Allylation of Orthoquinones Towards Annulated Polycyclic Aromatic Systems

Abstract
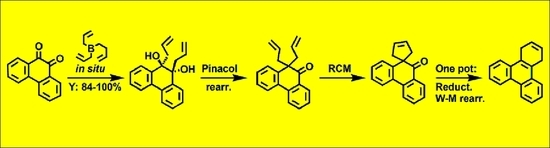
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of 1,2-Diols via Addition of Allyl Substituents to Carbonyl Groups in Orthoquinones
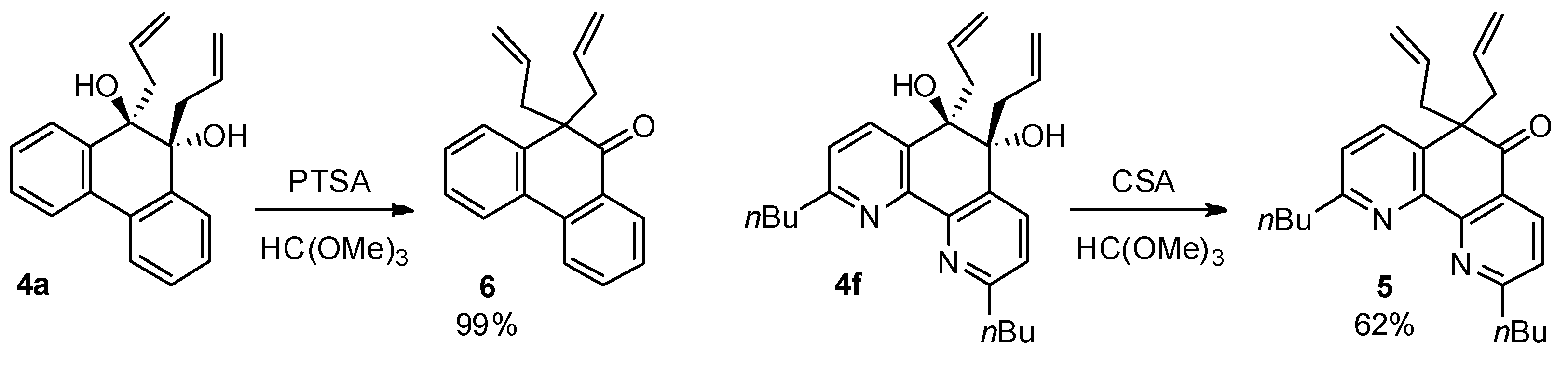
2.2. Pinacol Rearrangement of 1,2-Diol to α,α-Diallyl Ketone
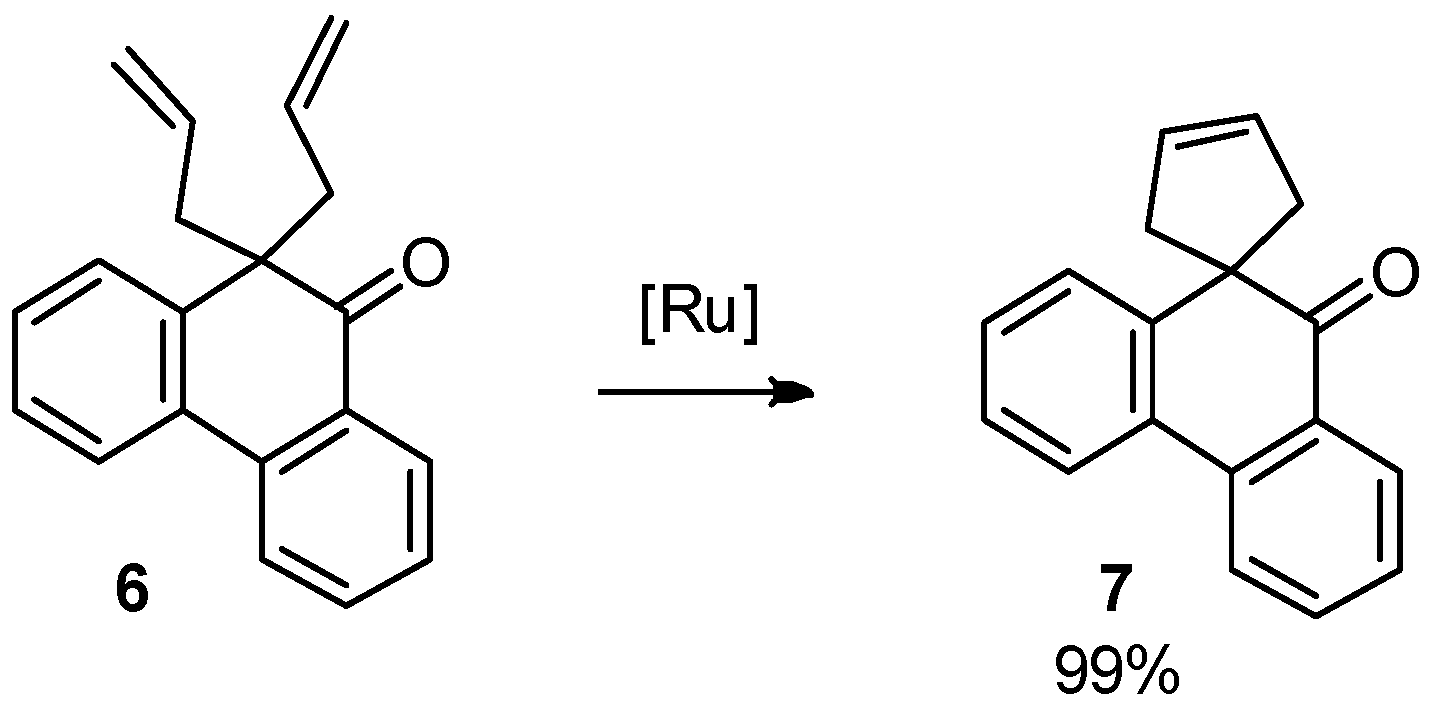
2.3. Ring Closing Metathesis (RCM)
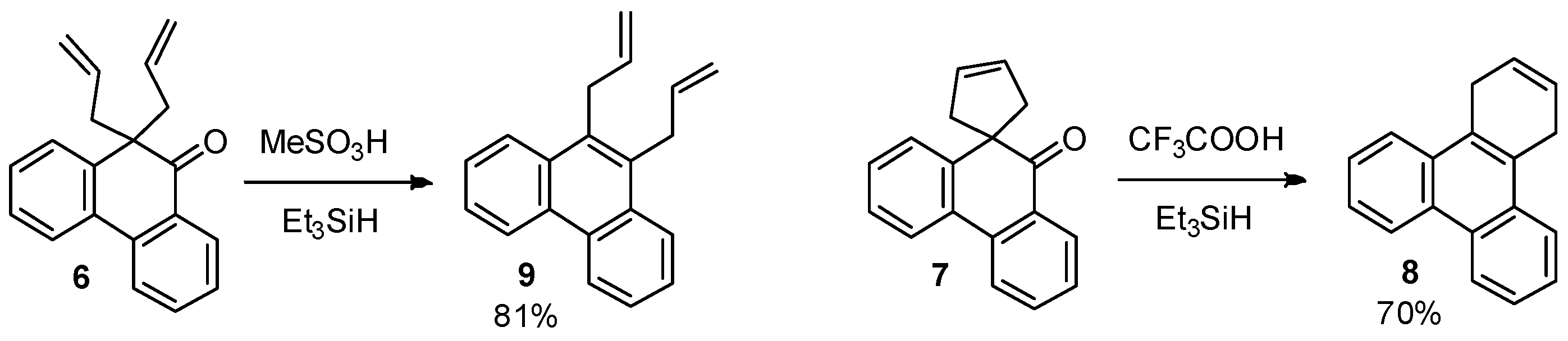
2.4. One-pot Reduction of Keto Group and Wagner-Meerwein Rearrangement
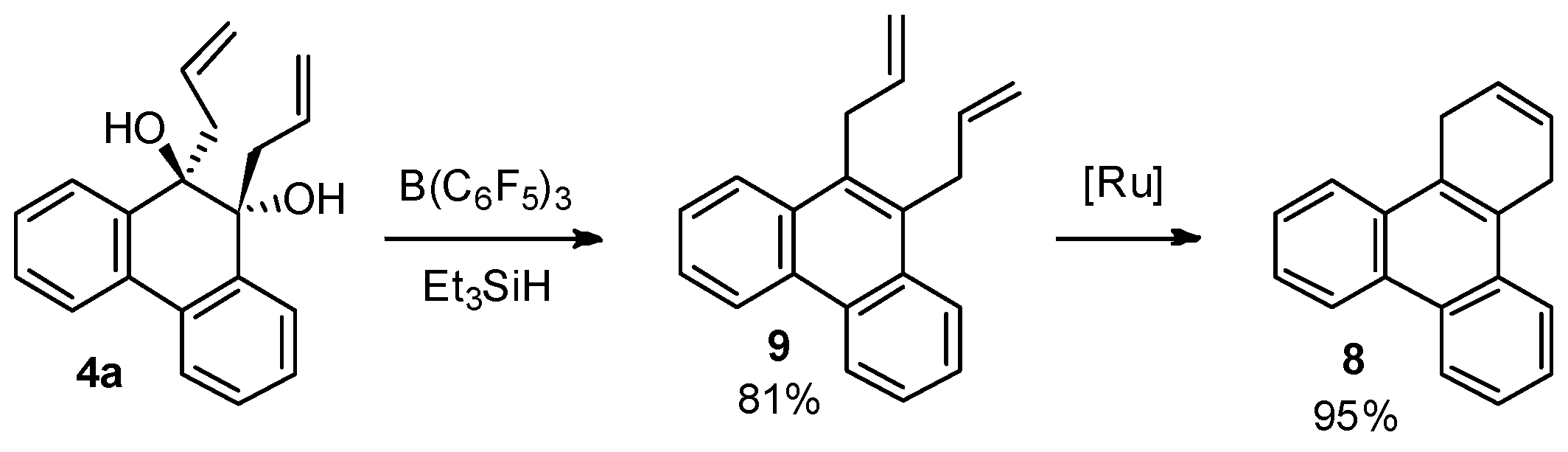
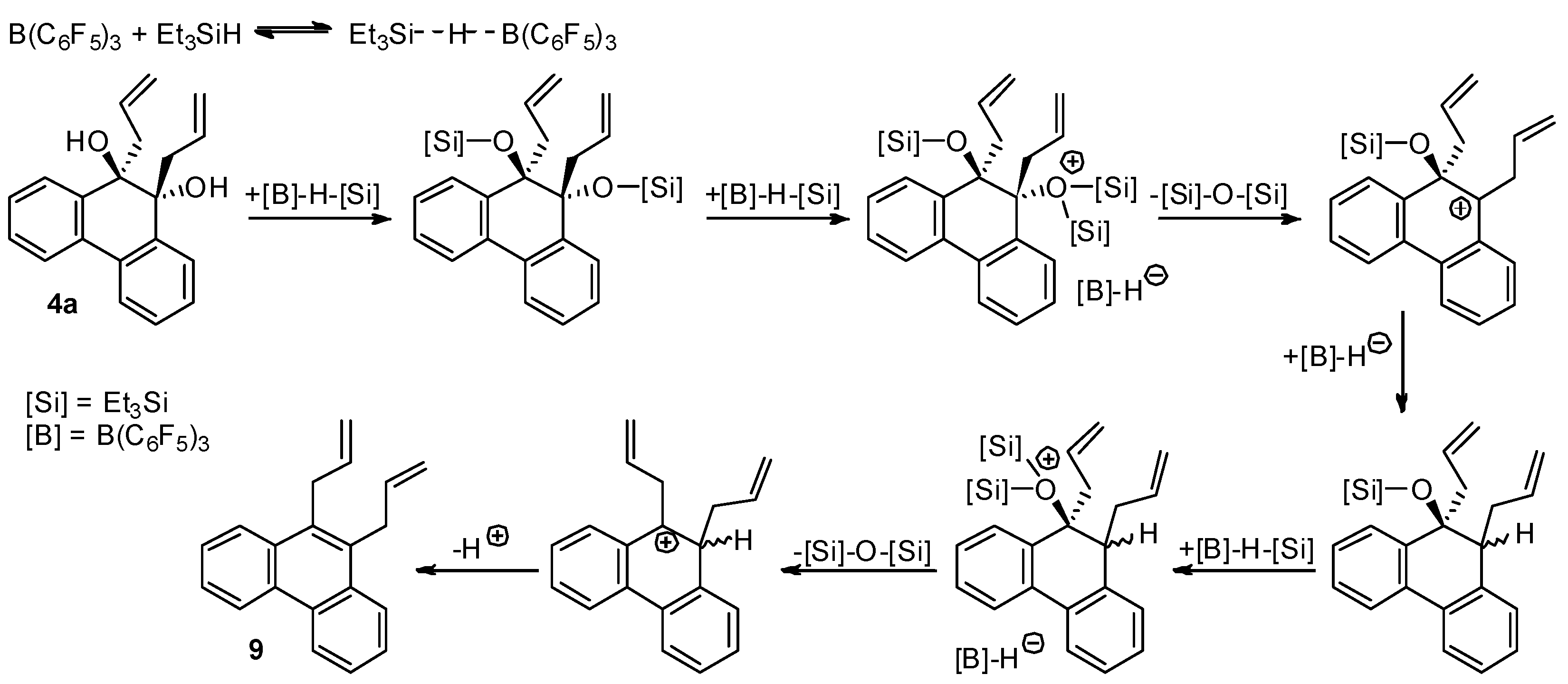
2.5. Direct Dehydroxylation of 9,10-Diallyl-9,10-dihydrophenanthrene-9,10-diol (4a) to 9,10-Diallylphenanthrene (9)
3. Materials and Methods
3.1. General Methods
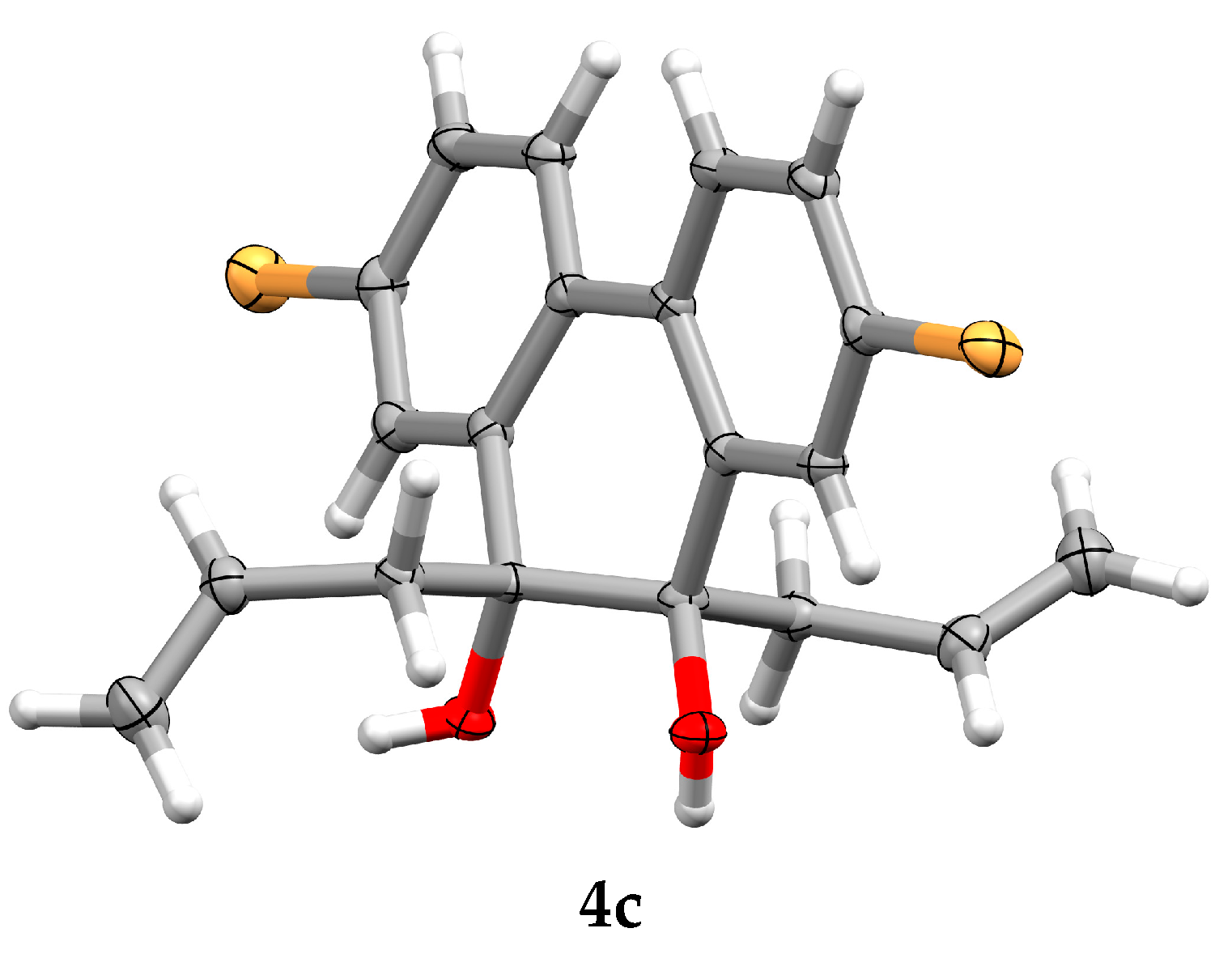
3.2. General Procedure for In Situ Triallylborane Generation and Their Reaction with the Quinone Carbonyl Groups
3.3. Pinacol Rearrangement of 9,10-Diallyl-9,10-dihydrophenanthrene-9,10-diol (4a)
3.4. Pinacol Rearrangement of 5,6-Diallyl-2,9-di-n-butyl-5,6-dihydro-1,10-phenanthroline-5,6-diol (4f)
3.5. Ring Closing Metathesis of 10,10-Diallylphenanthren-9(10H)-one (6)
3.6. One-Pot Reduction and Wagner-Meerwein Rearrangement of 10,10-Diallylphenanthren-9(10H)-one (6)
