Ultrasonically Modified Amended-Cloud Point Extraction for Simultaneous Pre-Concentration of Neonicotinoid Insecticide Residues

Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of Ultrasonically Modified Amended-Cloud Point Extraction Procedure
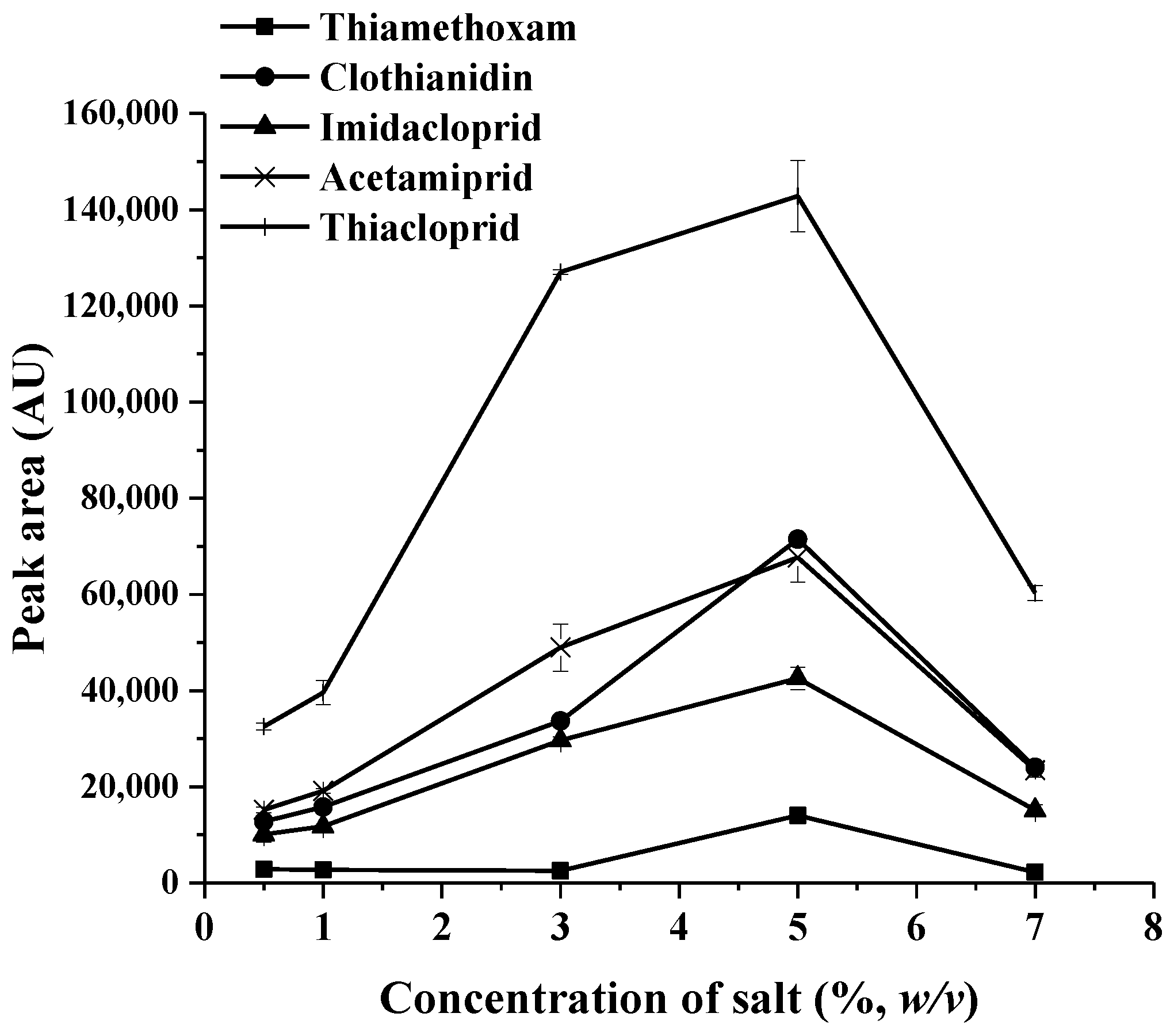
2.1.1. Effect of Salt Addition
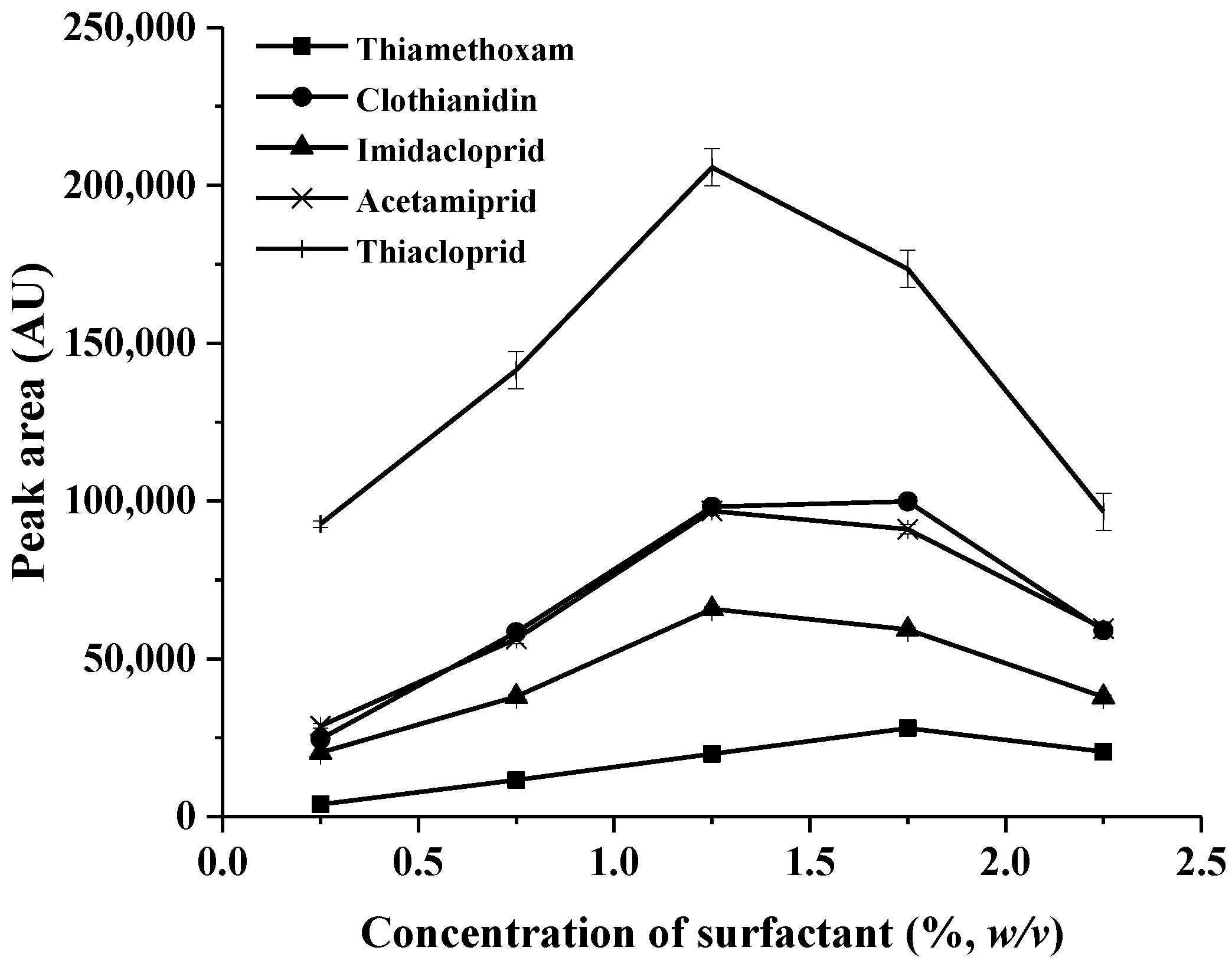
2.1.2. Effect of Concentration of TritonTM X-114
2.1.3. Effect of Kind and Concentration of Back Extraction Agent
2.1.4. Effect of Incubation Temperature and Time
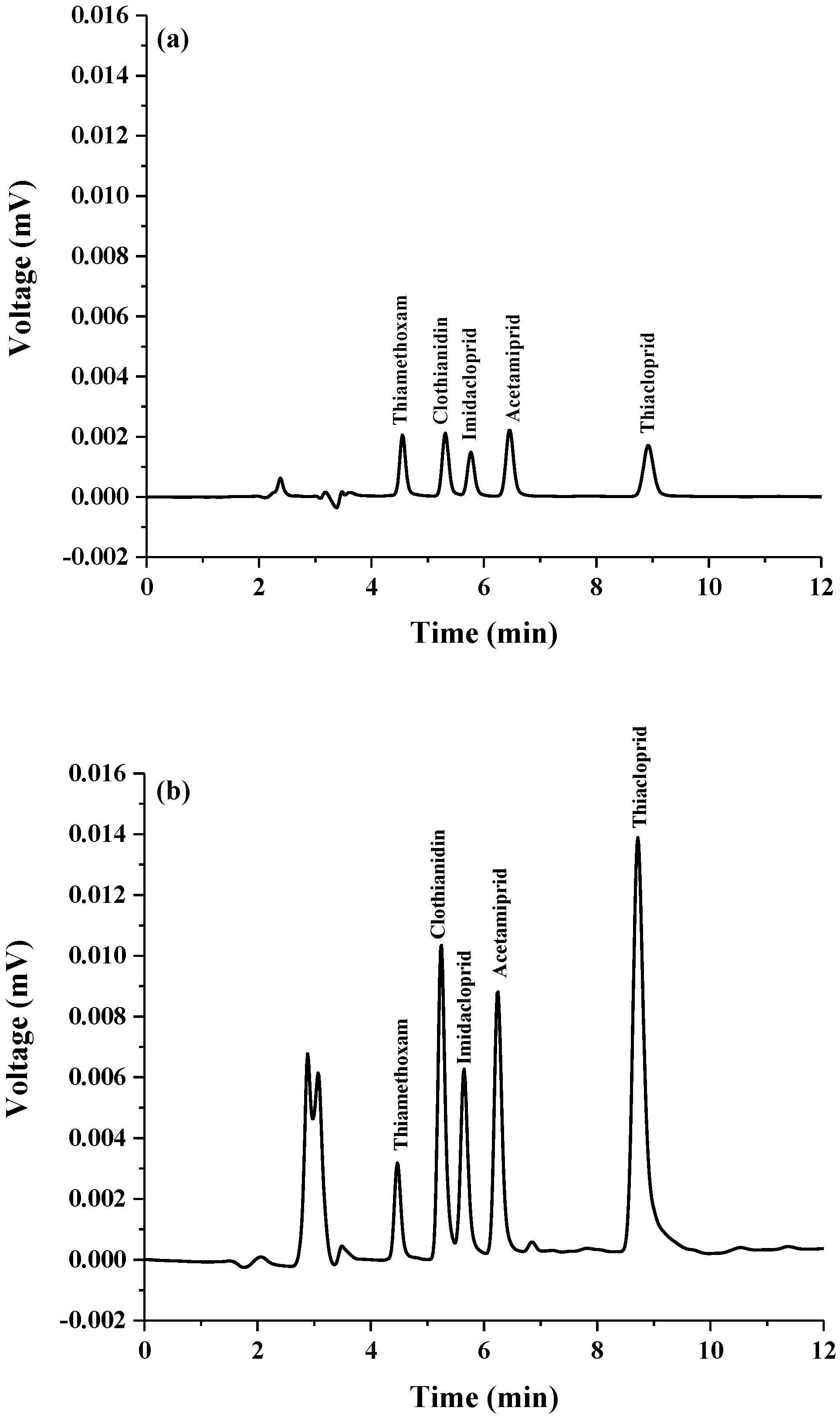
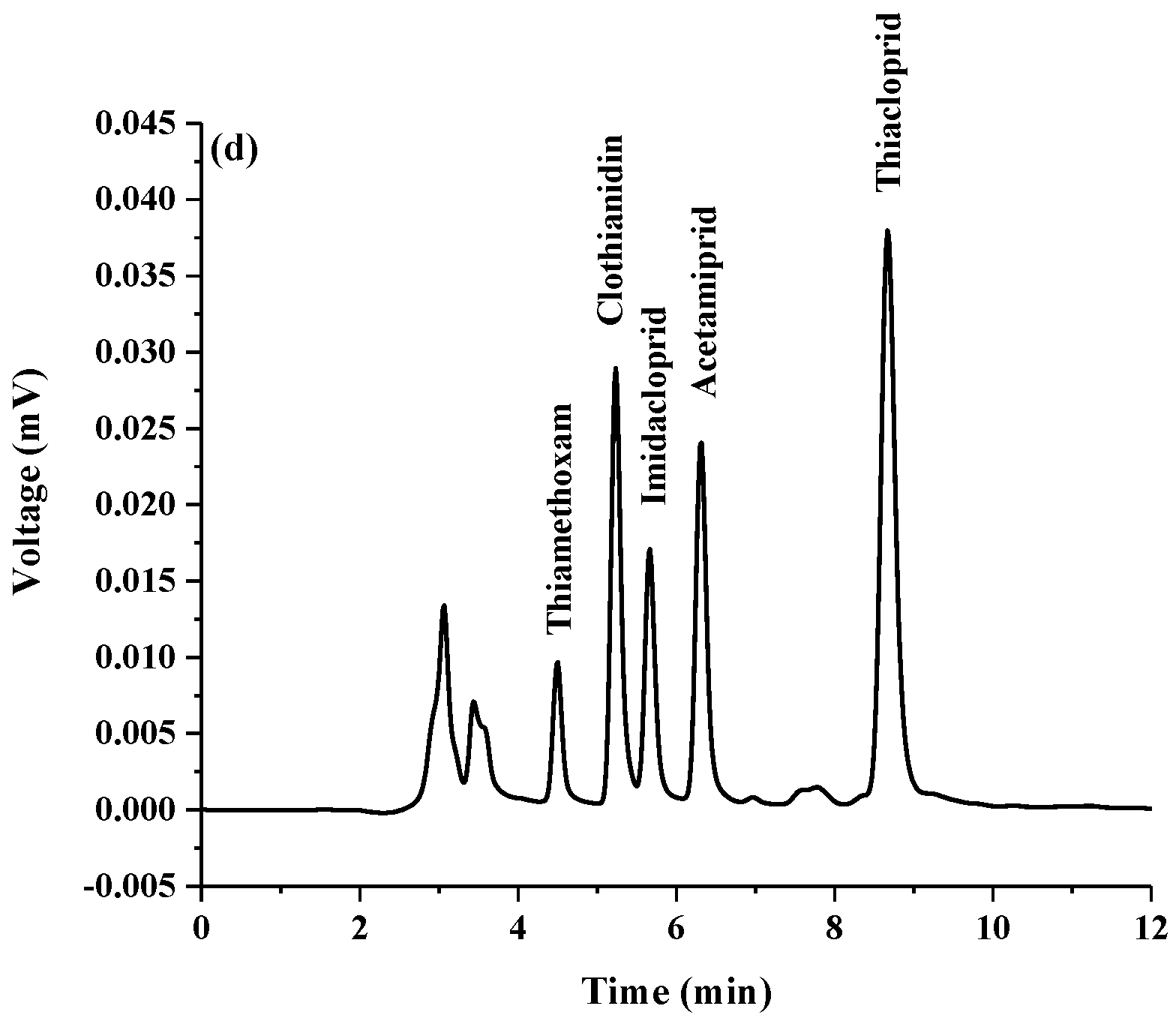
2.2. Analytical Performance of the Proposed Method
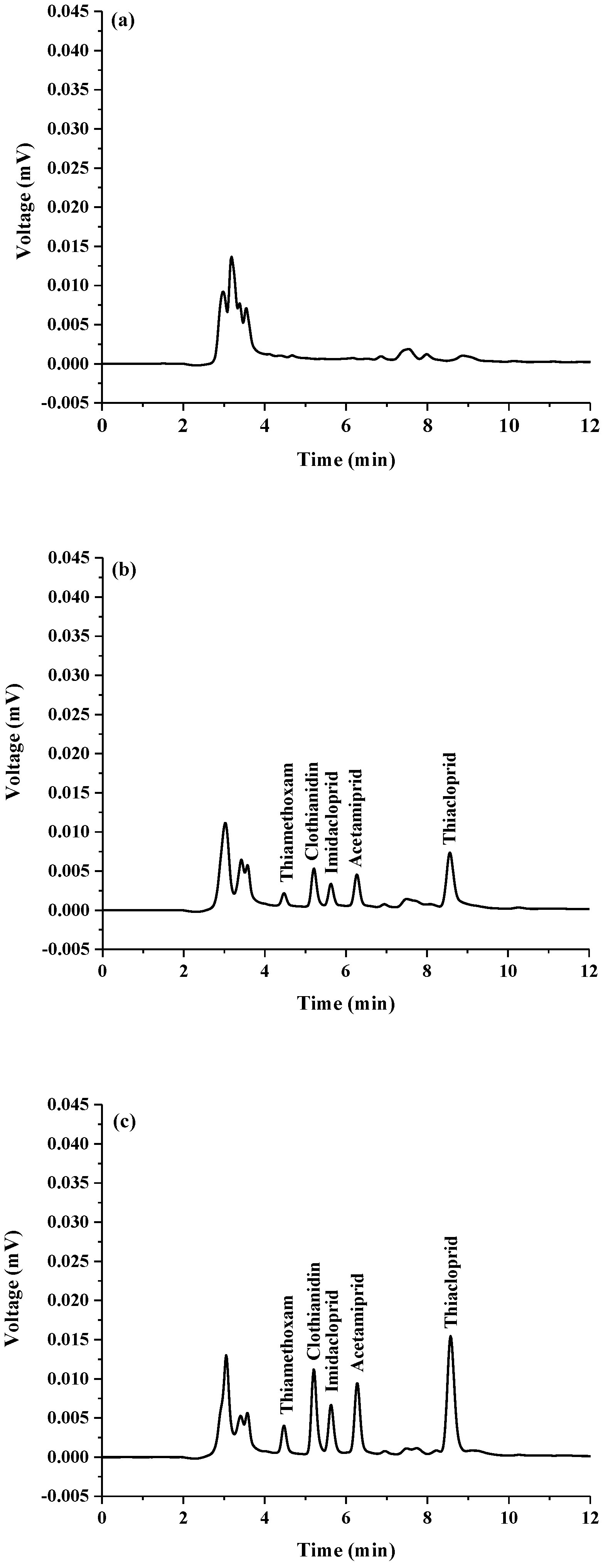
2.3. Application to Real Samples
2.4. Comparison with Other Sample Preparation Methods
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instrumentation
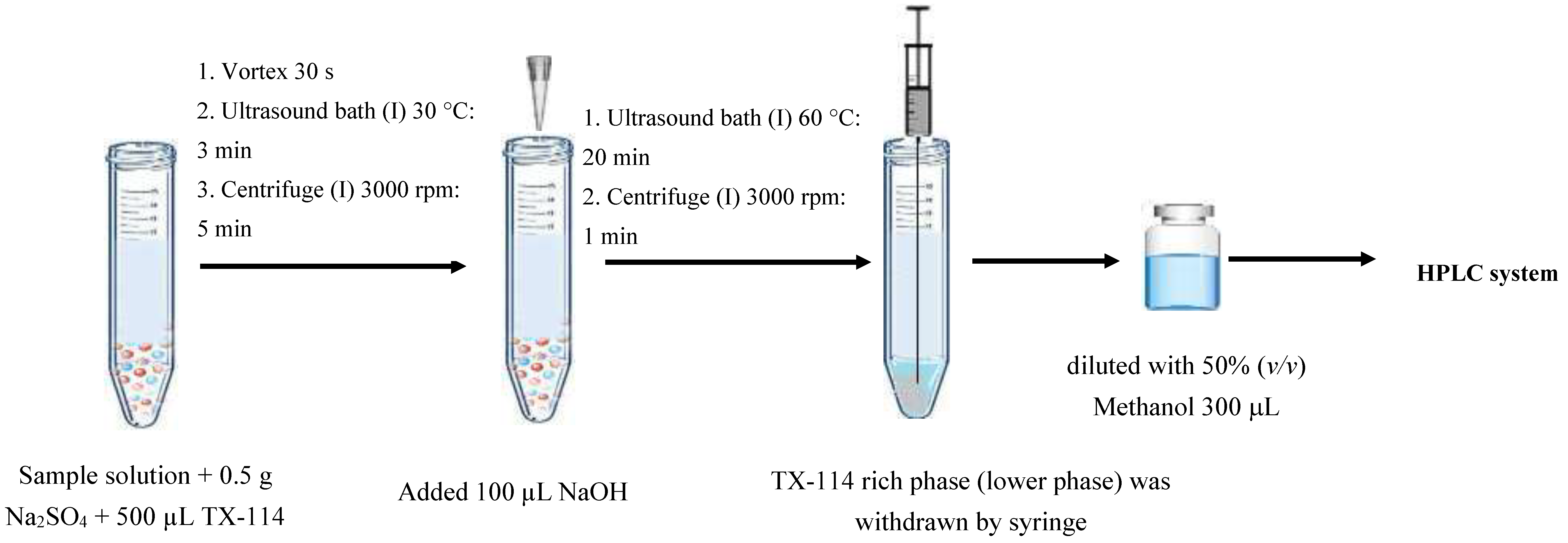
3.3. Ultrasonically-Modified Amended-Cloud Point Extraction Procedure
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Rodríguez-Cabo, T.; Casado, J.; Rodríguez, I.; Ramil, M.; Cela, R. Selective extraction and determination of neonicotinoid insecticides in wine by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A. 2016, 1460, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Taliansky-Chamudis, A.; Gómez-Ramírez, P.; León-Ortega, M.; García-Fernández, A.J. Validation of a QuECheRS method for analysis of neonicotinoids in small volumes of blood and assessment of exposure in Eurasian eagle owl (Bubo bubo) nestlings. Sci. Total Environ. 2017, 595, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Jovanov, P.; Guzsvány, V.; Lazić, S.; Franko, M.; Sakač, M.; Šarić, L.; Kos, J. Development of HPLC-DAD method for determination of neonicotinoids in honey. J. Food Compos. Anal. 2015, 40, 106–113. [Google Scholar] [CrossRef]
