A Ligand-Based Virtual Screening Method Using Direct Quantification of Generalization Ability


Abstract
:
1. Introduction
2. Results
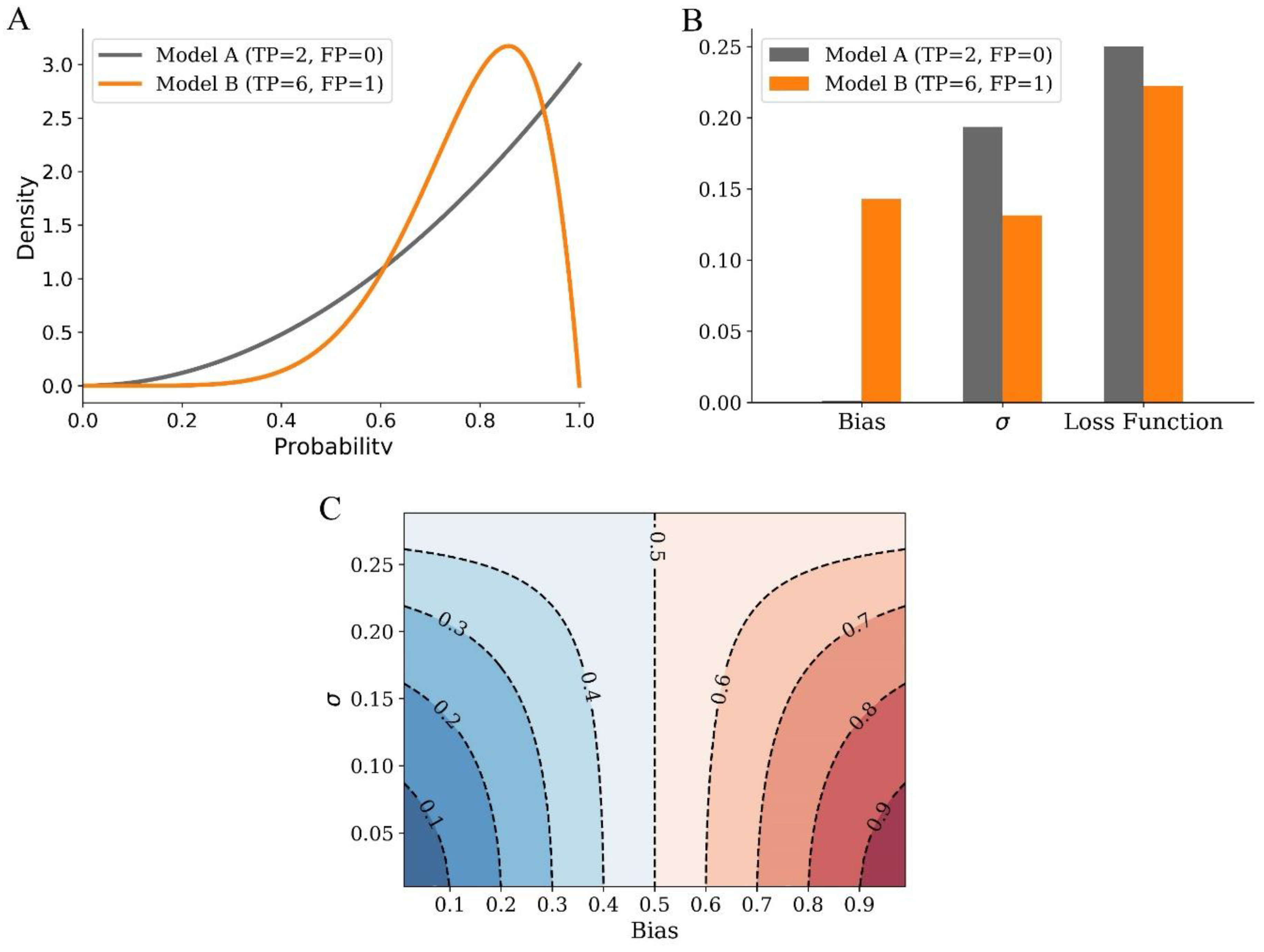
2.1. Bias and Variance Analysis of LBS
2.2. A Simulation Study of LBS
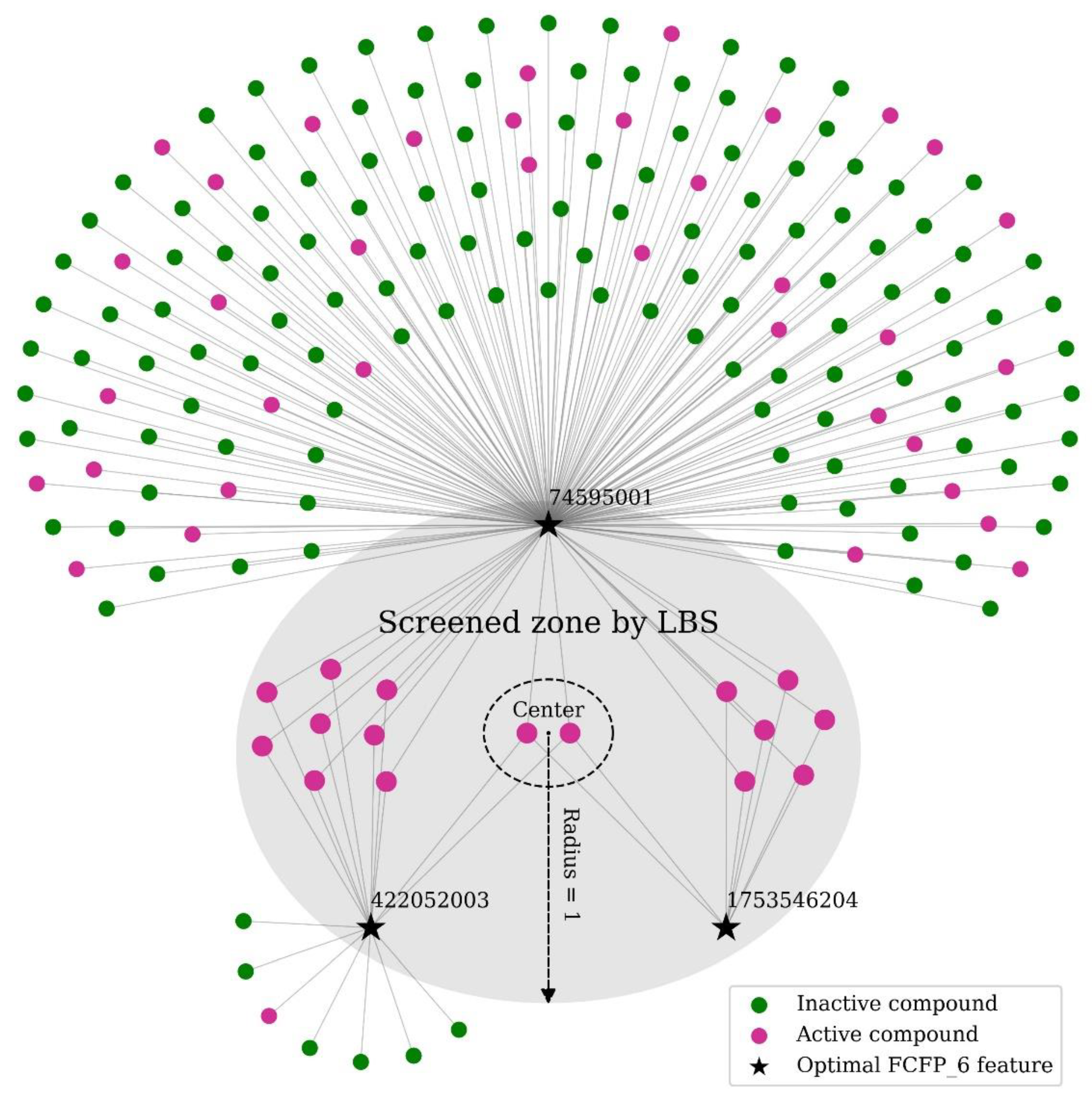
2.3. Application of LBS for Compound Screening in Real Datasets
3. Discussion
4. Materials and Methods
4.1. Local Beta Screening
4.2. Algorithm Implementation
4.3. Datasets
4.4. Molecular Docking
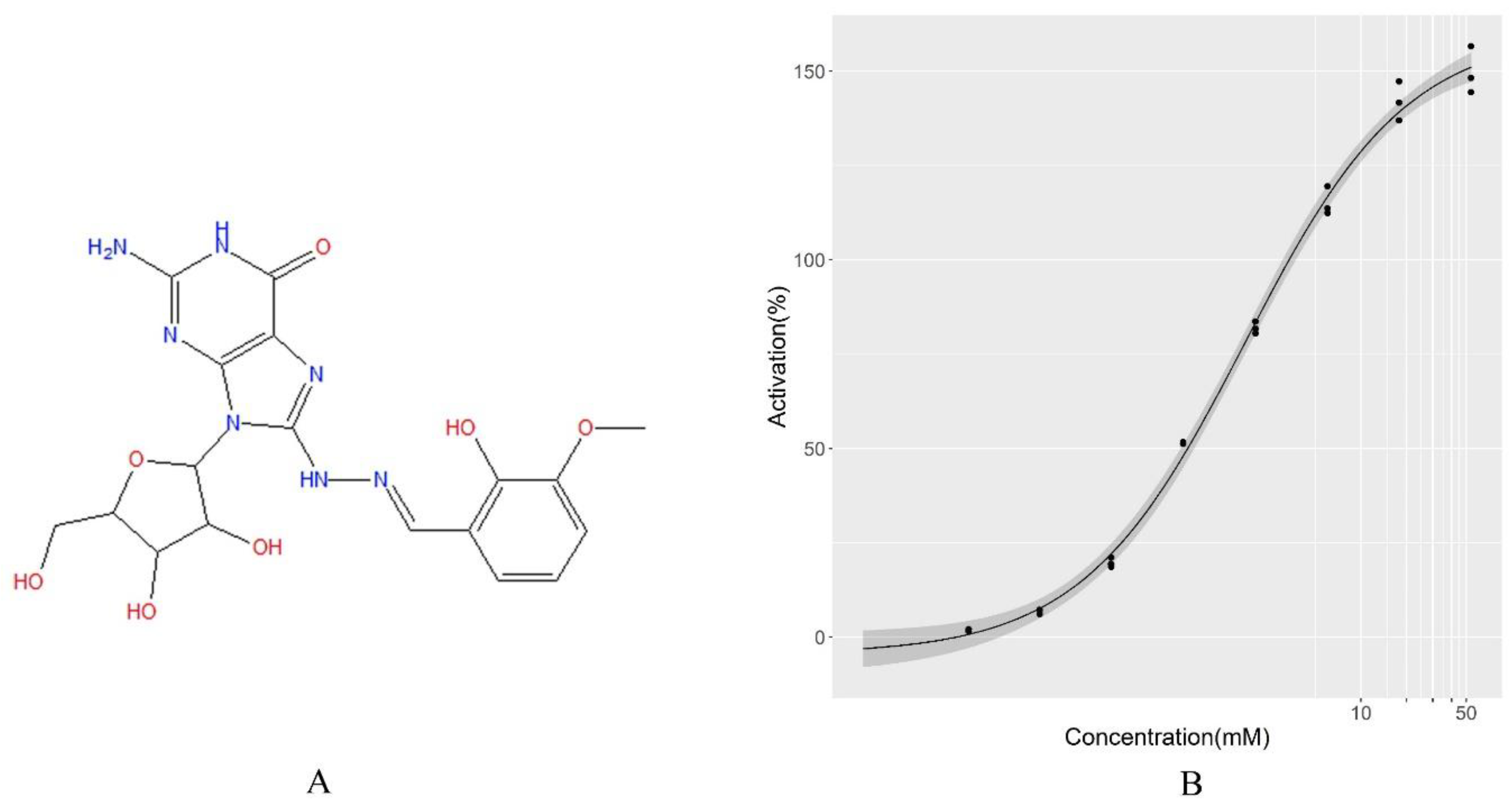
4.5. Experimental Validation of the Screening Results by LBS
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Bajorath, J. Integration of virtual and high throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G. Virtual screening: An endless staircase. Nat. Rev. Drug Discov. 2010, 9, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.L. Virtual screening in lead discovery: A viewpoint. Molecules 2002, 7, 51–62. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. Rusting of the lock and key model for protein-ligand binding. Science 1991, 254, 954–955. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Maggiora, G.M. Concepts and Application of Molecular Similarity; John Wiley & Sons: New York, NY, USA, 1990. [Google Scholar]