3.7. One-Pot Reduction and Wagner-Meerwein Rearrangement of 10′H-Spiro[cyclopent[3]ene-1,9′-phenanthren]-10′-one (7)
3.8. Direct Dehydroxylation of 9,10-Diallyl-9,10-dihydrophenanthrene-9,10-diol (4a) to 9,10-Diallylphenanthrene (9)
3.9. Ring Closing Metathesis of 9,10-Diallylphenanthrene (9)
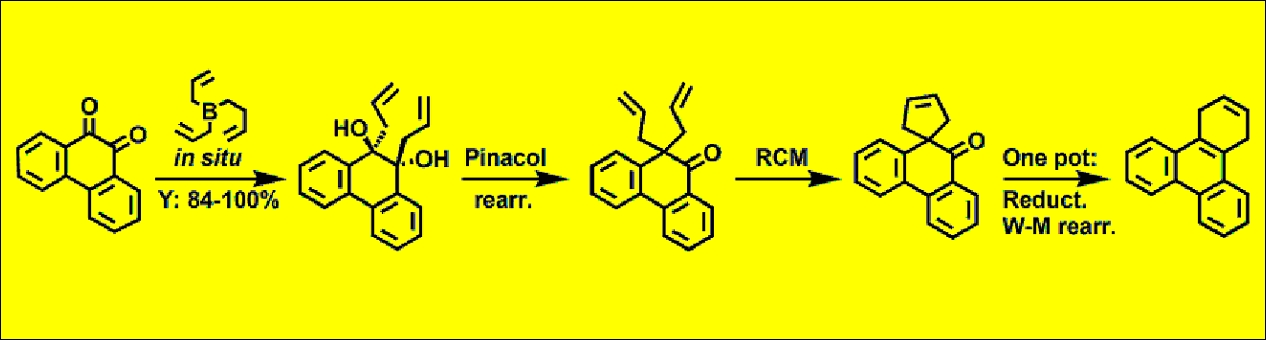
3.10. X-ray Diffraction Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nielsen, C.B.; Holliday, S.; Chen, H.Y.; Cryer, S.J.; McCulloch, I. Non-Fullerene Electron Acceptors for Use in Organic Solar Cells. Acc. Chem. Res. 2015, 48, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.S. (Ed.) Organic Nanophotonics, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2015; ISBN 78-3-662-45082-6. [Google Scholar]
- Miao, Q. (Ed.) Polycyclic Arenes and Heteroarenes: Synthesis, Properties, and Applications, 1st ed.; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2016; ISBN 9783527338474. [Google Scholar] [CrossRef]
- Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117, 3479–3716. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Pisula, W.; Müllen, K. Large polycyclic aromatic hydrocarbons: Synthesis and discotic organization. Pure Appl. Chem. 2009, 81, 2203–2224. [Google Scholar] [CrossRef]