- Biever, R.C.; Hoberg, J.R.; Jacobson, B.; Dionne, E.; Sulaiman, M.; McCahon, P. ICON rice seed treatment toxicity to crayfish (Procambarus clarkii) in experimental rice paddies. Environ. Toxicol. Chem. 2003, 22, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Li, X.; Wang, X.; Shen, J.; Ding, S. Determination of neonicotinoid insecticides residues in bovine tissues by pressurized solvent extraction and liquid chromatography–tandem mass spectrometry. J. Chromatogr. B. 2011, 879, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, X.; Wang, J.; Cui, J.; Dong, A.J.; Zhao, H.T.; Zhang, L.W.; Wang, Z.Y.; Xu, R.B.; Li, W.J.; et al. Multi-residue method for determination of seven neonicotinoid insecticides in grains using dispersive solid-phase extraction and dispersive liquid–liquid micro-extraction by high performance liquid chromatography. Food Chem. 2012, 134, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, X.; Yin, X.; Wang, C.; Wang, Z. Dispersive liquid–liquid microextraction combined with sweeping micellar electrokinetic chromatography for the determination of some neonicotinoid insecticides in cucumber samples. Food Chem. 2012, 133, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Rancan, M.; Rossi, S.; Sabatini, A.G. Determination of Thiamethoxam residues in honeybees by high performance liquid chromatography with an electrochemical detector and post-column photochemical reactor. J. Chromatogr. A 2006, 1123, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Subhani, Q.; Huang, Z.-P.; Zhu, Z.-Y.; Liu, L.-Y.; Zhu, Y. Analysis of insecticide thiacloprid by ion chromatography combined with online photochemical derivatisation and fluorescence detection in water samples. Chin. Chem. Lett. 2014, 25, 415–418. [Google Scholar] [CrossRef]
