoma cell proliferation, migration and invasion by upregulating miRNA-29c. Biol. Open 2017, 6, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Van der Pauw, M.T.; den Bos, T.V.; Everts, V.; Beertsen, W. Phagocytosis of Fibronectin and Collagens Type I, III, and V by Human Gingival and Periodontal Ligament Fibroblasts In Vitro. J. Periodontol. 2001, 72, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Götz, C.; Pfeiffer, R.; Tigges, J.; Ruwiedel, K.; Hübenthal, U.; Merk, H.F.; Krutmann, J.; Edwards, R.J.; Abel, J.; Pease, C.; et al. Xenobiotic metabolism capacities of human skin in comparison with a 3D-epidermis model and keratinocyte-based cell culture as in vitro alternatives for chemical testing: Phase II enzymes. Exp. Dermatol. 2012, 21, 364–369. [Google Scholar] [CrossRef]
- Cheng, Y. Dendrimer-Based Drug Delivery Systems: From Theory to Practice; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 978-0-470-46005-4. [Google Scholar]
- Zhang, J.; Liu, D.; Zhang, M.; Sun, Y.; Zhang, X.; Guan, G.; Zhao, X.; Qiao, M.; Chen, D.; Hu, H. The cellular uptake mechanism, intracellular transportation, and exocytosis of polyamidoamine dendrimers in multidrug-resistant breast cancer cells. Int. J. Nanomed. 2016, 11, 3677–3690. [Google Scholar] [CrossRef]
- Albertazzi, L.; Serresi, M.; Albanese, A.; Beltram, F. Dendrimer Internalization and Intracellular Trafficking in Living Cells. Mol. Pharm. 2010, 7, 680–688. [Google Scholar] [CrossRef]
- Kitchens, K.M.; Foraker, A.B.; Kolhatkar, R.B.; Swaan, P.W.; Ghandehari, H. Endocytosis and interaction of poly (amidoamine) dendrimers with Caco-2 cells. Pharm. Res. 2007, 24, 2138–2145. [Google Scholar] [CrossRef]
- Kitchens, K.M.; Kolhatkar, R.B.; Swaan, P.W.; Ghandehari, H. Endocytosis inhibitors prevent poly(amidoamine) dendrimer internalization and permeability across Caco-2 cells. Mol. Pharm. 2008, 5, 364–369. [Google Scholar] [CrossRef]
- Yellepeddi, V.K.; Kumar, A.; Palakurthi, S. Biotinylated poly(amido)amine (PAMAM) dendrimers as carriers for drug delivery to ovarian cancer cells in vitro. Anticancer Res. 2009, 29, 2933–2943. [Google Scholar]
- Zempleni, J. Uptake, Localization, and Noncarboxylase Roles of Biotin. Annu. Rev. Nutr. 2005, 25, 175–196. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Aspects Med. 2010, 31, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Kan, M.; Qiao, P.; Pan, Y.; Wang, Z.; Xiao, X.; Li, J.; Chen, L. Celecoxib induces apoptosis via a mitochondria-dependent pathway in the H22 mouse hepatoma cell line. Mol. Med. Rep. 2014, 10, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, B.; Ahrens, T.; Regenass, U. Optical Assays in Drug Discovery. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, Netherlands, 2007; pp. 577–598. ISBN 978-0-08-045044-5. [Google Scholar]
- Zimmermann, M.; Meyer, N. Annexin V/7-AAD staining in keratinocytes. Methods Mol. Biol. 2011, 740, 57–63. [Google Scholar]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target Ther. 2018, 3, 18. [Google Scholar] [CrossRef]