- Grzybowski, M.; Skonieczny, K.; Butenschön, H.; Gryko, D.T. Comparison of Oxidative Aromatic Coupling and the Scholl Reaction. Angew. Chem. Int. Ed. 2013, 52, 9900–9930. [Google Scholar] [CrossRef] [PubMed]
- Kawasumi, K.; Zhang, Q.; Segawa, Y.; Scott, L.T.; Itami, K. A grossly warped nanographene and the consequences of multiple odd-membered-ring defects. Nat. Chem. 2013, 5, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Narita, A.; Wang, X.Y.; Fengb, X.; Müllen, K. New advances in nanographene chemistry. Chem. Soc. Rev. 2015, 44, 6616–6643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhard, D.; Rominger, F.; Mastalerz, M. Synthesis of Triphenylene-Based Triptycenes via Suzuki–Miyaura Cross-Coupling and Subsequent Scholl Reaction. J. Org. Chem. 2015, 80, 9342–9348. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, T.; Segawa, Y.; Itami, K. Synthesis, Structures, and Properties of π-Extended Double Helicene: A Combination of Planar and Nonplanar π-Systems. J. Am. Chem. Soc. 2015, 137, 7763–7768. [Google Scholar] [CrossRef] [PubMed]
- Little, M.S.; Yeates, S.G.; Alwattar, A.A.; Heard, K.W.J.; Raftery, J.; Edwards, A.C.; Parry, A.V.S.; Quayle, P. Insights into the Scholl Coupling Reaction: A Key Transformation of Relevance to the Synthesis of Graphenes and Related Systems. Eur. J. Org. Chem. 2017, 1694–1703. [Google Scholar] [CrossRef]