- Vichapong, J.; Burakham, R.; Srijaranai, S. Vortex-assisted surfactant-enhanced-emulsification liquid–liquid microextraction with solidification of floating organic droplet combined with HPLC for the determination of neonicotinoid pesticides. Talanta 2013, 117, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Jovanov, P.; Guzsvány, V.; Franko, M.; Lazić, S.; Sakač, M.; Milovanović, I.; Nedeljković, N. Development of multiresidue DLLME and QuEChERS based LC–MS/MS method for determination of selected neonicotinoid insecticides in honey liqueur. Food Res. Int. 2014, 55, 11–19. [Google Scholar] [CrossRef]
- Regueiro, J.; Álvarez, G.; Mauriz, A.; Blanco, J. High throughput analysis of amnesic shellfish poisoning toxins in bivalve molluscs by dispersive solid-phase extraction and high-performance liquid chromatography using a monolithic column. Food Chem. 2011, 127, 1884–1891. [Google Scholar] [CrossRef]
- Pfaunmiller, E.L.; Paulemond, M.L.; Dupper, C.M.; Hage, D.S. Affinity monolith chromatography: A review of principles and recent analytical applications. Anal. Bioanal. Chem. 2013, 405, 2133–2145. [Google Scholar] [CrossRef] [PubMed]
- Guiochon, G. Monolithic columns in high-performance liquid chromatography. J. Chromatogr. A. 2007, 1168, 101–168. [Google Scholar] [CrossRef] [PubMed]
- Unger, K.K.; Skudas, R.; Schulte, M.M. Particle packed columns and monolithic columns in high-performance liquid chromatography-comparison and critical appraisal. J. Chromatogr. A. 2008, 1184, 393–415. [Google Scholar] [CrossRef] [PubMed]