- Willett, P. Enhancing the effectiveness of ligand-based virtual screening using data fusion. QSAR Comb. Sci. 2006, 25, 1143–1152. [Google Scholar] [CrossRef]
- Hert, J.; Willett, P.; Wilton, D.J. New methods for ligand-based virtual screening: Use of data fusion and machine learning to enhance the effectiveness of similarity searching. J. Chem. Inf. Model 2006, 46, 462–470. [Google Scholar] [CrossRef]
- Rish, I. An empirical study of the naïve bayes classifier. IJCAI 2001, 3, 41–46. [Google Scholar]
- Cover, T.M.; Hart, P. –neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Vapnik, V.N. The Nature of Statistical Learning Theory; Springer: New York, NY, USA, 1995. [Google Scholar]
- Ho, T.K. Random decision forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; pp. 278–282. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Jasial, W.; Gilberg, E.; Blaschke, T.; Bajorath, J. Machine learning distinguishes with high accuracy between pan assay interference compounds that are promiscuous or represent. J. Med. Chem. 2018, 61, 10255–10264. [Google Scholar] [CrossRef] [PubMed]
- Merget, B.; Turk, S.; Eid, S.; Rippmann, F.; Fulle, S. Profiling prediction of kinase inhibitors: Toward the virtual assay. J. Med. Chem. 2017, 60, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Efron, B.; Gong, G. A leisurely look at the bootstrap, the jackknife, and cross-validation. Am. Stat. 1983, 37, 36–48. [Google Scholar]
- Kim, C.; Cheon, M.; Kang, M.; Chang, I. A simple and exact laplacian clustering of complex networking phenomena: application to gene expression profiles. Proc. Natl. Acad. Sci. USA 2008, 105, 4083–4087. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Varian, H.R. Bootstrap tutorial. Math. J. 2005, 9, 768–775. [Google Scholar]
- Anderssen, E.; Dyrstad, K.; Westad, F.; Martens, H. Reducing over-optimism in variable selection by cross-model validation. Chemometr. Intell. Lab. 2006, 84, 69–74. [Google Scholar] [CrossRef]
- Efron, B. Estimating the error rate of a prediction rule: Improvement on cross-validation. J. Am. Stat. Assoc. 1983, 78, 316–331. [Google Scholar] [CrossRef]
- Bradley, D. Dealing with a data dilemma. Nat. Rev. Drug Discov. 2008, 7, 632–633. [Google Scholar]
- Trunk, G.V. A problem of dimensionality: A simple example. IEEE T. Pattern Anal. 1979, 3, 306–307. [Google Scholar] [CrossRef]
- Yin, L.Z.; Ge, Y.; Xiao, K.L.; Wang, X.H.; Quan, X.J. Feature selection for high-dimensional imbalanced data. Neurocomputing 2013, 105, 3–11. [Google Scholar] [CrossRef]
- Schierz, A.C. Virtual screening of bioassay data. J. Cheminform. 2009, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhao, Y.X.; Yang, L.M.; Zheng, Y.T.; Tang, Y.; Shen, X.; Jiang, H.L. Symmetrical 1-pyrrolidineacetamide showing anti-HIV activity through a new binding site on HIV-1 integrase. Acta Pharmacol. Sin. 2008, 29, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Eidahl, J.O.; Shkriabai, N.; Zhao, Z.; McKee, C.J.; Hess, S.; Burke, T.R.; Kvaratskhelia, M. An allosteric mechanism for inhibiting HIV-1 integrase with a small molecule. Mol. Pharmacol. 2009, 76, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Shkriabai, N.; Patil, S.S.; Hess, S.; Budihas, S.R.; Craigie, R.; Burke, T.R., Jr.; Le Grice, S.F.; Kvaratskhelia, M. Identification of an inhibitor-binding site to HIV-1 integrase with affinity acetylation and mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 6894–6899. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.S.; Learned-Miller, E.; McCallum, A. Active bias: Training More Accurate Neural Networks by Emphasizing High Variance Samples. Available online: https://arxiv.org/abs/1704.07433 (accessed on 6 January 2018).