- Nishihara, Y.; Suetsugu, M.; Saito, D.; Kinoshita, M.; Iwasaki, M. Synthesis of Cyclic 1-Alkenylboronates via Zr-Mediated Double Functionalization of Alkynylboronates and Sequential Ru-Catalyzed Ring-Closing Olefin Metathesis. Org. Lett. 2013, 15, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Shahlai, K.; Hart, H. A method for the synthesis of angular iptycenes. J. Org. Chem. 1989, 54, 2615–2620. [Google Scholar] [CrossRef]
- Velikorodov, A.V.; Babaitsev, D.D.; Mochalin, V.B. Some Heterocyclization Reactions of N,N′-Dimethoxycarbonyl-o-benzoquinone Diimine. Russ. J. Org. Chem. 2003, 39, 1200–1201. [Google Scholar] [CrossRef]
- Blomberg, C.; Grootveld, H.H.; Gerner, T.H.; Bickelhaupt, F. Radical formation during reactions of Grignard reagents with quinones. J. Organomet. Chem. 1970, 24, 549–553. [Google Scholar] [CrossRef]
- Wege, D. Abnormal addition of vinylmagnesium bromide to 9,10-phenanthraquinone. Aust. J. Chem. 1971, 24, 1531–1535. [Google Scholar] [CrossRef]
- Dallacker, F.; Leidig, H. Darstellung des 4,6,7,9-Tetramethoxy-3,10-perylenchinons. Chem. Ber. 1979, 112, 2672–2679. [Google Scholar] [CrossRef]
- Mikhailov, B.M. Methods of Synthesis and Properties of Allylboranes. Russ. Chem. Rev. 1976, 45, 557–572. [Google Scholar] [CrossRef]
