Recent Advances in Enantioselective Photochemical Reactions of Stabilized Diazo Compounds


Abstract
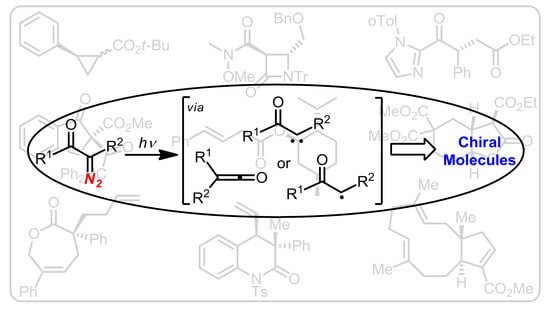
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
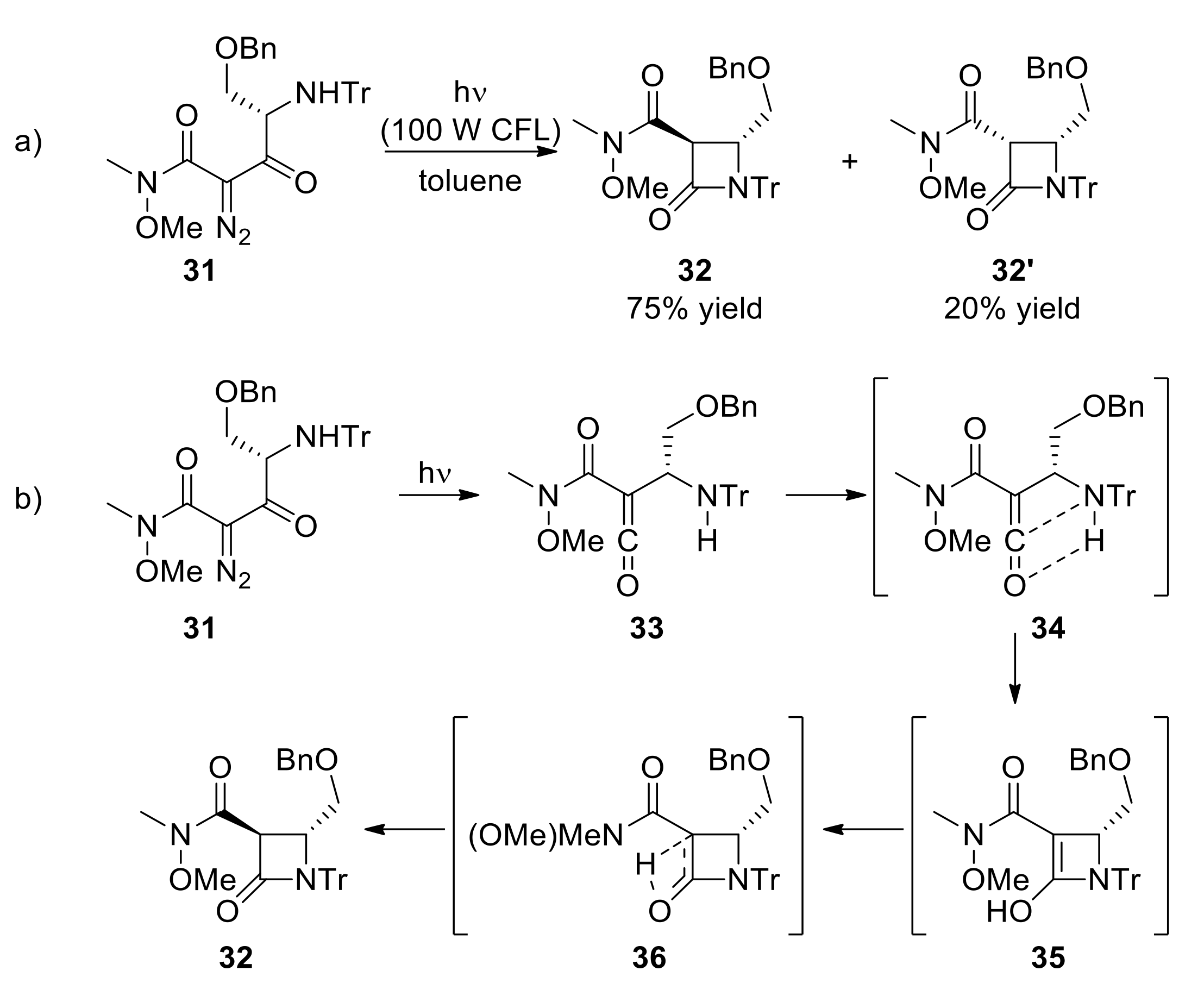{kind=link}
{kind=link}
{kind=link}
{kind=link}
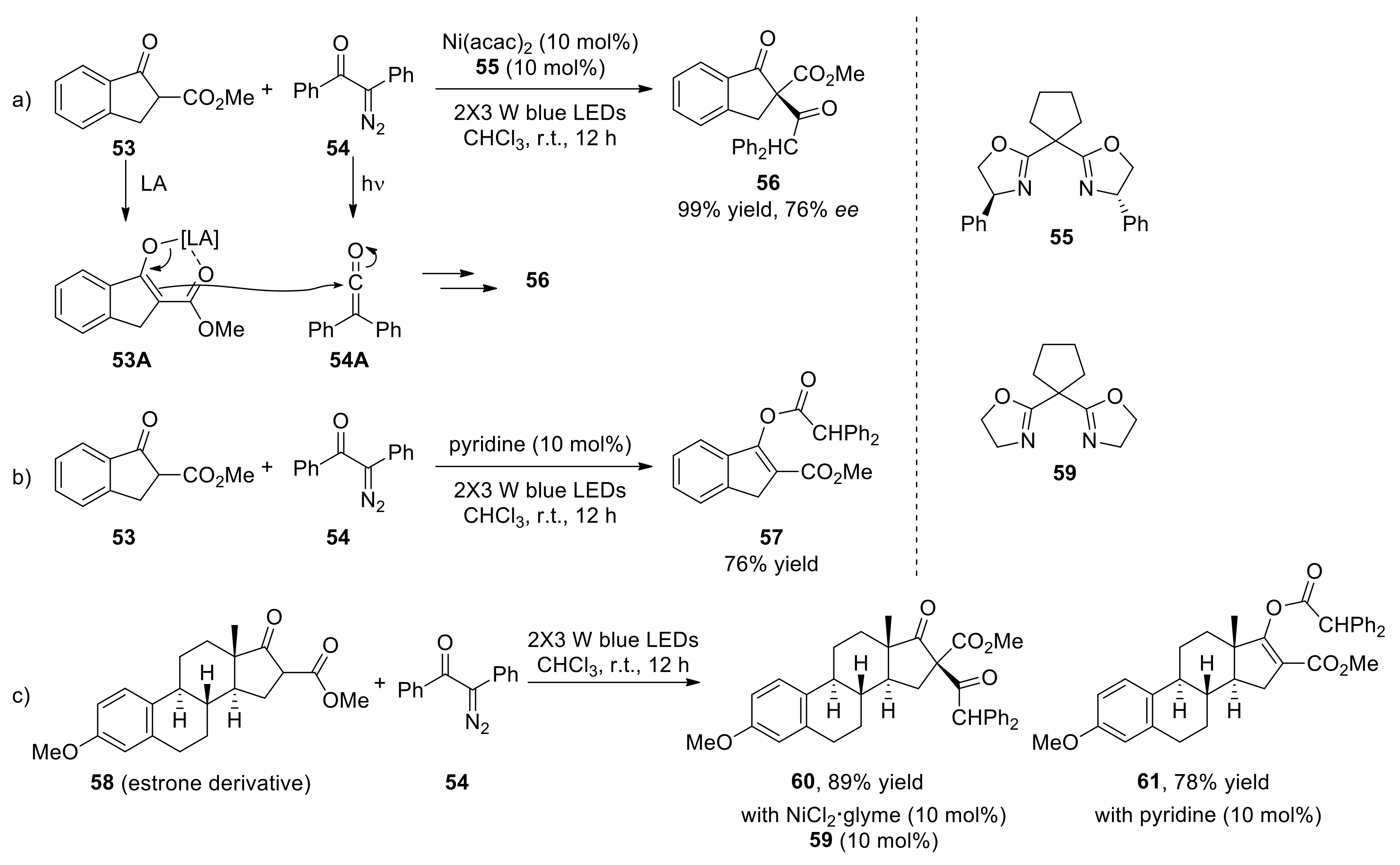{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Asymmetric Cyclopropanation
2.1. Intermolecular Asymmetric Cyclopropanation
2.2. Intramolecular Asymmetric Cyclopropanation
3. Asymmetric Reactions Based on In Situ Wolff Rearrangement
3.1. Nucleophilic Addition Based on the In Situ Wolff Rearrangement
3.1.1. Oxygen Nucleophilic Addition
3.1.2. Nitrogen Nucleophilic Addition
3.1.3. Carbon Nucleophilic Addition
3.1.4. Nucleophilic Addition Based on Ring Contraction via In Situ Wolff Rearrangement
3.2. Cycloaddition Reactions Based on the In Situ Wolff Rearrangement
3.2.1. Formal [2 + 2] Cycloaddition Reaction
3.2.2. Formal [4 + 2] Cycloaddition Reaction
3.2.3. Formal [5 + 2] Cycloaddition Reaction
3.2.4. Formal [3 + 2] Cycloaddition Reaction
4. Asymmetric Alkylation
5. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Curtius, T. Ueber die Einwirkung von salpetriger Säure auf salzsauren Glycocolläther. Ber. Dtsch. Chem. Ges. 1883, 16, 2230–2231. [Google Scholar] [CrossRef]
- Regitz, G.; Maas, M. Diazo Compounds: Properties and Synthesis; Elsevier: Amsterdam, The Netherlands, 1986. [Google Scholar]
- Doyle, M.P.; Forbes, D.C. Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev. 1998, 98, 911–936. [Google Scholar] [CrossRef] [PubMed]
- Wee, A.G.H. Rhodium(II)-Catalyzed Reaction of Diazo compounds in the Service of Organic Synthesis of Natural and Non-Natural Products. Curr. Org. Synth. 2006, 3, 499–555. [Google Scholar] [CrossRef]
- Candeias, N.; Afonso, C. Developments in the Photochemistry of Diazo Compounds. Curr. Org. Chem. 2009, 13, 763–787. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhang, Y.; Wang, J. Recent Developments in Copper-Catalyzed Reactions of Diazo Compounds. Chem. Commun. 2012, 48, 10162–10173. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef] [PubMed]