- Seebunrueng, K.; Santaladchaiyakit, Y.; Soisungnoen, P.; Srijaranai, S. Catanionic surfactant ambient cloud point extraction and high-performance liquid chromatography for simultaneous analysis of organophosphorus pesticide residues in water and fruit juice samples. Anal. Bioanal. Chem. 2011, 401, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Santalad, A.; Srijaranai, S.; Burakham, R.; Glennon, J.D.; Deming, R.L. Cloud-point extraction and reversed-phase high-performance liquid chromatography for the determination of carbamate insecticide residues in fruits. Anal. Bioanal. Chem. 2009, 394, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Arain, S.S.; Kazi, T.G.; Arain, J.B.; Afridi, H.I.; Brahman, K.D.; Naeemullah. Preconcentration of toxic elements in artificial saliva extract of different smokeless tobacco products by dual-cloud point extraction. Microchem. J. 2014, 112, 42–49. [Google Scholar] [CrossRef]
- Yin, X.-B. Dual-cloud point extraction as a preconcentration and clean-up technique for capillary electrophoresis speciation analysis of mercury. J. Chromatogr. A. 2007, 1154, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhong, S.; Fang, K.; Qian, Z.; Chen, J. Determination of cadmium (II), cobalt (II), nickel (II), lead (II), zinc (II), and copper (II) in water samples using dual-cloud point extraction and inductively coupled plasma emission spectrometry. J. Hazard. Mater. 2012, 239–240, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.-B.; Guo, J.-M.; Wei, W. Dual-cloud point extraction and tertiary amine labeling for selective and sensitive capillary electrophoresis-electrochemiluminescent detection of auxins. J. Chromatogr. A. 2010, 1217, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Nong, C.; Niu, Z.; Li, P.; Wang, C.; Li, W.; Wen, Y. Dual-cloud point extraction coupled to high performance liquid chromatography for simultaneous determination of trace