- Winternitz, P.F.; Carotti, A.A. The Thermal Decomposition of Trialkylboranes. J. Am. Chem. Soc. 1960, 82, 2430–2433. [Google Scholar] [CrossRef]
- Schröder, S.; Thiele, K.H. Koordinationsverbindungen von Bortrialkylen. II. Zur Komplexbildung zwischen Bortriallylen und tertiären Aminen. Z. Anorg. Allg. Chem. 1977, 428, 225–230. [Google Scholar] [CrossRef]
- Cole, T.E.; Quintanilla, R.; Rodewald, S. A Simple and Convenient Method for Synthesizing Trialkoxyboranes and Trialkoxyboroxins Using Calcium Hydride as a Drying Agent. Synth. React. Inorg. Met. Org. Chem. 1990, 20, 55–63. [Google Scholar] [CrossRef]
- Mikhailov, B.M.; Vaver, V.A. Organoboron compounds. Communication 56. Synthesis of trialkylborines from metaboric esters and their conversion into dialkylborinic esters. Bull. Acad. Sci. USRR Div. Chem. Sci. 1960, 9, 796–799. [Google Scholar] [CrossRef]
- Kita, Y.; Yoshida, Y.; Mihara, S.; Fang, D.F.; Higuchi, K.; Furukawa, A.; Fujioka, H. Efficient pinacol rearrangement mediated by trimethyl orthoformate. Tetrahedron Lett. 1997, 38, 8315–8318. [Google Scholar] [CrossRef]
- Bunton, C.A.; Hadwick, T.; Llewellyn, D.R.; Pocker, Y. 73. Tracer studies in alcohols. Part III. Intermediates in the pinacol–pinacone rearrangement. J. Chem. Soc. 1958, 403–408. [Google Scholar] [CrossRef]