- Candeias, N.R.; Paterna, R.; Gois, P.M.P. Homologation Reaction of Ketones with Diazo Compounds. Chem. Rev. 2016, 116, 2937–2981. [Google Scholar] [CrossRef]
- Galkina, O.S.; Rodina, L.L. Photochemical transformations of diazocarbonyl compounds: Expected and novel reactions. Russ. Chem. Rev. 2016, 85, 537–555. [Google Scholar] [CrossRef]
- Ciszewski, L.W.; Rybicka-Jasińska, K.; Gryko, D. Recent developments in photochemical reactions of diazo compounds. Org. Biomol. Chem. 2019, 17, 432–448. [Google Scholar] [CrossRef]
- Liu, X.; Huang, Y.; Meng, X.; Li, J.; Wang, D.; Chen, Y.; Tang, D.; Chen, B. Recent developments in the synthesis of N-containing heterocycles via C-H/N-H bond functionalizations and oxidative cyclization. Synlett 2019, 30, 1026–1036. [Google Scholar]
- Hua, T.B.; Xiao, C.; Yang, Q.Q.; Chen, J.R. Recent advances in asymmetric synthesis of 2-substituted indoline derivatives. Chin. Chem. Lett. 2019. [Google Scholar] [CrossRef]
- Maas, G. New Syntheses of Diazo Compounds. Angew. Chem. Int. Ed. 2009, 48, 8186–8195. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; McKervey, M.A. Organic Synthesis with α-Diazo Carbonyl Compounds. Chem. Rev. 1994, 94, 1091–1160. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J. Recent Studies on the Reactions of α-Diazocarbonyl Compounds. Tetrahedron 2008, 64, 6577–6605. [Google Scholar] [CrossRef]
- Ding, K.; Dai, L.X. Organic Chemistry-Breakthroughs and Perspectives; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Brimioulle, R.; Lenhart, M.S.D.; Maturi, M.S.M.M.; Bach, T. Enantioselective Catalysis of Photochemical Reactions. Angew. Chem. Int. Ed. 2015, 54, 3872–3890. [Google Scholar] [CrossRef] [PubMed]
- Meggers, E. Asymmetric catalysis activated by visible light. Chem. Commun. 2015, 51, 3290–3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Lu, Z. Catalytic enantioselective organic transformations via visible light photocatalysis. Org. Chem. Front. 2015, 2, 179–190. [Google Scholar] [CrossRef]
- Yoon, T.P. Photochemical Stereocontrol Using Tandem Photoredox-Chiral Lewis Acid Catalysis. Acc. Chem. Res. 2016, 49, 2307–2315. [Google Scholar] [CrossRef]
- Zou, Y.Q.; Hrmann, F.M.; Bach, T. Iminium and enamine catalysis in enantioselective photochemical reactions. Chem. Soc. Rev. 2018, 47, 278–290. [Google Scholar] [CrossRef]