sulfonamide antimicrobials in urine and water samples. J. Chromatogr. B. 2017, 1051, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Arain, S.A.; Kazi, T.G.; Afridi, H.I.; Abbasi, A.R.; Panhwar, A.H.; Naeemullah; Shanker, B.; Arain, M.B. Application of dual-cloud point extraction for the trace levels of copper in serum of different viral hepatitis patients by flame atomic absorption spectrometry: A multivariate study. Spectrochim. Acta A. 2014, 133, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, G.; Han, Q. Determination of trace uranium by resonance fluorescence method coupled with photo-catalytic technology and dual cloud point extraction. Spectrochim. Acta A. 2016, 169, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Santaladchaiyakit, Y.; Srijaranai, S. A simplified ultrasound-assisted cloud-point extraction method coupled with high performance liquid chromatography for residue analysis of benzimidazole anthelmintics in water and milk samples. Anal. Methods 2012, 4, 3864–3873. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.-H.; Zhang, Y.-Y.; Liu, W.-T.; Bi, K.-S. Determination of arbidol in rat plasma by HPLC–UV using cloud-point extraction. J. Chromatogr. B. 2007, 856, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, Y.-Q.; He, B.; Yuan, C.-G.; Shen, D.-Z.; Shao, J.; Jiang, G.-B. Determination of estrogens in water by HPLC–UV using cloud point extraction. Talanta 2006, 70, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Appusamy, A.; John, I.; Ponnusamy, K.; Ramalingam, A. Removal of crystal violet dye from aqueous solution using triton X-114 surfactant via cloud point extraction. Eng. Sci. Technol. Int. J. 2014, 17, 137–144. [Google Scholar] [CrossRef]