- Bengio, Y.; LeCun, Y. Scaling Learning Alogrithms towards AI. In Large-Scale Kernel Machines; MIT Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Fan, Y.; Tian, F.; Qin, T.; Bian, J. Learning What Data to Learn. Available online: https://arxiv.org/abs/1702.08635 (accessed on 28 February 2017).
- Pasumarthi, R.K.; Wang, X.; Li, C.; Bruch, S.; Bendersky, M.; Najork, M.; Pfeifer, J.; Golbandi, N.; Anil, R.; Wolf, S. TF-Ranking: Scalable tensorflow library for learning-to-rank. Available online: https://arxiv.org/abs/1812.00073 (accessed on 30 November 2018).
- Jarvelin, K.; Kekalainen, J. Cumulated gain-based evaluation of IR techniques. ACM T. Inform. Syst. 2002, 20, 422–446. [Google Scholar] [CrossRef]
- Moya, M.M.; Hush, D.R. Network constraints and multi-objective optimization for one-class classification. Neural Networks 1996, 9, 463–474. [Google Scholar] [CrossRef]
- Wold, S.; Esbensen, K.; Geladi, P. Principle component analysis. Chemometr. Intell. Lab. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Phuong, T.M.; Lin, Z.; Altman, R.B. Choosing SNPs using feature selection. J. Bioinform. Comput. Biol. 2006, 4, 241–257. [Google Scholar] [CrossRef]
- Manning, C.D.; Raghavan, P.; Schutze, H. Introduction to Information Retrieval; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM T. Intel. Syst. Tec. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- PubChem BioAssay Database. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1053131 (accessed on 30 January 2018).
- PubChem BioAssay Database. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1053171 (accessed on 30 January 2018).
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Guha, R.; Howard, M.T.; Hutchison, G.R.; Murray-Rust, P.; Rzepa, H.; Steinbeck, C.; Wegner, J.; Willighagen, E.L. The blue obelisk-interoperability in chemical informatics. J. Chem. Inf. Model 2006, 46, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.; Engelman, A.; et al. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J. Gamma Function and Related Functions. Handbook of Mathematical Functions with Formulas, Graphs, and Mathematical Tables; Dover Publications: New York, NY, USA, 1972; p. 258. [Google Scholar]
Sample Availability: Not Applicable. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pubchem CID | Molecular Formula | Molecular Weight | EC50 |
|---|---|---|---|
| 5291488 | C21H20O11 | 448.38 | 3.62 |
| 5378597 | C21H20O12 | 464.39 | 1.36 |
| 135427812 | C18H21N7O7 | 447.41 | 0.71 |
| 3656702 | C14H11FN2O4 | 290.25 | 4.88 |
| 4137880 | C13H18N2O7 | 314.30 | 1.26 |
| 46397461 | C19H18O12 | 438.35 | 1.26 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, W.; Guo, D. A Ligand-Based Virtual Screening Method Using Direct Quantification of Generalization Ability. Molecules 2019, 24, 2414. https://doi.org/10.3390/molecules24132414
Dai W, Guo D. A Ligand-Based Virtual Screening Method Using Direct Quantification of Generalization Ability. Molecules. 2019; 24(13):2414. https://doi.org/10.3390/molecules24132414
Chicago/Turabian StyleDai, Weixing, and Dianjing Guo. 2019. "A Ligand-Based Virtual Screening Method Using Direct Quantification of Generalization Ability" Molecules 24, no. 13: 2414. https://doi.org/10.3390/molecules24132414