- Silvi, M.; Melchiorre, P. Enhancing the potential of enantioselective organocatalysis with light. Nature 2018, 554, 41–49. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, L.Q.; Yu, D.G.; Zhu, C.J.; Xiao, W.J. Visible light-driven organic photochemical synthesis in China. Sci. China Chem. 2019, 62, 24–57. [Google Scholar] [CrossRef]
- Aratani, T. Cyclopropanation. In Comprehensive Catalysis III; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin, Germany, 1999. [Google Scholar]
- Lebel, H.; Marcoux, J.F.; Molinaro, C.; Charette, A.B. Stereoselective Cyclopropanation Reactions. Chem. Rev. 2003, 103, 977–1050. [Google Scholar] [CrossRef] [PubMed]
- Pfaltz, A.; Lydon, K.M.; McKervey, M.A.; Charette, A.B.; Lebel, H. Cyclopropanation and C-H Insertion Reactions. In Comprehensive Catalysis II; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin, Germany, 1999; Chapter 16. [Google Scholar]
- Ito, Y.N.; Katsuki, T. Asymmetric Catalysis of New Generation Chiral Metallosalen Complexes. Bull. Chem. Soc. Jpn. 1999, 72, 603–619. [Google Scholar] [CrossRef]
- Liu, X.; Manzur, C.; Novoa, N.; Celedon, S.; Carrillo, D.; Hamon, J.R. Multidentate unsymmetrically-substituted Schiff bases and their metal complexes: Synthesis, functional materials properties, and applications to catalysis. Coord. Chem. Rev. 2018, 357, 144–172. [Google Scholar] [CrossRef]
- Liu, X.; Hamon, J.R. Recent developments in penta-, hexa-and heptadentate Schiff base ligands and their metal complexes. Coord. Chem. Rev. 2019, 389, 94–118. [Google Scholar] [CrossRef]
- Uchida, T.; Irie, R.; Katsuki, T. Chiral (ON)Ru-Salen Catalyzed Cyclopropanation: High Cis- and Enantioselectivity. Synlett 1999, 10, 1163–1165. [Google Scholar] [CrossRef]
- Uchida, T.; Irie, R.; Katsuki, T. Highly cis- and Enantioface-Selective Cyclpropanation Using (R,R)-Ru-Salen Complex: Solubility Dependent Enantioface Selection. Synlett 1999, 10, 1793–1795. [Google Scholar] [CrossRef]
- Uchida, T.; Irie, R.; Katsuki, T. cis- and Enantio-selective Cyclopropanation with Chiral (ON+)Ru-Salen Complex as a Catalyst. Tetrahedron 2000, 56, 3501–3509. [Google Scholar] [CrossRef]