- Khan, M.; Kazi, T.G.; Afridi, H.I.; Bilal, M.; Akhtar, A.; Ullah, N.; Khan, S.; Talpur, S. Application of ultrasonically modified cloud point extraction method for simultaneous enrichment of cadmium and lead in sera of different types of gallstone patients. Ultrason. Sonochem. 2017, 39, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kazi, T.G.; Shah, F.; Afridi, H.I.; Baig, J.A.; Soomro, A.S. Cloud point extraction and flame atomic absorption spectrometric determination of cadmium and nickel in drinking and wastewater samples. J. AOAC Int. 2013, 96, 447–452. [Google Scholar]
- Vichapong, J.; Burakham, R.; Srijaranai, S.; Grudpan, K. Room temperature imidazolium ionic liquid: A solvent for extraction of carbamates prior to liquid chromatographic analysis. Talanta 2011, 84, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Sunganthi, A.; Bhuvaneswari, K.; Ramya, M. Determination of neonicotinoid insecticide residues in sugarcane juice using LCMSMS. Food Chem. 2018, 241, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Farajzadeh, M.A.; Bamorowat, M.; Mogaddam, M.R.A. Ringer tablet-based ionic liquid phase microextraction: Application in extraction and preconcentration of neonicotinoid insecticides from fruit juice and vegetable samples. Talanta 2016, 160, 211–216. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Insecticide | Linear Equation | Linearity (µg mL−1) | r2 | Limit of Detection (LOD) (µg mL−1) | Limit of Quantification (LOQ) (µg mL−1) | % Relative Standard Deviation (RSD) | Enrichment Factor (EF) | |
|---|---|---|---|---|---|---|---|---|
| tR | Peak Area | |||||||
| Thiamethoxam | y = 239217x + 1546.9 | 0.005–0.7 | 0.9978 | 0.001 | 0.003 | 1.02 | 3.77 | 100 |
| Clothianidin | y = 834910x + 1822.5 | 0.005–0.7 | 0.9997 | 0.001 | 0.005 | 0.96 | 3.89 | 200 |