- West, C.T.; Donnelly, S.J.; Kooistra, D.A.; Doyle, M.P. Silane reductions in acidic media. II. Reductions of aryl aldehydes and ketones by trialkylsilanes in trifluoroacetic acid. Selective method for converting the carbonyl group to methylene. J. Org. Chem. 1973, 38, 2675–2681. [Google Scholar] [CrossRef]
- Drosos, N.; Cheng, G.J.; Ozkal, E.; Cacherat, B.; Thiel, W.; Morandi, B. Catalytic Reductive Pinacol-Type Rearrangement of Unactivated 1,2-Diols through a Concerted, Stereoinvertive Mechanism. Angew. Chem. Int. Ed. 2017, 56, 13377–13381. [Google Scholar] [CrossRef] [PubMed]
- Gevorgyan, V.; Rubin, M.; Benson, S.; Liu, J.-X.; Yamamoto, Y. A Novel B(C6F5)3-Catalyzed Reduction of Alcohols and Cleavage of Aryl and Alkyl Ethers with Hydrosilanes. J. Org. Chem. 2000, 65, 6179–6186. [Google Scholar] [CrossRef] [PubMed]
- Kimuraa, M.; Fukasakaa, M.; Tamaru, Y. Palladium-Catalyzed, Triethylborane-Promoted C-Allylation of Naphthols and Benzene Polyols by Direct Use of Allyl Alcohols. Synthesis 2006, 3611–3616. [Google Scholar] [CrossRef]
- Ishi-i, T.; Yaguma, K.; Kuwahara, R.; Taguri, Y.; Mataka, S. Self-Assembling of n-Type Semiconductor. Tri(phenanthrolino)hexaazatriphenylenes with a Large Aromatic Core. Org. Lett. 2006, 8, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Brunner, K.; Dijken, A.; Börner, H.; Bastiaansen, J.J.A.M.; Kiggen, N.M.M.; Langeveld, B.M.W. Carbazole Compounds as Host Materials for Triplet Emitters in Organic Light-Emitting Diodes: Tuning the HOMO Level without Influencing the Triplet Energy in Small Molecules. J. Am. Chem. Soc. 2004, 126, 6035–6042. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, R.; Wu, Y.H.; Lin, H.C. Novel Thieno-imidazole Based Probe for Colorimetric Detection of Hg2+ and Fluorescence Turn-on Response of Zn2+. Org. Lett. 2012, 14, 2564–2567. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, D.; Harris, F.W. Ruthenium(III) Chloride Catalyzed Oxidation of Pyrene and 2,7-Disubstitued Pyrenes: An Efficient, One-Step Synthesis of Pyrene-4,5-diones and Pyrene-4,5,9,10-tetraones. J. Org. Chem. 2005, 70, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Zöphel, L.; Beckmann, D.; Enkelmann, V.; Chercka, D.; Riegera, R.; Müllen, K. Asymmetric pyrene derivatives for organic field-effect transistors. Chem. Commun. 2011, 47, 6960–6962. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-S.; Paquette, L.A. A Convenient Method for Determining the Concentration of Grignard Reagents. Synth. Commun. 1994, 24, 2503–2506. [Google Scholar] [CrossRef]