- Saha, B.; Uchida, T.; Katsuki, T. Intramolecular Asymmetric Cyclopropanation with (Nitroso)(salen)-ruthenium(II) Complexes as Catalysts. Synlett 2001, 12, 114–116. [Google Scholar] [CrossRef]
- Saha, B.; Uchida, T.; Katsuki, T. Highly Enantioselective Intramolecular Cyclopropanation of Alkenyl Diazo Ketones Using Ru(salen) as Catalyst. Chem. Lett. 2002, 31, 846–847. [Google Scholar] [CrossRef]
- Doyle, M.P.; Eismont, M.Y.; Zhou, Q.L. Enantiocontrol in intramolecular cyclopropanation of diazoketones. Conformational control of metal carbene alignment. Russ. Chem. Bull. 1997, 46, 955–958. [Google Scholar] [CrossRef]
- Müller, P.; Maitreiean, E. Rhodium(II)- and Copper(I)-Catalyzed Intramolecular Carbon-Hydrogen Bond Insertions with Metal Carbenoids Derived from Diazo Ketones. Collect. Czech. Chem. Commun. 1999, 64, 1807–1826. [Google Scholar] [CrossRef]
- Kirmse, W. 100 Years of the Wolff Rearrangement. Eur. J. Org. Chem. 2002, 2002, 2193–2256. [Google Scholar] [CrossRef]
- Yang, H.; Foster, K.; Stephenson, C.R.J.; Brown, W.; Roberts, E. Asymmetric Wolff Rearrangement. Reactions with α-Alkylated-α-diazoketones: Stereoselective Synthesis of α-Substituted-β-amino Acid Derivatives. Org. Lett. 2000, 2, 2177–2179. [Google Scholar] [CrossRef] [PubMed]
- Pinho, V.D.; Gutmann, B.; Kappe, C.O. Continuous flow synthesis of β-amino acids from α-amino acids via Arndt-Eistert homologation. RSC Adv. 2014, 4, 37419–37422. [Google Scholar] [CrossRef]
- Pinho, V.D.; Burtoloso, A.C.B. Total synthesis of (-)-indolizidine 167B via an unusual Wolff rearrangement from an α,β-unsaturated diazoketone. Tetrahedron Lett. 2012, 53, 876–878. [Google Scholar] [CrossRef]
- Bernardim, B.; Hardman-Baldwin, A.M.; Burtoloso, A.C.B. α,β-Unsaturated Diazoketones as Platforms in the Asymmetric Synthesis of Hydroxylated Alkaloids. Total Synthesis of 1-Deoxy-8,8a-diepicastanospermine and 1,6-Dideoxyepicastanospermine and Formal Synthesis of Pumiliotoxin 251D. J. Org. Chem. 2012, 77, 9926–9931. [Google Scholar] [CrossRef] [PubMed]
- Bernardim, B.; Hardman-Baldwin, A.M.; Burtoloso, A.C.B. LED lighting as a simple, inexpensive, and sustainable alternative for Wolff rearrangements. RSC Adv. 2015, 5, 13311–13314. [Google Scholar] [CrossRef]