| Imidacloprid | y = 505089x − 178.27 | 0.005–0.7 | 0.9999 | 0.0003 | 0.001 | 1.26 | 4.12 | 333 |
| Acetamiprid | y = 805535x + 347.71 | 0.005–0.7 | 0.9999 | 0.0005 | 0.002 | 1.67 | 4.54 | 20 |
| Thiacloprid | y = 2000006x + 5418.3 | 0.005–0.7 | 0.9992 | 0.002 | 0.006 | 1.92 | 0.01 | 100 |
| Analyte | Spiked (µg mL−1) | Surface Water I (n = 3) | Surface Water II (n = 3) | Surface Water III (n = 3) | |||
|---|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| Thiamethoxam | 0.00 | ND | - | ND | - | ND | - |
| 0.05 | 101.89 | 2.45 | 89.50 | 1.21 | 98.12 | 0.95 | |
| 0.10 | 115.33 | 2.61 | 111.52 | 0.70 | 101.64 | 6.63 | |
| 0.25 | 116.72 | 2.14 | 115.52 | 1.84 | 117.56 | 4.20 | |
| Clothianidin | 0.00 | ND | - | ND | - | ND | - |
| 0.05 | 92.45 | 8.21 | 89.08 | 1.74 | 68.78 | 1.77 | |
| 0.10 | 112.22 | 1.57 | 100.31 | 1.55 | 91.50 | 9.63 | |
| 0.25 | 113.08 | 4.09 | 119.63 | 0.86 | 106.43 | 4.82 | |
| Imidacloprid | 0.00 | ND | - | ND | - | ND | - |
| 0.05 | 90.43 | 9.13 | 85.30 | 1.02 | 95.97 | 3.48 | |
| 0.10 | 106.23 | 1.34 | 95.04 | 1.18 | 90.64 | 9.35 | |
| 0.25 | 106.90 | 2.73 | 110.15 | 0.75 | 98.67 | 4.84 | |
| Acetamiprid | 0.00 | ND | - | ND | - | ND | - |
| 0.05 | 90.27 | 3.89 | 87.76 | 0.26 | 82.31 | 2.24 | |
| 0.10 | 110.31 | 0.64 | 100.07 | 1.06 | 92.19 | 9.08 | |
| 0.25 | 112.78 | 1.64 | 111.41 | 0.31 | 103.14 | 4.06 | |
| Thiacloprid | 0.00 | ND | - | ND | - | ND | - |
| 0.05 | 82.40 | 5.45 | 64.54 | 2.50 | 60.11 | 9.28 | |
| 0.10 | 88.64 | 0.55 | 88.64 | 3.99 | 88.64 | 13.71 | |
| 0.25 | 95.71 | 0.34 | 102.46 | 3.09 | 84.24 | 6.76 | |
| Extraction Method | Analytical Technique | Linear Range | Recovery (%) | LOD | LOQ | EFs | Reference |
|---|---|---|---|---|---|---|---|
| DLLME | HPLC-DAD | LOQ–100.0 µg kg−1 | 73.4–118.3 | 1.5–2.5 µg kg−1 | 5.0–7.5 µg kg−1 | 66.6–105.9 | [3] |