- Ansell, M.F.; Murray, R.A. The reactions of some ortho-naphthoquinones with 2,3-dimethylbutadiene. J. Chem. Soc. C 1971, 1429–1437. [Google Scholar] [CrossRef]
- Marcinow, Z.; Sygula, A.; Rabideau, P.W. Metal-ammonia reduction of triphenylene. J. Org. Chem. 1988, 53, 3603–3606. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro Software System, version 1.171. 38.43d; Rigaku Corporation: Oxford, UK, 2015. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
Sample Availability: Samples are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Product | Yield (%) b |
|---|---|---|---|
| 1 |  |  | 93 |
| 2 |  |  | 85 |
| 3 |  |  | 94 |
| 4 |  |  | 100 |
| 5 |  |  | 97 |
| 6 |  |  | 84 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kędziorek, M.; Dobrzańska, L. Allylation of Orthoquinones Towards Annulated Polycyclic Aromatic Systems. Molecules 2018, 23, 2043. https://doi.org/10.3390/molecules23082043
Kędziorek M, Dobrzańska L. Allylation of Orthoquinones Towards Annulated Polycyclic Aromatic Systems. Molecules. 2018; 23(8):2043. https://doi.org/10.3390/molecules23082043
Chicago/Turabian StyleKędziorek, Mariusz, and Liliana Dobrzańska. 2018. "Allylation of Orthoquinones Towards Annulated Polycyclic Aromatic Systems" Molecules 23, no. 8: 2043. https://doi.org/10.3390/molecules23082043