- Zhang, W.; Romo, D. Transformation of Fused Bicyclic and Tricyclic β-Lactones to Fused γ-Lactones and 3(2H)-Furanones via Ring Expansions and O-H Insertions. J. Org. Chem. 2007, 72, 8939–8942. [Google Scholar] [CrossRef]
- Vaske, Y.S.M.; Mahoney, M.E.; Konopelski, J.P.; Rogow, D.L.; McDonald, W.J. Enantiomerically Pure trans-β-Lactams from α-Amino Acids via Compact Fluorescent Light (CFL) Continuous-Flow Photolysis. J. Am. Chem. Soc. 2010, 132, 11379–11385. [Google Scholar] [CrossRef]
- Gerstenberger, B.S.; Lin, J.; Mimieux, Y.S.; Brown, L.E.; Oliver, A.G.; Konopelski, J.P. Structural Characterization of an Enantiomerically Pure Amino Acid Imidazolide and Direct Formation of the β-Lactam Nucleus from an α-Amino Acid. Org. Lett. 2008, 10, 369–372. [Google Scholar] [CrossRef]
- Dong, C.; Mo, F.; Wang, J. Highly Diastereoselective Addition of the Lithium Enolate of α-Diazoacetoacetate to N-Sulfinyl Imines: Enantioselective Synthesis of 2-Oxo and 3-Oxo Pyrrolidines. J. Org. Chem. 2008, 73, 1971–1974. [Google Scholar] [CrossRef]
- Mo, F.; Li, F.; Qiu, D.; Wang, J. Enantioselective synthesis of condensed and transannular ring skeletons containing pyrrolidine moiety. Tetrahedron 2010, 66, 1274–1279. [Google Scholar] [CrossRef]
- Mo, F.; Li, F.; Qiu, D.; Zhang, Y.; Wang, J. Studies toward the Synthesis of (R)-(+)-Harmicine. Chin. J. Chem. 2012, 30, 2297–2302. [Google Scholar] [CrossRef]
- France, S.; Wack, H.; Taggi, A.E.; Hafez, A.M.; Wagerle, T.R.; Shah, M.H.; Dusich, C.L.; Lectka, T. Catalytic, Asymmetric α-Chlorination of Acid Halides. J. Am. Chem. Soc. 2004, 126, 4245–4255. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ding, W.; Zhou, Q.Q.; Wei, Y.; Lu, L.Q.; Xiao, W.J. Catalyst-Controlled Regioselective Acylation of β-Ketoesters with α-Diazo Ketones Induced by Visible Light. Org. Lett. 2018, 20, 7278–7282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Koreeda, M. Total Synthesis of (+)-Acanthodoral by the Use of a Pd-Catalyzed Metal-ene Reaction and a Nonreductive 5-exo-Acyl Radical Cyclization. Org. Lett. 2004, 6, 537–540. [Google Scholar] [CrossRef]
- Snyder, S.A.; Corey, E.J. Concise Total Syntheses of Palominol, Dolabellatrienone, β-Araneosene, and Isoedunol via an Enantioselective Diels-Alder Macrobicyclization. J. Am. Chem. Soc. 2006, 128, 740–742. [Google Scholar] [CrossRef]