| QuEChERS | HPLC-DAD | LOQ–100.0 µg kg−1 | 73.8–89.9 | 2.0–2.5 µg kg−1 | 5.0–10.0 µg kg−1 | 72.2–85.2 | [3] |
| DSPE (QuEChERS) | LC-MS/MS | 0.005–0.5 µg mL−1 | 62.0–129.93 | 0.0007–0.002 µg mL−1 | 0.002–0.005 µg mL−1 | - | [32] |
| DLLME | LC-MS/MS | LOQ–100.0 µg L−1 | 69.2–113.4 | 0.5–1.5 µg L−1 | 1.0–5.0 µg mL−1 | 67.8–95.0 | [12] |
| QuEChERS | LC-MS/MS | LOQ–100.0 µg L−1 | 71.8–94.9 | 1.0–2.5 µg L−1 | 2.5–10.0 µg mL−1 | 51.3–96.2 | [12] |
| DSPE and DLLME | HPLC-DAD | 0.02–4.50 µg mL−1 | 76–123 | 0.002–0.005 µg mL−1 | 0.007–0.018 µg mL−1 | - | [6] |
| RTIL-LPME | HPLC-DAD | 0.41–5000 ng mL−1 | 69.2–113.4 | 0.12–0.33 ng mL−1 | 0.41–1.1 ng mL−1 | 655–843 | [33] |
| Amended CPE | HPLC-UV | 0.005–0.7 µg mL−1 | 60.12–117.57 | 0.0003–0.002 µg mL−1 | 0.001–0.006 µg mL−1 | 20–333 | This work |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kachangoon, R.; Vichapong, J.; Burakham, R.; Santaladchaiyakit, Y.; Srijaranai, S. Ultrasonically Modified Amended-Cloud Point Extraction for Simultaneous Pre-Concentration of Neonicotinoid Insecticide Residues. Molecules 2018, 23, 1165. https://doi.org/10.3390/molecules23051165
Kachangoon R, Vichapong J, Burakham R, Santaladchaiyakit Y, Srijaranai S. Ultrasonically Modified Amended-Cloud Point Extraction for Simultaneous Pre-Concentration of Neonicotinoid Insecticide Residues. Molecules. 2018; 23(5):1165. https://doi.org/10.3390/molecules23051165
Chicago/Turabian StyleKachangoon, Rawikan, Jitlada Vichapong, Rodjana Burakham, Yanawath Santaladchaiyakit, and Supalax Srijaranai. 2018. "Ultrasonically Modified Amended-Cloud Point Extraction for Simultaneous Pre-Concentration of Neonicotinoid Insecticide Residues" Molecules 23, no. 5: 1165. https://doi.org/10.3390/molecules23051165