- Zamaraeva, M.V.; Sabirov, R.Z.; Maeno, E.; Ando-Akatsuka, Y.; Bessonova, S.V.; Okada, Y. Cells die with increased cytosolic ATP during apoptosis: A bioluminescence study with intracellular luciferase. Cell Death Differ. 2005, 12, 1390–1397. [Google Scholar] [CrossRef]
- Los, M.; Mozoluk, M.; Ferrari, D.; Stepczynska, A.; Stroh, C.; Renz, A.; Herceg, Z.; Wang, Z.-Q.; Schulze-Osthoff, K. Activation and Caspase-mediated Inhibition of PARP: A Molecular Switch between Fibroblast Necrosis and Apoptosis in Death Receptor Signaling. MBoC 2002, 13, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Nzengue, Y.; Steiman, R.; Guiraud, P. Characterization of the cell death induced by cadmium in HaCaT and C6 cell lines. Free Radic. Res. 2008, 42, 142–153. [Google Scholar] [CrossRef]
- Henseleit, U.; Rosenbach, T.; Kolde, G. Induction of apoptosis in human HaCaT keratinocytes. Arch. Dermatol. Res. 1996, 288, 676–683. [Google Scholar] [CrossRef]
- El Darzi, E.; Bazzi, S.; Daoud, S.; Echtay, K.S.; Bahr, G.M. Differential regulation of surface receptor expression, proliferation, and apoptosis in HaCaT cells stimulated with interferon-γ, interleukin-4, tumor necrosis factor-α, or muramyl dipeptide. Int. J. Immunopathol. Pharmacol. 2017, 30, 130–145. [Google Scholar] [CrossRef]
- Lehman, T.A.; Modali, R.; Boukamp, P.; Stanek, J.; Bennett, W.P.; Welsh, J.A.; Metcalf, R.A.; Stampfer, M.R.; Fusenig, N.; Rogan, E.M. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis 1993, 14, 833–839. [Google Scholar] [CrossRef]
- Henseleit, U.; Zhang, J.; Wanner, R.; Haase, I.; Kolde, G.; Rosenbach, T. Role of p53 in UVB-Induced Apoptosis in Human HaCaT Keratinocytes. J. Investig. Dermatol. 1997, 109, 722–727. [Google Scholar] [CrossRef] [Green Version]
- Barlow, M.; Edelman, M.; Glick, R.D.; Steinberg, B.M.; Soffer, S.Z. Celecoxib inhibits invasion and metastasis via a cyclooxygenase 2-independent mechanism in an in vitro model of Ewing sarcoma. J. Pediatr. Surg. 2012, 47, 1223–1227. [Google Scholar] [CrossRef]
- Behr, C.A.; Hesketh, A.J.; Barlow, M.; Glick, R.D.; Symons, M.; Steinberg, B.M.; Soffer, S.Z. Celecoxib inhibits Ewing sarcoma cell migration via actin modulation. J. Surg. Res. 2015, 198, 424–433. [Google Scholar] [CrossRef]
- Patel, M.I.; Subbaramaiah, K.; Du, B.; Chang, M.; Yang, P.; Newman, R.A.; Cordon-Cardo, C.; Thaler, H.T.; Dannenberg, A.J. Celecoxib inhibits prostate cancer growth: Evidence of a cyclooxygenase-2-independent mechanism. Clin. Cancer Res. 2005, 11, 1999–2007. [Google Scholar] [CrossRef]
- Lee, E.J.; Choi, E.M.; Kim, S.R.; Park, J.H.; Kim, H.; Ha, K.S.; Kim, Y.M.; Kim, S.S.; Choe, M.; Kim, J.-I.; et al. Cyclooxygenase-2 promotes cell proliferation, migration and invasion in U2OS human osteosarcoma cells. Exp. Mol. Med. 2007, 39, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.-Y.; Li, X.-Q.; Xu, X.-T.; Wang, L.-L.; Qin, S.-B.; Tu, Y. Celecoxib inhibits the proliferation and migration of human glioblastoma SHG-44 cells in vitro. Tumor 2010, 30, 913–916. [Google Scholar]