- Chapman, L.M.; Beck, J.C.; Wu, L.; Reisman, S.E. Enantioselective Total Synthesis of (+)-Psiguadial B. J. Am. Chem. Soc. 2016, 138, 9803–9806. [Google Scholar] [CrossRef] [Green Version]
- Chapman, L.M.; Beck, J.C.; Lacker, C.R.; Wu, L.; Reisman, S.E. Evolution of a Strategy for the Enantioselective Total Synthesis of (+)-Psiguadial B. J. Org. Chem. 2018, 83, 6066–6085. [Google Scholar] [CrossRef]
- Beck, J.C.; Lacker, C.R.; Chapman, L.M.; Reisman, S.E. A modular approach to prepare enantioenriched cyclobutanes: Synthesis of (+)-rumphellaone A. Chem. Sci. 2019, 10, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Podlech, J. Stereoselective synthesis of Aminoalkyl-Substituted β-Lactams via cycloaddition of ketenes Generated from α-Amino Acids. Synlett 1996, 7, 582–584. [Google Scholar] [CrossRef]
- Podlech, J.; Linder, M.R. Cycloadditions of Ketenes Generated in the Wolff Rearrangement. Stereoselective Synthesis of Aminoalkyl-Substituted β-Lactams from α-Amino Acids. J. Org. Chem. 1997, 62, 5873–5883. [Google Scholar] [CrossRef]
- Podlech, J.; Steurer, S. A Chiral, Oxidatively Cleavable Auxiliary in β-Lactam Synthesis-Double Diastereocontrol with p-Methoxyphenethyl-Substituted Imines. Synthesis 1999, 31, 650–654. [Google Scholar] [CrossRef]
- Linder, M.R.; Podlech, J. Synthesis of β-Lactams from Diazoketones and Imines: The Use of Microwave Irradiation. Org. Lett. 2001, 3, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.R.; Podlech, J. Synthesis of Peptidomimetics Containing a β-Lactam Moiety Using Peptidic Diazoketones and Imines in a Staudinger Reaction. Org. Lett. 1999, 1, 869–871. [Google Scholar] [CrossRef]
- Linder, M.R.; Frey, W.U.; Podlech, J. Diazoketones as precursors in β-lactam synthesis. New insights into the mechanism of the photochemically induced Staudinger reaction. J. Chem. Soc. Perkin Trans. 1 2001, 1, 2566–2577. [Google Scholar] [CrossRef]
- Li, M.M.; Wei, Y.; Liu, J.; Chen, H.W.; Lu, L.Q.; Xiao, W.J. Sequential Visible-Light Photoactivation and Palladium Catalysis Enabling Enantioselective [4+2] Cycloadditions. J. Am. Chem. Soc. 2017, 139, 14707–14713. [Google Scholar] [CrossRef]
- Li, T.R.; Wang, Y.N.; Xiao, W.J.; Lu, L.Q. Transition-metal-catalyzed cyclization reactions using vinyl and ethynyl benzoxazinones as dipole precursors. Tetrahedron Lett. 2018, 59, 1521–1530. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Xiao, W.J. Catalytic Asymmetric Synthesis of Chiral Dihydrobenzofurans through a Formal [4+1] Annulation Reaction of Sulfur Ylides and In Situ Generated ortho-Quinone Methides. Eur. J. Org. Chem. 2017, 2017, 233–236. [Google Scholar]
- Wei, Y.; Liu, S.; Li, M.M.; Li, Y.; Lan, Y.; Lu, L.Q.; Xiao, W.J. Enantioselective Trapping of Pd-Containing 1,5-Dipoles by Photogenerated Ketenes: Access to 7-Membered Lactones Bearing Chiral Quaternary Stereocenters. J. Am. Chem. Soc. 2019, 141, 133–137. [Google Scholar] [CrossRef]
- Liu, J.; Li, M.M.; Qu, B.L.; Lu, L.Q.; Xiao, W.J. Photoinduced Wolff rearrangement/Pd-catalyzed [3+2] cycloaddition sequence: An unexpected route to tetrahydrofurans. Chem. Commun. 2019, 55, 2031–2034. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Webster, R.D.; Harms, K.; Meggers, E. Asymmetric Catalysis with Organic Azides and Diazo Compounds Initiated by Photoinduced Electron Transfer. J. Am. Chem. Soc. 2016, 138, 12636–12642. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, X.; Meggers, E.; Houk, K.N. Origins of Enantioselectivity in Asymmetric Radical Additions to Octahedral Chiral-at-Rhodium Enolates: A Computational Study. J. Am. Chem. Soc. 2017, 139, 17902–17907. [Google Scholar] [CrossRef] [PubMed]





























© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, T.-B.; Yang, Q.-Q.; Zou, Y.-Q. Recent Advances in Enantioselective Photochemical Reactions of Stabilized Diazo Compounds. Molecules 2019, 24, 3191. https://doi.org/10.3390/molecules24173191
Hua T-B, Yang Q-Q, Zou Y-Q. Recent Advances in Enantioselective Photochemical Reactions of Stabilized Diazo Compounds. Molecules. 2019; 24(17):3191. https://doi.org/10.3390/molecules24173191
Chicago/Turabian StyleHua, Ting-Bi, Qing-Qing Yang, and You-Quan Zou. 2019. "Recent Advances in Enantioselective Photochemical Reactions of Stabilized Diazo Compounds" Molecules 24, no. 17: 3191. https://doi.org/10.3390/molecules24173191