- Seufert, S.; Coras, R.; Tränkle, C.; Zlotos, D.P.; Blümcke, I.; Tatenhorst, L.; Heneka, M.T.; Hahnen, E. PPAR Gamma Activators: Off-Target Against Glioma Cell Migration and Brain Invasion. PPAR Res. 2008, 2008, 513943. [Google Scholar] [CrossRef]
- Kim, Y. Regulation of Cell Proliferation and Migration in Glioblastoma: New Therapeutic Approach. Front. Oncol. 2013, 3, 53. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-P.; Peng, Y.-B.; Zhang, Y.-F.; Wang, Y.; Yu, W.-R.; Yao, M.; Fu, X.-J. Reactive Oxygen Species Mediated Prostaglandin E2 Contributes to Acute Response of Epithelial Injury. Oxid. Med. Cell Longev. 2017, 2017, 4123854. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Z.-G.; Shi, Z.; Shao, D.; Li, O.; Li, W.; Li, Z.-J.; Wang, K.-Z.; Chen, L. Inhibitory effect of celecoxib in lung carcinoma by regulation of cyclooxygenase-2/cytosolic phospholipase A2 and peroxisome proliferator-activated receptor gamma. Mol. Cell. Biochem. 2011, 355, 233–240. [Google Scholar] [CrossRef]
- Ramer, R.; Walther, U.; Borchert, P.; Laufer, S.; Linnebacher, M.; Hinz, B. Induction but not inhibition of COX-2 confers human lung cancer cell apoptosis by celecoxib. J. Lipid Res. 2013, 54, 3116–3129. [Google Scholar] [CrossRef] [Green Version]
- Roehm, N.W.; Rodgers, G.H.; Hatfield, S.M.; Glasebrook, A.L. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods 1991, 142, 257–265. [Google Scholar] [CrossRef]
- Uram, Ł.; Szuster, M.; Misiorek, M.; Filipowicz, A.; Wołowiec, S.; Wałajtys-Rode, E. The effect of G3 PAMAM dendrimer conjugated with B-group vitamins on cell morphology, motility and ATP level in normal and cancer cells. Eur. J. Pharm. Sci. 2017, 102, 275–283. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds G3-BCL are available from the authors. |








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uram, Ł.; Misiorek, M.; Pichla, M.; Filipowicz-Rachwał, A.; Markowicz, J.; Wołowiec, S.; Wałajtys-Rode, E. The Effect of Biotinylated PAMAM G3 Dendrimers Conjugated with COX-2 Inhibitor (celecoxib) and PPARγ Agonist (Fmoc-L-Leucine) on Human Normal Fibroblasts, Immortalized Keratinocytes and Glioma Cells in Vitro. Molecules 2019, 24, 3801. https://doi.org/10.3390/molecules24203801
Uram Ł, Misiorek M, Pichla M, Filipowicz-Rachwał A, Markowicz J, Wołowiec S, Wałajtys-Rode E. The Effect of Biotinylated PAMAM G3 Dendrimers Conjugated with COX-2 Inhibitor (celecoxib) and PPARγ Agonist (Fmoc-L-Leucine) on Human Normal Fibroblasts, Immortalized Keratinocytes and Glioma Cells in Vitro. Molecules. 2019; 24(20):3801. https://doi.org/10.3390/molecules24203801
Chicago/Turabian StyleUram, Łukasz, Maria Misiorek, Monika Pichla, Aleksandra Filipowicz-Rachwał, Joanna Markowicz, Stanisław Wołowiec, and Elżbieta Wałajtys-Rode. 2019. "The Effect of Biotinylated PAMAM G3 Dendrimers Conjugated with COX-2 Inhibitor (celecoxib) and PPARγ Agonist (Fmoc-L-Leucine) on Human Normal Fibroblasts, Immortalized Keratinocytes and Glioma Cells in Vitro" Molecules 24, no. 20: 3801. https://doi.org/10.3390/molecules24203801